
Abstract
This paper presents an application of ultra-high performance liquid chromatography-quadrupole time-of-flight mass spectrometry (UHPLC-QTOF-MS) for simultaneous screening and identification of 427 pesticides in fresh fruit and vegetable samples. Both full MS scan mode for quantification, and an artificial-intelligence-based product ion scan mode information-dependent acquisition (IDA) providing automatic MS to MS/MS switching of product ion spectra for identification, were conducted by one injection. A home-in collision-induced-dissociation all product ions accurate mass spectra library containing more than 1700 spectra was developed prior to actual application. Both qualitative and quantitative validations of the method were carried out. The result showed that 97.4 % of the pesticides had the screening detection limit (SDL) less than 50 μg kg−1 and more than 86.7 % could be confirmed by accurate MS/MS spectra embodied in the home-made library. Meanwhile, calibration curves covering two orders of magnitude were performed, and they were linear over the concentration range studied for the selected matrices (from 5 to 500 μg kg−1 for most of the pesticides). Recoveries between 80 and 110 % in four matrices (apple, orange, tomato, and spinach) at two spiked levels, 10 and 100 μg kg−1, was 88.7 or 86.8 %. Furthermore, the overall relative standard deviation (RSD, n = 12) for 94.3 % of the pesticides in 10 μg kg−1 and 98.1 % of the pesticides in 100 μg kg−1 spiked levels was less than 20 %. In order to validate the suitability for routine analysis, the method was applied to 448 fruit and vegetable samples purchased in different local markets. The results show 83.3 % of the analyzed samples have positive findings (higher than the limits of identification and quantification), and 412 commodity-pesticide combinations are identified in our scope. The approach proved to be a cost-effective, time-saving and powerful strategy for routine large-scope screening of pesticides.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Pesticide use has increased agricultural production worldwide and thereby contributed to food security [1]. With the expected 30 % increase of world population to 9.2 billion by 2050, there is a projected demand to increase food production by 70 % according to the calculation by Popp et al. [2]. Hence, there will be a continuing need for pesticide-based solutions for pest control and food security in the future to increase food production to meet the increasing food supply [1, 2]. More than 1100 pesticides have been widely used in various combinations at different stages of cultivation and postharvest storage. Despite their usefulness, pesticides could pose potential risks to food safety and the environment. Pesticide residues could possibly remain in foodstuff, such as fruits and vegetables, which pose a potential threat to human and animal health. For this reason, most countries or regions have drafted rigorous regulations to control the use of pesticides in crop production and to monitor their levels for compliance. The USA [3, 4], Canada [5, 6], European Union [7, 8], and many other governments have set national chemical/pesticide residues monitoring programs and have conducted health risk assessment of pesticide residues in food and established maximum residue limits (MRLs) for domestic use and international trade of foods. In China, the newly implemented national food safety standard (GB2763-2014) has established about 3650 MRLs including 387 kinds of pesticides residues on 284 species of food [9]. However, pesticide residues are one of the most frequently detected xenobiotics in food analysis, especially in enormously consumed fruits and vegetables all over world.
The traditional technique in modern determination for pesticide is gas chromatography-mass spectrometry (GC-MS) or/and liquid chromatography-mass spectrometry (LC-MS) [10–14]. Triple-quadrupole tandem mass spectrometry (QqQ-MS/MS) in multiple selected reaction monitoring (SRM) mode is especially predominant owing to its selectivity and sensitivity [15–21]. Typically, the common practical analytical approach to multiple reaction monitoring (MRM) applied in many routine laboratories usually consists of selecting around 100–200 targeted compounds. Any significant increase to this number is uncommon for the reason of cost-effectiveness [22]. Moreover, those target detections are blind to any other contaminant present in the sample. Nevertheless, the SRM technique could not provide specific structural information for confirmation and the number of compounds that could be monitored in a single run has seriously limited the screening capability. Meanwhile, those routine low-resolution mass analyzers may be insufficient to distinguish exogenetic chemicals from numerous natural compounds, especially in complex matrices, for instance, Ferrer et al. have found more than 2500 natural compounds in bell peppers [23] and more than 4000 in red peppers [24]. Benefitting from the rapid development of instrument techniques, high-resolution mass spectrometry (HRMS), such as time-of-flight (TOF), quadrupole time-of-flight (QTOF), or orbitrap, permits for large-scale screening and identification of different compounds (pesticides, metabolites, etc.) in varied matrices (such as food [22, 25–29] and water [26]). Furthermore, those new HRMS techniques with full-scan mode possess the capability of non-target screening and data tracing. García-Reyes et al. discussed an approach of identification of non-target chlorinated pesticides in complex food extracts combining high sensitivity of LC-TOF-MS full-scan spectra and accurate mass measurements, and several chlorinated pesticides in different market-purchased fruit and vegetable extracts were identified without the use of standards [30]. Férnandez-Alba and his research group developed and evaluated a rapid automated screening method for determining nearly 300 pesticide residues in food [31], and almost 400 organic micro-contaminants in water samples using LC-(Q)TOF-MS based on the use of an accurate-mass database [32]. A similar strategy was also reported by Diaz et al. [33], they validated a wide-scope screening of organic contaminants (around 1100 organic contaminants) in natural and waste water based on the use of LC-QTOF-MS. They also explored the potential of UHPLC-(Q)TOF-MS to detect and identify/elucidate organic contaminants in both target and non-target approaches in food, human urine, as well as urban wastewater samples [34]. Most recently, a new approach for the analysis of pharmaceuticals in water, simultaneous quantitative screening of target, and the qualitative analysis of non-target in just one run by LC-QTOF-MS was described by Férnandez-Alba [35], and satisfactory results regarding sensitivity and linearity of method were obtained. The LC-(Q)TOF-MS system used for the above works has demonstrated to obtain mass spectra with a relatively high degree of mass accuracy (<2 ppm) at sufficient mass resolutions in most cases, regardless of the matrices or the concentration level, which is one of the most promising tools when moving towards non-targeted approaches. Furthermore, this common TOF-MS target and non-target screening strategies based on MS1 (molecular ion mass spectra of compound) database searching are exposed to false negatives and false positives that need to be clarified in the confirmatory step. Consequently, a MS/MS analysis of the detected compounds is indispensable for confirmation. For this purpose, libraries of MS/MS spectra can be very useful for rapid confirmation. Due to the different operational conditions, and the existence in the market of different interfaces, ionization, voltages applied, mobile phase compositions, etc., introduce diverse variables into the analytical measurement that make the development of standardized fragment mass spectral libraries unfeasible, which is still a significant bottleneck encountered in tandem mass spectral library search and rapid analysis. Though building empirical in-house libraries containing as many compounds as possible is the best option at the moment [22].
