
Abstract
Alkyl moieties which can retain target analytes due to their lipophilicity are important in sample preparation. In this work, hexadecyl-functionalized magnetic core-shell microspheres (Fe3O4@SiO2-C16) was successfully prepared by one-pot sol–gel method and used for magnetic solid-phase extraction of polychlorinated biphenyls (PCBs) in environmental water samples. Optimized preparation method was achieved by altering the adding moment of hexadecyl-silane. The resultant materials were systematically characterized by scanning electron microscope, transmission electron microscope, Fourier transform infrared spectroscopy, energy dispersive X-ray spectrometry, tensionmeter, and vibrating sample magnetometer. The results demonstrated that the optimized adsorbent exhibited core-shell structure, superparamagnetic (66 emu/g), and extremely hydrophobic (water contact angle of 122°) properties. To evaluate the extraction performance, the prepared material coupled with gas chromatography-triple quadrupole mass spectrometry (GC-MS/MS) was applied to determinate PCBs. The extraction conditions were optimized. Under the optimal conditions, the proposed method showed a good linearity range of 1–100 ng L−1 with correlation coefficients (R) of 0.9989–0.9993. Based on a signal-to-noise ratio of 3 and 10, the limits of detection (LODs) and limits of quantification (LOQs) were in the range 0.14–0.27 and 0.39–0.91 ng L−1, respectively. The intra- and inter-day relative standard deviations (RSDs) were less than 9.06%. The absolute recoveries of PCBs in spiked real water samples were in the range of 75.17 to 101.20%. Additionally, reusability and batch-to-batch reproducibility of the resultant material were acceptable with RSDs less than 5.64 and 3.25%, respectively.

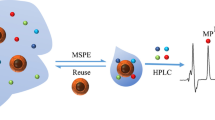
The synthesis procedure of Fe3O4@SiO2-C16 and determination of PCBs in water sample 129 × 50 mm (300 × 300 DPI)
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Magnetic solid-phase extraction (MSPE) as a new mode of SPE has gained increasing attention in sample preparation in recent years [1–3]. Compared with traditional column SPE, magnetic adsorbents are commonly incubated directly with a sample, which not only avoid the cumbersome packing of adsorbent into column and time-consuming loading of large-volume samples but also shortens the extraction time and enhances extraction efficiency due to the increased contact between adsorbents and analytes [3, 4]. Moreover, suspended magnetic adsorbents can be facilely isolated from the solution by applying an external magnetic field, successfully circumventing high-speed centrifugation, or tedious filtration process [5, 6]. Consequently, lots of functionalized magnetic materials have been developed and used.
In traditional SPE, alkyl moieties (e.g., C18) bonded silica is one of the most widely used adsorbents and can be applied for the pretreatment of most hydrophobic analytes based on their lipophilic character [7–9]. Hence, alkyl-functionalized magnetic silica spheres are also favored by a number of researchers. For instance, C18-functionalized magnetic silica particles have been synthesized and applied for extraction and preconcentration of ergosterol in cigarettes [10], endocrine disruptors in milk samples [11], volatile organic metabolites in human urine [12], and polycyclic aromatic hydrocarbons, pesticides, and plasticizers in aqueous samples [13–16]. Additionally, C18/C8-functionalized magnetic silica was demonstrated to be an efficient capture probe for purification of veterinary drug residues [17]. In the preparation of these materials, magnetic fraction (usually Fe3O4) was first coated with silica by the hydrolysis and condensation of tetraethoxysilane, and then further modified with alkyl-siloxane or trichlorosilane under harsh conditions, such as heating, nitrogen protection, or usage of toxic reagent (e.g., toluene). Such kind of time-consuming and cumbersome process prompted us to explore other more convenient, rapid, and gentle preparation method for synthesizing effective hydrophobic adsorbents.
Polychlorinated biphenyls (PCBs) are a group of chlorinated hydrocarbons in which various degrees of hydrogen atoms on a biphenyl were substituted by chlorine atoms. PCBs were widely used decades ago, but its manufacturing has been banned in late 1970s because of its high toxicity, resistance to degradation, bioaccumulation, and long-distance transfer [18]. As such, PCBs were classified as probably carcinogenic to humans by the International Agency for Research on Cancer, and also included in Stockholm Convention as persistent organic pollutants in 2001 [19]. Although prohibitions on the use were carried out many years ago, many studies have shown that PCBs are ubiquitous in the environment [20, 21]. Consequently, the content of PCBs in different matrices such as environmental waters is limited under legislation in many countries and organizations [22]. Therefore, analytical methods for determining trace amount of PCBs in environment are of great significance.
Owing to the trace and ultra-trace levels of PCBs in environmental waters, sample preparation before their detection is inevitable [23–25]. Various sample preparation methods have been reported for the extraction and enrichment of PCBs, such as liquid-liquid microextraction (LLME) [26, 27], solid-phase microextraction (SPME) [28–31], and SPE [32, 33], among which SPE is still one of the preferred pretreatment technique due to its simplicity, effectivity, and versatility. In recent years, with MSPE becoming a very useful alternative to traditional SPE, a variety of functionalized magnetic adsorbents have been explored for the enrichment of PCBs, including Fe3O4@MIL-100 [34], magnetic graphene composite [35–37], magnetic multiwalled carbon nanotube [38], Fe3O4@chitosan-poly(m-phenylenediamine) [39], and palmitate- or oleate-coated magnetic nanoparticles [40]. Nevertheless, complex synthetic procedures of materials restricted their routine usage. In this sense, simple, rapid, and efficient methods for the pretreatment of PCBs in the environmental waters are increasingly in demand.
Conventional detection methods for PCBs are mainly chromatographic techniques such as high-performance liquid chromatography (HPLC) and gas chromatography (GC) coupled with ultraviolet (UV), electron capture detection (ECD), or mass spectrometry (MS) [29, 31, 36, 38]. HPLC-UV and GC-ECD are lacking of selectivity for target analytes, and might be easily interfered by other substances causing inaccurate results [29, 31]. Although GC-MS is a selective detector and lots of researches were carried out with the equipment, the analytical results still have certain uncertainty, especially when the to-be-detected sample is very complicated [36, 38]. To acquire reliable results, tandem mass spectrometry especially GC-MS/MS has been used for the determination of various organic pollutants including PCBs based on its powerful qualitative and quantitative abilities [34, 35, 40].
Therefore, the aim of this study was to develop an inexpensive and effective sample preparation for the accurate determination of PCBs in real water samples. Hexadecyl-functionalized magnetic microsphere (Fe3O4@SiO2-C16) was prepared with a convenient, rapid, and gentle one-pot method and used as magnetic adsorbent for the extraction and preconcentration of PCBs prior to GC-MS/MS determination. The preparation process was optimized and the prepared magnetic microspheres were systematically characterized. Seven PCBs including PCB28, PCB52, PCB101, PCB118, PCB138, PCB153, and PCB180 were selected as model analytes to evaluate the extraction performance of Fe3O4@SiO2-C16. Various experimental parameters affecting the extraction efficiencies were investigated. Reusability and reproducibility of Fe3O4@SiO2-C16 were also explored. Moreover, the performances of PCBs analysis were compared among other MSPE-GC-MS/MS methods and US EPA 3520C method and US EPA 3535 method. Finally, the application of the prepared materials for the determination of PCBs in environmental water samples was successfully demonstrated.
Experimental
Reagents and materials
Ferric chloride (FeCl3·6H2O), ethylene glycol (EG), ethylene diamine (ED), sodium acetate (NaAc), ethanol (EtOH), aqueous ammonia solution (NH3·H2O, 25 wt%), and hydrochloric acid (HCl) were all of analytical reagent grade and supplied by Shanghai General Chemical Reagent Factory, China. Tetraethyl orthosilicate (TEOS, 98%) and hexadecyltrimethoxysilane (C16-TMOS, 85%) were purchased from Shanghai Aladdin Chemical Reagent Co., Ltd., China. Acetone, n-hexane, and dichloromethane of GC grade were purchased from TEDIA Company (Fairfield, OH, USA). Anhydrous sodium sulfate (Na2SO4) obtained from Sigma-Aldrich (Oakville, ON, Canada) was dried at 450 °C for 4 h before use. Ultrapure water produced with a Heal Force water purification system (Cannex Analytic Instrument, Shanghai, China) was used. PCBs standard solution (10 mg L−1, in isooctane) and 13C-PCB 141 (internal standard, I.S.) were obtained from o2si (Charleston, SC, USA). PCBs standard solution contains 2,4,4′-trichlorobiphenyl (PCB 28), 2,2′,5,5′-tetrachlorobiphenyl (PCB 52), 2,2′,4,5,5′-pentachlorobiphenyl (PCB 101), 2,3′,4,4′,5-pentachlorobiphenyl (PCB 118), 2,2′,3,4,4′,5-hexachlorobiphenyl (PCB 138), 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB 153), and 2,2′,3,4,4′,5,5′-heptachlorobiphenyl (PCB 180). The PCBs and I.S. stock solutions were diluted in acetone at a concentration of 0.05 mg L−1. All the stock solutions were stored at −4 °C in darkness.
The rain water, river water, and pond water were chosen for real environmental samples. The rain water was collected from Wuhan after several sunny days; the river water was taken from Yangtze River, China; and the pond water was obtained from the campus of our college. All the water samples were filtered through a 0.45 μm membrane and stored in brown bottles at −4 °C until analysis.
Preparation of Fe3O4@SiO2-C16
The Fe3O4 was prepared by a solvothermal reduction method according to previous study [41]. FeCl3·6H2O (5 g) was dissolved in 100 mL EG. Then, 15 g NaAc and 50 mL ED were added in the solution and vigorously stirred for 30 min. The mixture was poured into a 200-mL teflon-lined stainless-steel autoclave and heated at 200 °C for 8 h. After cooling down to room temperature, the obtained Fe3O4 was washed with EtOH and water for several times and vacuum-dried at 60 °C for 6 h.