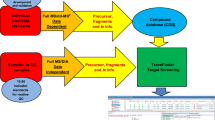
To our knowledge, only a few MS/MS libraries have been developed, but most of them are in the environmental field, fewer in such complex matrixes as food, especially in vegetables and fruits [34, 36–38]. Our previous studies [39, 40] have evaluated a two-injection approach for qualitative screening and identification of multi-pesticides residues in fresh fruits and vegetables employing HPLC-Q-TOF/MS technique based on a home-in MS/MS library for approximately 300 pesticides. Nevertheless, the disadvantage of this strategy is that the MS method of the second injection is based on the result of first injection data analysis which requires an analyst’s input and may not be available within a few hours depending on the other time commitment(s), which is a significant bottleneck for fast screening purpose. Furthermore, the idea of screening method is that the data evaluation should be done in an automated, fast, and simple way with large-scale target compounds [41]. This study goes a step forward into establishing the optimum working conditions in order to obtain reliable and more spectra to be included in the library for further non-target analysis. The all ions MS/MS strategy employing the latest generation UHPLC-QTOF-MS by ion extraction against a home-made library including as much as 420 pesticides is evaluated. An information-dependent acquisition (IDA) mode, an artificial intelligence-based product ion scan providing automatic MS to MS/MS switching, is conducted to obtain MS/MS spectra of the most intense precursor ions (without previous selection) in a run, and the identity of the analytes by MS/MS library searching is confirmed. Four hundred twenty-seven toxicological chemicals were investigated with UHPLC-QTOF-MS before the actual sample screening to establish the accurate MS/MS spectra library and XIC list. The applicability of the home-made spectra library and XIC list leading to true results was evaluated with solvent standard mixture and spiked matrices. In addition, the quantitative performance of the IDA approach with UPLC-QTOF-MS is also validated, including linearity and recovery for the most frequently detected pesticides. Finally, 448 fresh fruit and vegetable extracts were investigated by the proposed technique and 75 relevant pesticides or metabolites were distinctly identified.
Materials and Methods
Chemicals and reagents
Pesticide (as listed in Table S1, see Electronic Supplementary Material, ESM) analytical standards of high purity (>98 %) included in this study were purchased from Dr. Ehrenstorfer (Ausburg, Germany) and from Sigma-Aldrich (Steinheim, Germany). Individual pesticide standard stock solutions of all compounds (approximately 1 mg mL−1) were prepared in pure methanol or acetonitrile, depending on the solubility of each individual compound. The working standard solution was prepared by mixing the appropriate amounts of each standard solutions and diluting them with methanol to a final concentration of 1 mg L−1. The stock solutions and mixed working solutions were stored at 4 °C in a freezer, and were used for no more than 1 year for the former and 1 month for the latter. HPLC-grade acetonitrile and methanol were obtained from Dima Technology INC (CA, USA). Formic acid was purchased from Anaqua Chemicals Supply (TX, USA). Ammonium acetate (NH4OAc) was bought from Dima Technology INC (Richmond Hill, ON, L4B 3N6 Canada). Anhydrous sodium sulfate (Na2SO4, Analytical reagent) and sodium chloride (NaCl, Analytical reagent) were obtained from Damao Chemical Factory and Fengchuan Chemical Reagent Science and Technology (Tianjin, China).
Sampling and sample preparation
Four hundred forty-eight fresh fruit and vegetable commodities were randomly purchased from local markets from 18 Sep, 2015 to 20 Apr. 2016. The edible parts of samples were cut into small pieces without any pretreatment and were triturated with a chopper. The homogenized were preserved in a refrigerator at −18 °C. Before using, the samples were thawed at 5 °C overnight.
An off-line solid phase extraction (SPE), already reported elsewhere, was used for the extraction and clean-up of the samples [40]. Concisely, a portion of 20 g sample previously homogenized was mixed with 40 mL of acetonitrile and 5 g sodium chloride. The mixture was blended for 1 min with high-speed blender Ultra-Turrax T25 at 10,000 rpm, and then centrifuged at 4200 rpm (equivalent to RCF 3155×g) for 5 min. The supernatants extracts (20 mL) of the extract were transferred into individual heart-shaped bottles and evaporated to 1 mL on a vacuum rotary evaporator at 40 °CA layer of anhydrous sodium sulfate (2 cm) was added to the Carbon/NH2 cartridge (500 mg/6 cc, Waters, Milford, MA, USA) to remove traces of water from the extract. The columns were conditioned with 4 mL acetonitrile/toluene (3:1, v:v) before adding the samples, then the extract was loaded onto the cartridge and the retained analytes were eluted with 25 mL of acetonitrile/toluene. (3:1, v:v). The entire volume of effluent was collected and concentrated to 0.5 mL at 40 °C with a rotary evaporator, then evaporated to dryness using a nitrogen evaporator. Finally, the residues were re-dissolved up to 2 mL with 0.1 % formic acid of water-acetonitrile (1:1, v:v) thoroughly. The extract was filtered through a 0.2 μm filter into a glass vial before the UPLC-QTOF-MS analysis. With this treatment, 1 mL sample extract represents 5 g of sample.