With the purpose of simplifying the synthetic procedures, one-pot sol-gel method was proposed for facile preparation of Fe3O4@SiO2-C16 in this work which integrating the immobilization of C16 with the sol–gel process of TEOS into only one process. One gram of resultant Fe3O4 was ultrasonically dispersed in 0.1 M HCl for about 10 min. The black precipitate was washed with deionized water for three times. Then, the washed Fe3O4 was dispersed in a mixture of 400 mL EtOH and 100 mL H2O in a 1000-mL three-necked bottle. After adding 4 mL NH3·H2O, the 1 mL TEOS which was dissolved in 20 mL EtOH was added in the mixture drop by drop with stirring vigorously at room temperature. Subsequently, the 0.5 mL C16-TMOS (dissolved in 20 mL EtOH) was added in the mixture drop by drop at different time intervals (0, 3, 6, and 10 h) after the addition of TEOS. After stirring vigorously at room temperature for another 6 h, the obtained magnetic microspheres were magnetically separated and washed with EtOH for several times, and then dried at 60 °C in vacuum. For clarity, the obtained Fe3O4@SiO2-C16 microspheres prepared by different time intervals of adding C16-TMOS were labeled as Fe3O4@SiO2-C16-1, Fe3O4@SiO2-C16-2, Fe3O4@SiO2-C16-3, and Fe3O4@SiO2-C16-4, respectively.
Instrumentation and analytical conditions
The morphology of the adsorbent was observed by field-emission scanning electron microscopy (FE-SEM, SU8010, Hitachi, Japan) and transmission electron microscope (TEM, CM12, Philips, Netherlands). Fourier transform infrared spectroscopy (FT-IR) spectra were performed with a Thermo Nicolet 670 FT-IR instrument (Boston, MA, USA). Energy dispersive X-ray (EDX) spectroscopy was also determined by SU8010 FE-SEM. The water contact angle was determined by X100 tensionmeter (KRUSS, German). Magnetization curves were determined with a PPMS-9 vibrating sample magnetometer (VSM, Quantum Design, San Diego, CA, USA).
PCBs samples were analyzed by gas chromatography (Agilent Technologics 7890B, USA) coupled with triple quadrupole mass spectrometry (Agilent Technologics 7000C, USA). The gas chromatography separation was carried out on a CP-Sil 8 CB capillary column (Agilent Technologies, 60 m × 250 μm × 0.12 μm). The oven temperature was programmed from 150 °C at 20°C min−1to 200 °C, then increased to 230 °C at a rate of 5 °C min−1 and held for 1 min, finally ramped at 10 °C/min to 290 °C and held for 5 min. The total analysis time was 20.5 min. The injection volume was 1.0 μL, and pulse splitless injection mode was used. Helium (purity ≥99.999%) was used as carrier gas and quench gas at a flow rate of 1.0 and 2.25 mL min−1, respectively. The temperatures of the injection port, ion source, and transfer line were 280, 250, and 280 °C, respectively. MS was operated in EI mode with standard electron energy of 70 eV. The solvent cut time was 5 min. Nitrogen (purity ≥99.999%) was used as collision gas at 1.5 mL min−1. Target analytes were quantified by multiple reaction monitoring (MRM) mode. The retention time, qualitative and quantitative ion pair, and collision energy were presented in Table S1 in the Electronic Supplementary Material (ESM).
MSPE procedure
Fifty milligrams of adsorbent was rinsed by 2 mL acetone and dispersed into 30 mL spiked PCBs solution (10 ng L−1) or real environmental waters. The mixture was sonicated for 10 min. With the aid of an external magnet, the adsorbent was isolated from the suspension immediately. The supernatant was discarded and the adsorbent was eluted with 3 mL of n-hexane by fierce vortex for 1 min. Similarly, the desorption solution was collected by the assist of an external magnet and dehydrated by some amount of anhydrous sodium sulfate. Subsequently, the solution was concentrated into 0.3 mL under a gentle stream of nitrogen. Finally, 1 μL of the concentrated solution was injected into GC-MS/MS instrument for analysis. The internal standard solution (13C-PCB 141) was added into the spiked water samples after MSPE procedure for calculating recovery. All experiments were repeated for three times.
Results and discussion
Characterization of materials
The morphologies of the prepared magnetic particles were observed by FE-SEM and TEM (Fig. 1). SEM images of Fe3O4, Fe3O4@SiO2-C16-1, Fe3O4@SiO2-C16-2, Fe3O4@SiO2-C16-3, and Fe3O4@SiO2-C16-4 are showed in panels a–e of Fig. 1, respectively. Fe3O4 particles were nearly spherical and homogeneous with a mean diameter of about 180 nm (Fig. 1a). After coating with TEOS and C16-TMOS, the sizes of the magnetic particles become larger with the mean diameters of 220 nm (Fig. 1b), 230 nm (Fig. 1c), 210 nm (Fig. 1d), and 210 nm (Fig. 1e), respectively. Although the particle size of Fe3O4@SiO2-C16 has not changed much under the four different time intervals, their morphology has distinct differences. The particles of Fe3O4@SiO2-C16-1 and Fe3O4@SiO2-C16-2 tend to crosslink with each other and are coated with some floccule (Fig. 1b, c). However, the particles of Fe3O4@SiO2-C16-3 and Fe3O4@SiO2-C16-4 are more dispersed and sphere-like (Fig. 1d, e). TEM image of Fe3O4@SiO2-C16-3 is further shown in Fig. 1f. Obvious core-shell structure could be seen in the particles, and the coating layer was about 20 nm. The results indicate that Fe3O4 can be successfully covered with a thin layer of coating under one-pot sol–gel procedure, and the time of adding C16-TMOS has great influence on the morphology of the resultant materials. It has to be mentioned here that only Fe3O4@SiO2-C16-3 was characterized in the following sections, which was abbreviated as Fe3O4@SiO2-C16.