Moreover, all solvents and reagents utilized were carefully checked to be analyte-free, by performing blank procedure analysis. To ensure the reliability of the process and the absence of background contamination, matrix blank and procedural blanks were carried out with each batch of samples. “Blank” vegetable and fruit extracts were used to prepare the matrix-matched standards for qualitative and quantitative validation purposes.
Instrument and software
The UHPLC system coupled to a hybrid quadrupole time-of-flight was applied to analyze the pesticides in fruit and vegetable samples. The separation of the analysts was performed on a UHPLC system (Nexera X2, Shimadzu, Kyoto, Japan) consisting of vacuum degasser, auto-sampler and a binary pump, equipped with a reversed-phase Zorbax SB-C18 analytical column (100 mm × 2.1 mm and 3.5 μm particle size).
The mass spectrometry detection was performed using a quadrupole time-of-flight mass spectrometry (Triple TOF™5600+, AB SCIEX, Redwood City, CA USA) in positive ionization mode with a Duo-Spray ion source. The QTOF was tuned and calibrated as recommended by the manufacturer in MS and MS/MS in high sensitivity mode, respectively. The automated calibration device system (CDS) was set to perform an external calibration every five samples using calibrate solution. The MS acquisition was performed in an automatic IDA method that consist of two experiments (Fig. 1a): the survey scan type (TOF-MS) that was a full-scan mass spectrum between m/z 100–950 and the information dependent acquisition type (MS/MS), a product ion scan in which the system selects ions automatically without any ion predefined by the user. Both the full-scan TOF-MS and MS/MS mode through IDA were operated within a single-run analysis. The resolving power is >40,000 (FWHM, full width at half-maximum, at m/z 609.28066) in MS and >35,000 in MS/MS (high-resolution mode, m/z 448.19660). However, large-scale analyte screening requires application of compromise conditions, such as collision energy or declustering potential rewarding as many compounds as possible. The source parameters were optimized as follows: ion spray voltage floating (ISVF), 5500 V; temperature, 400 °C with curtain gas (CUR) 30 psi; nebulizing gas (GS1), 35 psi; heater gas (GS2), 35 psi. In the full-scan TOF-MS experiment, declustering potential (DP) and collision energy (CE) were set at 80 V and 10 eV, respectively and the accumulation time 100 ms. IDA MS/MS was performed using the following switch criteria: 10 most intense ions, exclude isotopes within 4 Da, ion tolerance 50 mDa, and a collision energy of 35 eV with a spread of ±15 eV (CES), dynamic background subtract was activated. The accumulation time for each IDA experiment was 50 ms.


LC-QTOF-MS chromatograms. a The upper is IDA survey scan (TOF-MS) total ion chromatogram (TIC) corresponding to a tomato extract spiked at 0.1 mg kg−1 with the pesticide mixture; and the lower is sum of IDA dependent scan (n = 10); b LC retention time distribution of 427 pesticides; c Example of extracted ion chromatograms (XIC) (first column, up to down, isoproturon (206.14191, 6.85 min), diethyltoluamide (191.13101, 6.90 min), and karbutilate (279.15829, 5.76 min); second column, up to down, carbendazim (191.06948, 2.84 min), dimefuron (338.11457, 8.36 min), and 1,3-diphenyl urea (212.09496, 7.11 min))
Data acquisition was carried out using the software Analyst® TF (Version 1.6, ABSciex). The complex data were processed by the powerful data mining tool, Peak View (Version 1.0, ABSciex) with XIC Manager add-in. The XIC Manager consists of a table for defining a list of formulae (XIC list) to automatically generate extracted ion chromatograms (XIC), and to perform compound identification. Moreover, it allows for exploring and processing mass spectral data using the parameters of LC retention time, accurate mass. Meanwhile, purity score of each finding in XIC list was also obtained by matching an experimental MS/MS spectrum against library MS/MS spectrum by XIC Manager. Quantitation was carried out with MultiQuant ((Version 2.1, ABSciex) using the survey scan by extracting the narrow window extracted ion chromatogram of the molecular ion for each compound (typically extracted using a 10 mDa window) and the retention time (with a 0.5 min window).
An automatic high-speed blender (Ultra-Turrax T25, Janke & Kunkel GmbH &Co., Staufen, Germany), a low-speed centrifuge (KDC-40, USTC Chuangxin Co., Ltd. Zonkia branch, China), a rotary evaporator (R-215, BUCHI Labortechnik AG, Switzerland), and a nitrogen evaporator (Organomation Associates, EVAP 112, USA) were used for sample pretreatment and extraction. A Milli-Q-Plus ultra-pure water system from Millipore (Milford, MA, USA) was used throughout the study to obtain the HPLC-grade (18.2 MΩ•cm) water during the analysis. The electronic analytical balance used for sample weighing was obtained from Shimadzu (TXB622L, Shimadzu, Kyoto, Japan).