SEM images of Fe3O4 (a), Fe3O4@SiO2-C16-1 (b), Fe3O4@SiO2-C16-2 (c), Fe3O4@SiO2-C16-3 (d) and Fe3O4@SiO2-C16-4 (e) and TEM image of Fe3O4@SiO2-C16-3 (f)
FT-IR spectra of Fe3O4@SiO2-C16 as well as naked Fe3O4 were measured to investigate the characteristics of the resultant materials (Fig. 2a). The peaks at 3420 and 560 cm−1 that appeared in both curves can be attributed to the O–H and Fe–O vibrations, respectively. After coating with TEOS and C16-TMOS, the band at 1080 cm−1 is characteristic for Si–O, revealing the presence of SiO2. The peaks at 2851 and 2921 cm−1 are assigned as the stretching vibration of C-H group which indicates that alkyl group has been successfully modified on the surface of magnetic particles. The chemical composition of Fe3O4@SiO2-C16 was further examined by EDX. As shown in Fig. 2b, the peaks of Fe, Si, and O can be obviously observed. The peak of C can also be detected although its intensity is relatively weak. These results demonstrated that the strategy reported here is feasible for simultaneous coating of SiO2 and modification of hexadecyl group on the surface of Fe3O4 microparticles.

FT-IR spectra of Fe3O4 (a) and EDX spectrum of Fe3O4@SiO2-C16 (b)
The hydrophobicity of Fe3O4 and Fe3O4@SiO2-C16 was evaluated by water contact angle measurement. As shown in ESM Fig. S1, the water drop disperses into Fe3O4 immediately, showing that Fe3O4 is hydrophilic. Conversely, the contact angle of Fe3O4@SiO2-C16 is about 122°, which indicates that the Fe3O4@SiO2-C16 was hydrophobic. These results further prove the existence of hexadecyl group on the surface of the prepared material, and Fe3O4@SiO2-C16 has been successfully synthesized.
The magnetic properties of Fe3O4 and Fe3O4@SiO2-C16 were verified by VSM. Magnetization curves of the two materials are shown in Fig. S2 (see ESM). Both curves have no magnetic hysteresis loops, which reveal that they are superparamagnetic. The saturation magnetization values of Fe3O4 and Fe3O4@SiO2-C16 are calculated to be 83 and 66 emu g−1, respectively. Although saturation magnetization value decreases lightly after coating with TEOS and C16-TMOS, the magnetization of Fe3O4@SiO2-C16 is still stronger than most of magnetic adsorbents prepared in previous reports [34, 36, 39]. Therefore, Fe3O4@SiO2-C16 microspheres can be rapidly and efficiently separated from the aqueous solution with the help of an external magnet, which would benefit the process of sample preparation.
Comparison in the extraction efficiency of different adsorbents
The extraction efficiency of different adsorbents, including Fe3O4, Fe3O4@SiO2, Fe3O4@SiO2-C16-1, Fe3O4@SiO2-C16-2, Fe3O4@SiO2-C16-3, and Fe3O4@SiO2-C16-4, was compared by the recoveries of seven PCBs, Fe3O4@SiO2 of which was prepared under the same conditions without adding C16-TMOS. As shown in ESM Fig. S3, naked Fe3O4 has little extraction ability for PCBs. Fe3O4@SiO2 shows better extraction ability than Fe3O4, but the highest recovery of PCBs is still below 40%. On the contrary, all of Fe3O4@SiO2-C16 microspheres display good extraction capacities for PCBs with the recoveries higher than 64%. Additionally, it is interesting to find that the recoveries are varied among the four Fe3O4@SiO2-C16, and Fe3O4@SiO2-C16-3 showed the best extraction efficiency. It can be explained by the different densities of hexadecyl group on the surface of the four adsorbents caused by the various preparation processes. In this way, Fe3O4@SiO2-C16-3 was chosen for the following experiments, and it was written as Fe3O4@SiO2-C16 for short.
Optimization of MSPE conditions
To achieve the best extraction efficiency of seven indicator PCBs, several parameters including amount of the adsorbent, desorption solvent, extraction time, desorption time, and salt concentration were evaluated (Fig. 3). Because PCBs are non-polar compounds and pH has limited effect on their extraction, the effect of pH was not investigated in this work.

The effect of amount of the adsorbent (a), type of desorption solvent (b), volume of desorption solvent (c), extraction time (d), desorption time (e), and salt concentration (f) on the extraction efficiency of PCBs in ultrapure water (spiked at 10 ng L−1). Error bar shows the standard deviation (n = 3)
The amount of sorbents was evaluated by varying sorbent dosage at 10, 30, 50, and 70 mg. The result is shown in Fig. 3a. Generally, the recovery increased with the rise of adsorbent dosage. Therefore, from 10 to 30 mg, the recovery of all PCBs markedly increased. The recoveries obtained by 50 mg adsorbent were a little higher than 30 mg and almost the same as by 70 mg. Thus, 50 mg adsorbent (the recovery ranged from 88.5 to 98.4%) was used for the following experiments.
Considering the characteristics of adsorbent and PCBs, three kinds of desorption solvent (n-hexane, acetone, dichloromethane) were investigated. As shown in Fig. 3b, it is clearly that n-hexane achieved the best desorption efficiency among the studied solvents. It can be explained by the similar chain structure of n-hexane and C16-functionalized adsorbent. Hence, n-hexane was used as the optimal desorption solvent. Additionally, the optimal volume of n-hexane was also investigated. As shown in Fig. 3c, 3 mL of n-hexane was enough to obtain a good result, so it was employed in the following experiments.
The extraction time investigation was carried out by increasing the sonicated time. Figure 3d shows the effect of extraction time on the recovery. The recoveries of PCBs increased in the range from 5 to 10 min. Whereafter, there was no significant change from 10 to 20 min. It was clear that extraction equilibrium was achieved at 10 min. Hence, 10 min was chosen as the optimal extraction time.
The effect of desorption time was investigated by altering the vortex time from 1 to 5 min (Fig. 3e). It was found out that the desorption time had no significant influence on the recovery. The desorption equilibrium can be achieved fast after vortexing 1 min. So, 1 min was chosen as the desorption time for this study.
The changing of ionic strength can affect the solubility of target analytes in aqueous solution and their interaction with adsorbent. The concentration of NaCl from 0 to 80 mM was added in the extraction solution to evaluate the effect of ionic strength. The result shows that the recovery decreased dramatically with the increase of salt concentration (Fig. 3f). This result can be attributed to the “oil effect” [41, 42]. The existence of salt would reduce the interaction PCBs and Fe3O4@SiO2-C16, leading to the poor extraction efficiency. Therefore, no salt was added in the following experiments.
Reusability and reproducibility of Fe3O4@SiO2-C16
Reusability and reproducibility are important performance of in-house prepared magnetic adsorbent. To evaluate the recycling ability, the regenerated Fe3O4@SiO2-C16 washed twice by hexane after each MSPE was used again to extract PCBs under the optimized condition. As shown in ESM Table S2, the recoveries of PCBs have no obviously change among six recycles with the relative standard deviations (RSDs) less than 5.64%. The batch-to-batch reproducibility of the adsorbent was investigated by preparing three batches of Fe3O4@SiO2-C16 under the same condition. The RSDs of the recoveries of PCBs were less than 3.25% (Table S2, see ESM). All the results indicated that the prepared Fe3O4@SiO2-C16 was reusable and reproducible.
Analytical performance
The analytical performance of Fe3O4@SiO2-C16 was evaluated under the optimized condition. The linearity, limit of detection (LOD), limit of quantification (LOQ), and repeatability were investigated (Table 1 and ESM Table S3). For the construction of the calibration curves, triplicate measurements were performed, and the calibration curves were generated by plotting the mean peak area ratio (y, targeted analytes/I.S.) versus analytes concentration (x) in the range of 1–100 ng L−1. As shown in Table 1, the linear correlation coefficients (R) values of seven curves are higher than 0.9989. The LODs and LOQs based on a signal-to-noise ratio of 3 and 10 are in the range of 0.14–0.27 and 0.39–0.91 ng L−1, respectively.
The reproducibility of the method was determined by the intra-day and inter-day precisions (Table S3, see ESM). The intra-day and inter-day precisions were investigated at different concentrations (1, 10, and 50 ng L−1). The intra-day and inter-day RSDs were below 6.87 and 9.06%, respectively. The results demonstrated that the precision of the present method was acceptable.
The analytical performance of this method was compared to the previous reports applying MSPE-GC-MS/MS to analyze PCBs in environmental waters. According to Table 2, sensitivity of the current method was comparable with other approaches. However, the consumption of desorption solvent was less than the previous reported materials, and the dosage of water sample was moderate. Moreover, the saturation magnetization value of Fe3O4@SiO2-C16 was higher than most of other magnetic adsorbents, which favors the quick separation of adsorbents from the aqueous solution. In order to further evaluate the pretreatment of the proposed method, we did a comprehensive comparison with Environmental Protection Agency (EPA) pretreatment methods (EPA 3520C [43] and EPA 3535 [44]) which are recommended by the EPA 8082A method [45]. Compared to US EPA method 3520C liquid-liquid extraction which requires 1 L of the sample and uses 300–500 mL of methylene chloride to extract 18–24 h continuously, our method only need 30-mL sample, extract 10 min and use 3 mL of n-hexane to elute. As to US EPA method 3535 solid-phase extraction, the SPE method requires a special SPE device and SPE cartridges. The SPE device is not portable and need electricity. Besides, the water passes through the SPE cartridges (e.g., C18 SPE cartridge) at 3–5 mL min−1 and dried by vacuum for 30–60 min, resulting more than 50-min extraction [33, 46]. Moreover, this method may not be appropriate for aqueous samples with greater than 1% suspended solids, as such samples can be difficult to filter as a result of clogging the column. In contrast, none special device is essential in the proposed method except a small portable magnet which can be applied for sample preparation on site. The adsorbent were dispersed into water sample which made the extraction time shorten to 10 min and was appropriate for all type of water samples. Furthermore, one “blank” sample spiked with PCBs (standards concentration was 10.0 ng L−1) was separately analyzed in triplicate using the optimized MSPE method and the SPE and LLE methods. The recoveries of the PCBs using the improved MSPE method were 75.17–101.20%, with RSDs of less than 8.51%. The recoveries using LLE and SPE method were 69.70–87.90% (RSDs <14.53%) and 75.77–95.43% (RSDs <9.94%), respectively. By these comparisons, traditional SPE and LLE consume large amounts of chemicals and hazardous organic solvents and extend multistep extraction procedures, and the recoveries and RSDs using the improved MSPE method were superior to LLE method and comparable to SPE method. Therefore, the proposed method is promising for extraction of trace PCBs in water samples.