Building the MS2 spectral library
MS/MS spectra libraries are critical tools for small molecular identification in food analysis as well as other research areas like environmental monitoring, forensic science, and metabolomics. These repositories allow investigators to compare MS/MS data from research samples to MS/MS data from model compounds compiled in the library and thereby improve the efficiency and cost-effectiveness of untargeted studies. However, LC-MS/MS spectra libraries are considered too variable to be able to create universal libraries [35, 42] and the standardized fragment mass spectral libraries are still unavailable. Therefore, the generation of the MS/MS spectral library was performed using Analyst® and Microsoft Access software before the actual sample screening. The MS/MS spectra were acquired by introducing individual pesticide solutions (as listed in ESM Table S1), available from our laboratories’ collection, into the electrospray ion source of the instrument by flow injection directly into the source. The MS/MS spectra of each precursor at four different collision energies (20, 35, 50, and 35 ± 15 eV) were recorded. As such, the acquired MS/MS spectra, together with compound name, molecular formula, and molecular mass et al., were integrated into our in-house library correspondingly. The mass spectral library comprises more than 1700 MS/MS spectra.
Validation study
The main purpose of qualitative screening is to ensure the presence of an analyte in a sample at a certain concentration level. The increased interest and application of qualitative screening methods based on chromatography and MS has resulted in a need for more detailed and harmonized guidance and criteria for validation of such methods. Since 2010, EU guidance document on pesticide residue analysis have included some criteria and parameters to be considered in the validation of screening qualitative methods [43]. In this work, method validation has been performed following a similar strategy to our previous paper [40]. Based on this point, 2.5, 25, and 125 μL of the 1 mg L−1 mix-standard solution including 416 pesticides (the other 11 pesticides standard material, azinphos-ethyl, benodanil, bensulide, bromfenvinfos, heptenophos, isofenphos-oxon, butralin, demeton-S, pyributicarb, thiofanox sulfone, and bitertanol were used up prior to validation) were transferred into each vial, and then the solvent in the vial was evaporated to dryness using a nitrogen evaporator at room temperature. Then, 500 μL filtered blank extracts (equal to 2.5 g solid sample) prepared as mentioned above were added to each vial to obtain spiked extract with concentration at 5, 50, and 250 μg L−1 for the pesticides, corresponding to 1, 10, and 50 μg kg−1 in solid samples. Finally, a total of 18 spiked extracts, two matrices (apple and spinach) with three levels in triplicates, were prepared for qualitative validation and then analyzed by UHPLC-QTOF-MS as afore mentioned. As the goal of the qualitative screening is to detect and identify positive samples at a given concentration, the method was considered as satisfactorily validated. The screening detection limit (SDL) which is the lowest concentration level tested for which a compound was detected in all the samples, and limit of identification (LOI), which is defined as the lowest concentration for which the identification criteria are met is discussed [44, 45].
Moreover, the quantitative performance of the IDA approach was also discussed in this paper while being an excellent instrument for qualitative purposes in many domains. The linearity was evaluated by preparing calibration curves in seven calibration points, that is, 5, 10, 20, 50, 100, 200, and 500 μg kg−1, for the 106 most frequently occurring pesticides in blank tomato extracts. The curves were constructed for each compound by plotting peak area against the corresponding concentration using linear regression analysis. Recovery and relative standard deviations (RSD) of 106 most frequently occurring pesticides were also calculated by spiking samples of four different matrices (apple, orange, tomato, and spinach in triplicates) at two different levels, 10 and 100 μg kg−1. The pesticide mixtures were spiked to the blank fruit and vegetable samples and left for at least 20 min at room temperature before the extraction to ensure the appropriate distribution of the analytes in the matrix. Then the fortified samples were extracted and cleaned-up by the SPE protocol aforementioned. Each extract was analyzed with the UHPLC-QTOF-MS and the contents of spiked pesticides were determined by matrix-matched calibration curves.
Results and discussion
Optimization of UPLC-QTOF-MS
The ultra-high performance liquid chromatograph along with a 100-mm Zorbax SB-C18 analytical column with 3.5 μm particle (Agilent Technologies, USA) showed a satisfactory chromatographic performance to separate pesticides under the given gradient condition depicted elsewhere [40]. The flow rate was set to 0.4 mL min−1, and injection volume was 5 μL. The column temperature was held at 40 °C. The mobile phase consisted of (A) water with 0.1 % formic acid and 5 mmol L−1 ammonium acetate, and (B) acetonitrile, respectively. Gradient elution was performed in 25 min under the following conditions: 0 min, 99 % A; 3 min 70 %; 6 min, 60 % A and kept for 3 min, 15 min, 40%A; 19 min, 10%A and kept for 5 min. Then the system was programmed to regain its initial conditions and followed by a 5 min re-equilibration prior to the next injection.
As summarized in Fig. 1b, all of the pesticides were chromatographically separated in 25 min running time and most of the LC peaks were narrow and sharp with Gaussian distribution (Fig. 1c). In addition, a 5-min equilibration was conducted prior to each injection. The relative retention time (RT) of each pesticide in Table S1 (see ESM) was acquired by running a series of mixture reference standard solutions using survey scan type under liquid chromatography conditions as aforementioned. The first pesticide eluted from the column was the polar pesticide chlormequat chloride at 0.6 min, and the last one was pyridalyl at 20.35 min. Most pesticides (97.4 %) were eluted between 2 and 19 min. Only other four pesticides, mepiquat (0.71), cyromazine (0.72), daminozide (0.74), and methamidophos (1.84), were eluted before 2 min; and the other five, pyridaben (19.01), tribufos (19.05 min), aspon (19.10 min), bioresmethrin (19.28 min), and pyridate (19.77 min), were eluted after 18 min. The retention times, within- and between-batches, were reproducible for most of the pesticides, and the tolerance of retention time matching did not exceed 2.5 % relative to the retention time of a standard in the same batch.