Real sample analysis
To demonstrate the applicability of the proposed method, it was applied to analyze different environmental water samples, including rain water, river water, and pond water. The water samples and spiked solutions (10 ng L−1) were all analyzed in triple replicates, and the results were summarized in Table 3. PCB 28 and PCB52 were found to be 0.52 and 1.08 ng L−1 in river water, respectively. The absolute recoveries of spiked PCBs in three different water samples were in the range of 75.17–101.20% with RSDs ranging from 1.12 to 8.31%. The chromatograms detected and spiked PCBs in river water of GC-MS/MS were shown in Fig. 4.

The chromatograms of river water sample and river water sample spiked with 10 ng L−1 PCBs
Conclusions
In this work, we have demonstrated a facile one-pot sol–gel method for the synthesis of magnetic core-shell Fe3O4@SiO2-C16 microspheres. The preparation method was convenient, effective, gentle, and repeatable. The resulting Fe3O4@SiO2-C16 exhibited excellent sorption efficiency and reusability for the extraction of PCBs. Combined with GC-MS/MS, the overall method showed excellent sensitivity, accuracy, and precision. The method was successfully used to analyze PCBs in different environmental waters. The results also display great potential of Fe3O4@SiO2-C16 for the MSPE of other trace organic pollutants from environmental samples.
References
Wierucka M, Biziuk M. Application of magnetic nanoparticles for magnetic solid-phase extraction in preparing biological, environmental and food samples. TrAC Trends Anal Chem. 2014;59:50–8.
Huang DN, Deng CH, Zhang XM. Functionalized magnetic nanomaterials as solid-phase extraction adsorbents for organic pollutants in environmental analysis. Anal Methods-Uk. 2014;6(18):7130–41.
Kaur R, Hasan A, Iqbal N, Alam S, Saini MK, Raza SK. Synthesis and surface engineering of magnetic nanoparticles for environmental cleanup and pesticide residue analysis: a review. J Sep Sci. 2014;37(14):1805–25.
Li XS, Zhu GT, Luo YB, Yuan BF, Feng YQ. Synthesis and applications of functionalized magnetic materials in sample preparation. TrAC Trends Anal Chem. 2013;45:233–47.
Chen LG, Wang T, Tong J. Application of derivatized magnetic materials to the separation and the preconcentration of pollutants in water samples. TrAC Trends Anal Chem. 2011;30(7):1095–108.
Giakisikli G, Anthemidis AN. Magnetic materials as sorbents for metal/metalloid preconcentration and/or separation. A review. Anal Chim Acta. 2013;789:1–16.
Dziadas M, Nowacka M, Jesionowski T, Jelen HH. Comparison of silica gel modified with three different functional groups with C-18 and styrene-divinylbenzene adsorbents for the analysis of selected volatile flavor compounds. Anal Chim Acta. 2011;699(1):66–72.
Bang DY, Byeon SK, Moon MH. Rapid and simple extraction of lipids from blood plasma and urine for liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2014;1331:19–26.
Luo K, Gao Q, Hu JY. Derivatization method for sensitive determination of 3-hydroxybenzo[a]pyrene in human urine by liquid chromatography-electrospray tandem mass spectrometry. J Chromatogr A. 2015;1379:51–5.
Sha YF, Deng CH, Liu BZ. Development of C18-functionalized magnetic silica nanoparticles as sample preparation technique for the determination of ergosterol in cigarettes by microwave-assisted derivatization and gas chromatography/mass spectrometry. J Chromatogr A. 2008;1198:27–33.
Synaridou MES, Sakkas VA, Stalikas CD, Albanis TA. Evaluation of magnetic nanoparticles to serve as solid-phase extraction sorbents for the determination of endocrine disruptors in milk samples by gas chromatography mass spectrometry. J Chromatogr A. 2014;1348:71–9.
Qiao Z, Perestrelo R, Reyes-Gallardo EM, Lucena R, Cardenas S, Rodrigues J, et al. Octadecyl functionalized core-shell magnetic silica nanoparticle as a powerful nanocomposite sorbent to extract urinary volatile organic metabolites. J Chromatogr A. 2015;1393:18–25.