CE plays a key role in both the sensitivity and fragmentation patterns. It can provide valuable structural information (characteristic fragmentation for each pesticide), making attainable the accurate mass of each characteristic fragment ion together with its elemental composition, which can be used with the molecular ion for confident identification criteria. Prior to application of the screening, different CEs (20, 35, 50, and 35 ± 15 (CES) eV) were evaluated to acquire effective and plentiful MS/MS spectra of each precursor. As shown in Fig. 2a, b, lower CE induced fewer dissociation fragment spectra. Extremely low intensity (relative intensity < 5 % of the molecular ion) of few fragment ions were observed in fenpropimorph (m/z = 147.11546) and ancymidol (m/z = 81.04462 and 135.04394) at CE = 20 eV. But CE = 50 eV or higher led to too extensive fragments to identify the compound (Fig. 2c, d). Taking into account that the MS/MS spectrum for most of the pesticides should preferably consist of diagnostic fragment ions, a compromise value of the CE (35 ± 15 eV) was chosen for further experiments. However, because the fragmentation depends strongly on the specific structure, this predefined method is not always optimum and would be improved in the further research.

Accurate mass spectra in different collision energies embodied in the home-in library. a Fenpropimorph and b Ancymidol at CE = 20 eV; c Cadusafos and d Ethoprophos at CE = 50 eV; e Cadusafos and f Ethoprophos at CE = 35 ± 15 eV
Qualitative validation
The primitive goal of screening is focused on detectability, that is, to ensure the presence of an analyte in a sample at a certain concentration level. Most recently, SANCO documents laid down some criteria and parameters to be considered in the validation of screening methods on pesticide residue analysis [43]. In this paper, a similar qualitative validation in line with the suggestion of Diaz et al. has been conducted [33].
Positive findings of the target pesticides were determined according to the optimized searching parameters (a) accurate mass measurement of the extract ion with an error of ≤5 ppm; and (b) retention time of the analyte compared with that of a standard with an error of ≤2.5 %; and (c) difference in isotopic pattern ≤20 %. While purity score of matching library spectrum compared with acquired MS/MS spectrum was ≥70 %, a positive result was unambiguously identified/confirmed. The ability to confirm compounds using MS/MS library searching ensured that no false positives were reported [35]. The requirements for confirmation in EU Document No. SANCO/12495/2011 include two diagnostic ions having mass accuracy of <5 ppm. In general, QTOF-MS is able to achieve the goal that includes both precursor and product ions with accurate masses. Figure 3 shows an UHPLC-QTOF-MS screening result of an apple sample which included four positive analytes, carbendazim, pyraclostrobin, propargite, and pyridaben. The mass accuracy for the precursor ions of the non-targeted carbendazim, pyraclostrobin, propargite, and pyridaben, are −0.7, 0.7, 0.6 and −0.2 ppm, respectively. Whereas, the purity scores of the IDA MS/MS product ions in Fig. 3c2, d2, e2, and f2 against a home-in library are 100, 92.6, 91.2, and 89.8, respectively.

Identification of non-targeted positive analytes (carbendazim, pyraclostrobin, propargite, and pyridaben) in an apple sample using the XIC Manager based on accurate mass, LC retention time, isotopic profiling, and product ions information. (a) Extracted ion chromatogram (XIC) of the four analytes, (1)-carbendazim, (2)-pyraclostrobin, (3)-propargite, and (4)-pyridaben according to the retention time against (b) XIC Manger table with data on the 427 pesticides. (c1, d1, e1 and f1) are accurate mass spectrums (full MS scan) and isotopic profiling from chromatogram A at 2.84, 15.51, 18.39 and 18.91 min, respectively. (c2, d2, e2 and f2) are product ion spectrums (MS/MS) of carbendazim [M + H] + (m/z 192.07675), pyraclostrobin [M + H] + (m/z 388.10586), propargite [M + NH4]+ (m/z 368.18901), and pyridaben [M + H]+ (m/z 365.14489), respectively. Those MS/MS spectra also confirm the identity of the corresponding compounds
As can be seen from ESM Table S1, 85 % (n = 343, the screening and identification results of each analyte in selected matrix were summarized in ESM Table S2) of the compounds were automatically found at the spiked level of 1 μg kg−1, according to the proposed strategy above, the SDLs of those pesticides were no more than 1 μg kg−1. There were 45 pesticides detected at 10 μg kg−1, and 17 pesticides found at 50 μg kg−1 using the automatic screening method, thus having SDLs between 1–10 μg kg−1 and 10–50 μg kg−1, respectively. However, the other 11 pesticides SDLs (aldicarb, azinphos-methyl, demeton-S-methyl, diafenthiuron, fenazaquin, isomethiozin, lactofen, norflurazon, pyridalyl, rimsulfuron) were believed to be higher than 50 μg kg−1 due to their low response under ESI conditions. Meanwhile, more than 86 % of the detected results could be identified against accurate MS/MS spectra embodied in the home-made library with purity score ≥70 % with LOI no more than 50 μg kg−1. Nevertheless, the proposed technique is sensitive enough to detect those large scope pesticide residues in fruits and vegetables and that is better than our previous work [40].