Liu Y, Li HF, Lin JM. Magnetic solid-phase extraction based on octadecyl functionalization of monodisperse magnetic ferrite microspheres for the determination of polycyclic aromatic hydrocarbons in aqueous samples coupled with gas chromatography–mass spectrometry. Talanta. 2009;77(3):1037–42.
Maddah B, Shamsi J. Extraction and preconcentration of trace amounts of diazinon and fenitrothion from environmental water by magnetite octadecylsilane nanoparticles. J Chromatogr A. 2012;1256:40–5.
Ahmadi F, Rajabi M, Faizi F, Rahimi-Nasrabadi M, Maddah B. Magnetic solid-phase extraction of Zineb by C18-functionalised paramagnetic nanoparticles and determination by first-derivative spectrophotometry. Int J Environ Anal Chem. 2014;94(11):1123–38.
Yamini Y, Faraji M, Adeli M. Magnetic silica nanomaterials for solid-phase extraction combined with dispersive liquid-liquid microextraction of ultra-trace quantities of plasticizers. Microchim Acta. 2015;182(7–8):1491–9.
Zhang H, Liu QS, Yang CL, Lv JZ, Xie LQ, Tang MJ, et al. C18/C8-functionalized magnetic silica nanospheres (Fe3O4@Si-C8/C18) as capture probes for highly efficient and rapid purification of veterinary drug residues. Food Anal Methods. 2013;6(3):933–40.
Breivik K, Sweetman A, Pacyna JM, Jones KC. Towards a global historical emission inventory for selected PCB congeners—a mass balance approach-3. An update. Sci Total Environ. 2007;377(2–3):296–307. doi:10.1016/j.scitotenv.2007.02.026.
Perez RA, Albero B, Tadeo JL, Sanchez-Brunete C. Oleate functionalized magnetic nanoparticles as sorbent for the analysis of polychlorinated biphenyls in juices. Microchim Acta. 2016;183(1):157–65. doi:10.1007/s00604-015-1617-2.
Mai BX, Zeng EY, Luo XJ, Yang QS, Zhang G, Li XD, et al. Abundances, depositional fluxes, and homologue patterns of polychlorinated biphenyls in dated sediment cores from the Pearl River Delta, China. Environ Sci Technol. 2005;39(1):49–56. doi:10.1021/es049015d.
Barakat AO, Khairy M, Aukaily I. Persistent organochlorine pesticide and PCB residues in surface sediments of Lake Qarun, a protected area of Egypt. Chemosphere. 2013;90(9):2467–76.
Van den Berg M, Birnbaum LS, Denison M, De Vito M, Farland W, Feeley M, et al. The 2005 World Health Organization reevaluation of human and mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol Sci. 2006;93(2):223–41.
Font G, Manes J, Molto JC, Pico Y. Current developments in the analysis of water pollution by polychlorinated biphenyls. J Chromatogr A. 1996;733(1–2):449–71. doi:10.1016/0021-9673(95)00972-8.
Wolska L, Galer K, Gorecki T, Namiesnik J. Surface water preparation procedure for chromatographic determination of polycyclic aromatic hydrocarbons and polychlorinated biphenyls. Talanta. 1999;50(5):985–91. doi:10.1016/S0039-9140(99)00193-9.
Grzeskowiak T, Czarczynska-Goslinska B, Zgola-Grzeskowiak A. Current approaches in sample preparation for trace analysis of selected endocrine-disrupting compounds: focus on polychlorinated biphenyls, alkylphenols, and parabens. TrAC Trends Anal Chem. 2016;75:209–26.
Rezaei F, Bidari A, Birjandi AP, Hosseini MRM, Assadi Y. Development of a dispersive liquid-liquid microextraction method for the determination of polychlorinated biphenyls in water. J Hazard Mater. 2008;158(2–3):621–7. doi:10.1016/j.jhazmat.2008.02.005.
Hassan J, Shamsipur M. Extraction of ultra traces of polychlorinated biphenyls in aqueous samples using suspended liquid-phase microextraction and gas chromatography-electron capture detection. Environ Monit Assess. 2013;185(5):3637–44. doi:10.1007/s10661-012-2815-6.
Wu YY, Yang CX, Yan XP. Fabrication of metal-organic framework MIL-88B films on stainless steel fibers for solid-phase microextraction of polychlorinated biphenyls. J Chromatogr A. 2014;1334:1–8. doi:10.1016/j.chroma.2014.01.079.
Zheng HJ, Liu QW, Jia Q. Preparation of poly(butyl methacrylate-co-ethyleneglyceldimethacrylate) monolithic column modified with beta-cyclodextrin and nano-cuprous oxide and its application in polymer monolithic microextraction of polychlorinated biphenyls. J Chromatogr A. 2014;1343:47–54. doi:10.1016/j.chroma.2014.03.067.
Lin SC, Gan N, Qiao L, Zhang JB, Cao YT, Chen YJ. Magnetic metal-organic frameworks coated stir bar sorptive extraction coupled with GC-MS for determination of polychlorinated biphenyls in fish samples. Talanta. 2015;144:1139–45. doi:10.1016/j.talanta.2015.07.084.
Lei Y, He M, Chen BB, Hu B. Polyaniline/cyclodextrin composite coated stir bar sorptive extraction combined with high performance liquid chromatography-ultraviolet detection for the analysis of trace polychlorinated biphenyls in environmental waters. Talanta. 2016;150:310–8. doi:10.1016/j.talanta.2015.12.025.
Conka K, Drobna B, Kocan A, Petrik J. Simple solid-phase extraction method for determination of polychlorinated biphenyls and selected organochlorine pesticides in human serum. J Chromatogr A. 2005;1084(1–2):33–8. doi:10.1016/j.chroma.2004.11.029.
Yang FX, Jin SW, Meng DY, Xu Y. Solid phase extraction with pyrenebutyric acid-bonded silica for analysis of polychlorinated biphenyls in sewage water by gas chromatography–mass spectrometry. Chemosphere. 2010;81(8):1000–5. doi:10.1016/j.chemosphere.2010.09.016.
Chen XF, Ding N, Zang H, Yeung H, Zhao RS, Cheng CG, et al. Fe3O4@MOF core-shell magnetic microspheres for magnetic solid-phase extraction of polychlorinated biphenyls from environmental water samples. J Chromatogr A. 2013;1304:241–5. doi:10.1016/j.chroma.2013.06.053.
Cao XJ, Chen JY, Ye XM, Zhang FF, Shen LX, Mo WM. Ultrasound-assisted magnetic SPE based on Fe3O4-grafted graphene for the determination of polychlorinated biphenyls in water samples. J Sep Sci. 2013;36(21–22):3579–85. doi:10.1002/jssc.201300770.
Karamani AA, Douvalis AP, Stalikas CD. Zero-valent iron/iron oxide-oxyhydroxide/graphene as a magnetic sorbent for the enrichment of polychlorinated biphenyls, polyaromatic hydrocarbons and phthalates prior to gas chromatography–mass spectrometry. J Chromatogr A. 2013;1271:1–9. doi:10.1016/j.chroma.2012.11.018.
Zeng SL, Gan N, Weideman-Mera R, Cao YT, Li TH, Sang WG. Enrichment of polychlorinated biphenyl 28 from aqueous solutions using Fe3O4 grafted graphene oxide. Chem Eng J. 2013;218:108–15. doi:10.1016/j.cej.2012.12.030.
Zhang JB, Gan N, Pan MY, Lin SC, Cao YT, Wu DZ, et al. Separation and enrichment of six indicator polychlorinated biphenyls from real waters using a novel magnetic multiwalled carbon nanotube composite absorbent. J Sep Sci. 2015;38(5):871–82. doi:10.1002/jssc.201400696.
Liao QG, Wang DG, Luo LG. Chitosan-poly(m-phenylenediamine)@Fe3O4 nanocomposite for magnetic solid-phase extraction of polychlorinated biphenyls from water samples. Anal Bioanal Chem. 2014;406(29):7571–9. doi:10.1007/s00216-014-8215-8.
Perez RA, Albero B, Tadeo JL, Molero E, Sanchez-Brunete C. Application of magnetic iron oxide nanoparticles for the analysis of PCBs in water and soil leachates by gas chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2015;407(7):1913–24. doi:10.1007/s00216-014-8409-0.
Ding J, Gao QA, Luo D, Shi ZG, Feng YQ. n-Octadecylphosphonic acid grafted mesoporous magnetic nanoparticle: preparation, characterization, and application in magnetic solid-phase extraction. J Chromatogr A. 2010;1217(47):7351–8.
Zuin WG, Montero L, Bauer C, Popp P. Stir bar sorptive extraction and high-performance liquid chromatography-fluorescence detection for the determination of polycyclic aromatic hydrocarbons in Mate teas. J Chromatogr A. 2005;1091(1–2):2–10.
U.S. EPA. Method 3520C (SW-846): continuous liquid-liquid extraction, Revision 3, 1996.
U.S. EPA. Method 3535 (SW-846): solid-phase extraction, revision 0, 1996.
U.S. EPA. Method 8082A (SW-846): polychlorinated biphenyls (PCBs) by gas chromatography, revision 1, 2007.
Wang L, Wang X, Zhou J-B, Zhao R-S. Carbon nanotube sponges as a solid-phase extraction adsorbent for the enrichment and determination of polychlorinated biphenyls at trace levels in environmental water samples. Talanta. 2016;160:79–85. doi:10.1016/j.talanta.2016.07.005.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (21507116 and 41473095), China Postdoctoral Science Foundation (2015M572213 and 2016T90744), and Fundamental Research Funds for the Central Universities, China University of Geosciences (Wuhan) (CUGL160409).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The author(s) declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 319 kb)
Rights and permissions
About this article

Cite this article
Fan, YH., Zhang, SW., Qin, SB. et al. Facile preparation of hexadecyl-functionalized magnetic core-shell microsphere for the extraction of polychlorinated biphenyls in environmental waters. Anal Bioanal Chem 409, 3337–3346 (2017). https://doi.org/10.1007/s00216-017-0278-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-017-0278-x