Quantitative performance
As being an excellent instrument for qualitative purposes in many domains, the quantitative performance of the IDA approach with UHPLC-QTOF-MS was also pursued in this paper. The quantitative performance results are summarized in Table 1 and are illustrated in Fig. 4. As we can see in Table 1, the response of the detected is linear for most compounds in the range of 5–500 μg kg−1 (except about 39 pesticides with the range of 5–200 μg kg−1) with correlation coefficient (R 2) of 0.9894 (atrazine-desethyl and fenobucarb)–0.9998 (indoxacarb), and the matrix-matched regression equations (based on peak area in tomato extract) of the spiked pesticides are listed in Table 1. Recoveries were evaluated in four matrices (apple, orange, tomato and spinach) at two spiked levels, 10 and 100 μg kg−1 in triplicates (ESM Table S3). The total average recoveries (n = 12) ranged from 71.7 % (piperonyl butoxide) to 126.6 % (dimethenamid) at 10 μg kg−1 and from 61.6 % (emamectin) to 124.4 % (isoprocarb) at 100 μg kg−1 spiked levels. About 88.7 or 86.8 % of the pesticides in the selected fruits and vegetables matrices had recoveries between 80 and 110 % at 10 or 100 μg kg−1 spiked levels. Depending on the type of sample matrices, about 86.8 % (71.7 %) in apples, 91.4 % (80.0 %) in oranges, 79.2 % (72.6 %) in tomatoes, and 75.2 % (83.8 %) in spinaches of the pesticides had recoveries between 80 and 110 % at the 100 μg kg−1 (10 μg kg−1) spiked level; 94.3 % of the pesticides in 10 μg kg−1 and 98.1 % of the pesticides in 100 μg kg−1 spiked levels had a relative standard deviation (RSD) ≤ 20 % (n = 12, except ethoprophos and emamectin) among the spiked matrices. These results show that the accuracy is satisfactory for most of the pesticides. However, it should be noted that those pesticides, including ethoprophos (in spinach) and emamectin (in orange), are problematic pesticides by UHPLC-QTOF-MS as a result high/low recovery, and poor repeatability. Further study on extraction and/or the applications of different mass spectrometric techniques is necessary to obtain better quantitative results upon those pesticides. Quantification was performed using MultiQuant™ 2.1 software using the peak area of the precursor ion versus the concentration of the analyte for all test samples.

Overall recovery of apple, orange, tomato, and spinach sample at two spiked levels a 10 and b 100 μg kg−1 in triplicates
It is clear, however, that the quantitative results are additional to the valuable qualitative information obtained. Because the more time spent to record MS/MS spectra and identify unknowns, the less time is left to record the MS survey chromatogram [42]. But there is no conflict in the picture of screening analytical approaches. They all provide a qualitative result directing further analysis and generally also provide a first idea of the quantity of toxicant present. The latter should be confirmed in a dedicated quantitative analysis but also provides a quick reference to the severity of an intoxication, for example, in banned pesticides or in doping agents. Though a follow-up dedicated quantitative analysis may be required for the rigorous monitoring.
Survey of the library pesticides in market samples
In order to verify the performance of the method and its suitability for routine analysis, it was applied to market samples. Four hundred and forty-six fresh vegetable and fruit samples were randomly purchased from local markets in Xi’an, in the West of China, from Sep 18, 2015 to Apr 20, 2016. Twenty-one kinds of fresh vegetables and fruits in total (n = 448), including apple, banana, bitter melons, celery, cherry tomato, Chinese chives, courgettes, cucumber, eggplant, grape, hot pepper, kiwi fruit, lettuce leaf, mandarin, orange, peach, pear, pitayas, radish, sweet pepper, and tomato, were analyzed according to the protocol discussed above.
Several internal quality control measures have been taken to ensure the reliability of the analysis. During each submitted sequence, reagent blanks, full procedural blanks, and fortified extracts (at 100 μg kg−1) were analyzed to ensure the instrument and the procedure were in good condition. Reagent blanks, including acetonitrile and water, were examined at the beginning of each batch to make sure no analyte signals were found. Then, a full procedural blank was analyzed to ensure no laboratory contamination was introduced in the whole procedure. In order to ensure the analytes remain detectable in the whole submitted sequence, the fortified extracts were analyzed at the beginning and the end as internal quality controls.
To compensate for the matrix effects, known to occur frequently in pesticide residues detected in both GC and LC methods, the concentration of the positive result was quantified using a single-point external matrix-matched standard. The concentration of analyte present in the sample extract is calculated from the relative responses of the analyte in the blank extract and the spiked sample extract.
As a result, 16.7 % of the samples were blank in our scope, or contained pesticides at levels lower than the limits of identification and quantification, while 83.3 % (373 in 448) of them contained one or more of the pesticides studied. A total number of 1432 findings were identified according to the proposed screening method in involving 412 commodity-pesticide combinations, and as much as 75 kinds of pesticide in our scope. Part of the results data were shown in Table 2. In general, the top 10 most frequently detected compounds (number of findings) are carbendazim (175), diethyltoluamide (150), dimethomorph (98), difenoconazole (73), pyridaben (71), chlormequat Chloride (67), azoxystrobin (61), metalaxyl (59), imazalil (58), and imidacloprid (57). Compared with national food safety standard [8], maximum residue limits for up to 64 % of the findings has not been established in the country. Meanwhile, nine non-compliance samples were also identified in six commodity-pesticide combinations, that is, celery-phorate (1), kiwi fruit-forchlorfenuron (1), cucumber-carbofuran (1), tomato-carbofuran (1), hot pepper-carbofuran (2) and Chinese chives-carbofuran (2). Furthermore, direct spraying on fruit and vegetable products of phorate and carbofuran is prohibited. These should be given more cautious attention in future research.
Conclusions
The developed method allows qualitative and quantitative analysis of 427 pesticides in fruit and vegetable samples in UHPLC-QTOF-MS with the traditional SPE extraction. The obtained validation results demonstrated the great qualitative and quantitative strength of this newly developed UHPLC-QTOF-MS method, which additionally offers great potential for untargeted screening. This system is also confirmed as a time-competitive approach for routine analysis compared to other LC-MS quantitation methods. The method has been validated for routine analysis, and has been applied to real samples for the survey of pesticides in fruits and vegetables. As a result of these analyses, 412 commodity-pesticide combinations were identified and six of them were non-compliances. The strategy can therefore be used as an exploratory approach for unknown samples. If there is evidence of the presence of target compounds, after this screening step, a further sample treatment step maybe carried out if necessary.
References
Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, et al. Emerging fungal threats to animal, plant and ecosystem health. Nature. 2012;484:186–94. doi:10.1038/nature10947.
Popp J, Peto K, Nagy J. Pesticide productivity and food security. A review. Agron Sustain Dev. 2013;33(1):243–55. doi:10.1007/s13593-012-0105-x.
United States Department of Agriculture. Pesticide data program (PDP). http://www.ams.usda.gov/AMSv1.0/pdp. Accessed 10 Apr 2016.
U.S. Food and Drug Administration. Pesticide Program Residue Monitoring. http://www.fda.gov/food/foodborneillnesscontaminants/pesticides/ucm2006797.htm. Accessed 10 Apr 2016.
Health Canada. Maximum Residue Limits for Pesticides. http://www.hc-sc.gc.ca/cps-spc/pest/part/protect-proteger/food-nourriture/mrl-lmr-eng.php. Accessed 17 Apr 2016.
Canadian Food Inspection Agency (CFIA). National Chemical Residue Monitoring Program 2012–2013 Report. http://www.inspection.gc.ca/food/chemical-residues-microbiology/chemical-residues/eng/1324258929171/1324264923941. Accessed 17 Apr 2016.
EU-wide Pesticide Residues Monitoring. http://ec.europa.eu/food/fvo/specialreports/pesticides_index_en.htm. Accessed 10 Dec 2015.
EU - Pesticides database. http://ec.europa.eu/food/plant/pesticides/eu-pesticides-database. Accessed 10 Apr 2016.
National food safety standard - Maximum residue limits for pesticides in food. http://www.nhfpc.gov.cn/ewebeditor/uploadfile/2014/04/20140409103007373.pdf. Accessed 10 Apr 2016.
Pang GF, Liu YM, Fan CL, Zhang JJ, Cao YZ, Li XM, et al. Simultaneous determination of 405 pesticide residues in grain by accelerated solvent extraction then gas chromatography-mass spectrometry or liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem. 2006;384(6):1366–408. doi:10.1007/s00216-005-0237-9.
Pang GF, Fan CL, Liu YM, Cao YZ, Zhang JJ, Li XM, et al. Determination of residues of 446 pesticides in fruits and vegetables by three-cartridge solid-phase extraction-gas chromatography-mass spectrometry and liquid chromatography-tandem mass spectrometry. J AOAC Int. 2006;89(3):740–71.
Lian YJ, Pang GF, Shu HR, Fan CL, Liu YM, Feng J, et al. Simultaneous determination of 346 multiresidue pesticides in grapes by PSA-MSPD and GC-MS-SIM. J Agric Food Chem. 2010;58(17):9428–53. doi:10.1021/jf1019592.
Anastassiades M, Maštovská K, Lehotay SJ. Evaluation of analyte protectants to improve gas chromatographic analysis of pesticides. J Chromatogr A. 2003;1015(1–2):163–84. doi:10.1016/S0021-9673(03)01208-1.
Maštovská K, Lehotay SJ. Evaluation of common organic solvents for gas chromatographic analysis and stability of multiclass pesticide residues. J Chromatogr A. 2004;1040(2):259–72. doi:10.1016/j.chroma.2004.04.017.
Hayward DG, Wong JW, Shi F, Zhang K, Lee NS, DiBenedetto AL, et al. Multiresidue pesticide analysis of botanical dietary supplements using salt-out acetonitrile extraction, solid-phase extraction cleanup column, and gas chromatography–triple quadrupole mass spectrometry. Anal Chem. 2013;85(9):4686–93. doi:10.1021/ac400481w.
Mol HGJ, Zomer P, García López M, Fussell RJ, Scholten J, de Kok A, et al. Identification in residue analysis based on liquid chromatography with tandem mass spectrometry: experimental evidence to update performance criteria. Anal Chim Acta. 2015;873:1–13. doi:10.1016/j.aca.2015.03.007.
Pareja L, Cesio V, Heinzen H, Fernandez-Alba AR. Evaluation of various QuEChERS based methods for the analysis of herbicides and other commonly used pesticides in polished rice by LC-MS/MS. Talanta. 2011;83(5):1613–22. doi:10.1016/j.talanta.2010.11.052.
Tomasini D, Sampaio MRF, Caldas SS, Buffon JG, Duarte FA, Primel EG. Simultaneous determination of pesticides and 5-hydroxymethylfurfural in honey by the modified QuEChERS method and liquid chromatography coupled to tandem mass spectrometry. Talanta. 2012;99:380–6. doi:10.1016/j.talanta.2012.05.068.
Mol HGJ, van Dam RCJ, Steijger OM. Determination of polar organophosphorus pesticides in vegetables and fruits using liquid chromatography with tandem mass spectrometry: selection of extraction solvent. J Chromatogr A. 2003;1015(1–2):119–27. doi:10.1016/S0021-9673(03)01209-3.
Picó Y, Blasco C, Font G. Environmental and food applications of LC–tandem mass spectrometry in pesticide-residue analysis: an overview. Mass Spectrom Rev. 2004;23(1):45–85. doi:10.1002/mas.10071.
Pang GF, Fan CL, Liu YM, Cao YZ, Zhang JJ, Fu BL, et al. Multi-residue method for the determination of 450 pesticide residues in honey, fruit juice and wine by double-cartridge solid-phase extraction/gas chromatography-mass spectrometry and liquid chromatography-tandem mass spectrometry. Food Addit Contam. 2006;23(8):777–810. doi:10.1080/02652030600657997.
Gomez-Ramos MM, Ferrer C, Malato O, Aguera A, Fernandez-Alba AR. Liquid chromatography-high-resolution mass spectrometry for pesticide residue analysis in fruit and vegetables: screening and quantitative studies. J Chromatogr A. 2013;1287:24–37. doi:10.1016/j.chroma.2013.02.065.
Ferrer I, Fernandez-Alba A, Zweigenbaum JA, Thurman EM. Exact-mass library for pesticides using a molecular-feature database. Rapid Commun Mass Spectrom. 2006;20(24):3659–68. doi:10.1002/rcm.2781.
Thurman EM, Ferrer I, Zavitsanos P, Zweigenbaum JA. Analysis of isobaric pesticides in pepper with high-resolution liquid chromatography and mass spectrometry: complementary or redundant? J Agric Food Chem. 2013;61(10):2340–7. doi:10.1021/jf304642g.
Gomez-Ramos MD, Rajski L, Heinzen H, Fernandez-Alba AR. Liquid chromatography Orbitrap mass spectrometry with simultaneous full scan and tandem MS/MS for highly selective pesticide residue analysis. Anal Bioanal Chem. 2015;407(21):6317–26. doi:10.1007/s00216-015-8709-z.
Lacorte S, Fernandez-Albaz AR. Time of flight mass spectrometry applied to the liquid chromatographic analysis of pesticides in water and food. Mass Spectrom Rev. 2006;25(6):866–80. doi:10.1002/mas.20094.
Besil N, Ucles S, Mezcua M, Heinzen H, Fernandez-Alba AR. Negative chemical ionization gas chromatography coupled to hybrid quadrupole time-of-flight mass spectrometry and automated accurate mass data processing for determination of pesticides in fruit and vegetables. Anal Bioanal Chem. 2015;407(21):6327–43. doi:10.1007/s00216-015-8514-8.
Yoshioka N, Akiyama Y, Matsuoka T, Mitsuhashi T. Rapid determination of five post-harvest fungicides and metabolite in citrus fruits by liquid chromatography/time-of-flight mass spectrometry with atmospheric pressure photoionization. Food Control. 2010;21(2):212–6. doi:10.1016/j.foodcont.2009.04.001.
Cervera MI, Portolés T, López FJ, Beltrán J, Hernández F. Screening and quantification of pesticide residues in fruits and vegetables making use of gas chromatography–quadrupole time-of-flight mass spectrometry with atmospheric pressure chemical ionization. Anal Bioanal Chem. 2014:1–13. doi:10.1007/s00216-014-7853-1.
Garcia-Reyes JF, Ferrer I, Thurman EM, Molina-Diaz A, Fernandez-Alba AR. Searching for non-target chlorinated pesticides in food by liquid chromatography/time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2005;19(19):2780–8. doi:10.1002/rcm.2127.
Mezcua M, Malato O, García-Reyes JF, Molina-Díaz A, Fernández-Alba AR. Accurate-mass databases for comprehensive screening of pesticide residues in food by fast liquid chromatography time-of-flight mass spectrometry. Anal Chem. 2009;81(3):913–29. doi:10.1021/ac801411t.
Gomez MJ, Gomez-Ramos MM, Malato O, Mezcua M, Fernandez-Alba AR. Rapid automated screening, identification and quantification of organic micro-contaminants and their main transformation products in wastewater and river waters using liquid chromatography-quadrupole-time-of-flight mass spectrometry with an accurate-mass database. J Chromatogr A. 2010;1217(45):7038–54. doi:10.1016/j.chroma.2010.08.070.
Diaz R, Ibanez M, Sancho JV, Hernandez F. Qualitative validation of a liquid chromatography-quadrupole-time of flight mass spectrometry screening method for organic pollutants in waters. J Chromatogr A. 2013;1276:47–57. doi:10.1016/j.chroma.2012.12.030.
Diaz R, Ibanez M, Sancho JV, Hernandez F. Target and non-target screening strategies for organic contaminants, residues and illicit substances in food, environmental and human biological samples by UHPLC-QTOF-MS. Anal Methods. 2012;4(1):196–209. doi:10.1039/c1ay05385j.
Martinez Bueno MJ, Ulaszewska MM, Gomez MJ, Hernando MD, Fernandez-Alba AR. Simultaneous measurement in mass and mass/mass mode for accurate qualitative and quantitative screening analysis of pharmaceuticals in river water. J Chromatogr A. 2012;1256:80–8. doi:10.1016/j.chroma.2012.07.038.
Pavlic M, Libiseller K, Oberacher H. Combined use of ESI–QqTOF-MS and ESI–QqTOF-MS/MS with mass-spectral library search for qualitative analysis of drugs. Anal Bioanal Chem. 2006;386(1):69–82. doi:10.1007/s00216-006-0634-8.
Diaz R, Ibanez M, Sancho JV, Hernandez F. Building an empirical mass spectra library for screening of organic pollutants by ultra-high-pressure liquid chromatography/hybrid quadrupole time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2011;25(2):355–69. doi:10.1002/rcm.4860.
Zhang K, Wong JW, Yang P, Hayward DG, Sakuma T, Zou Y, et al. Protocol for an electrospray ionization tandem mass spectral product ion library: development and application for identification of 240 pesticides in foods. Anal Chem. 2012;84(13):5677–84. doi:10.1021/ac300844d.
Zhao Z, Shi Z, Kang J, Peng X, Cao X, Fan C, et al. Rapid screening and confirmation of 281 pesticide residues in apples, tomatos and cabbages by liquid chromatography/ quadrupole time-of-flight mass spectrometry. Chin J Chromatogr. 2013;31(4):372–9.
Wang Z, Chang Q, Kang J, Cao Y, Ge N, Fan C, et al. Screening and identification strategy for 317 pesticides in fruits and vegetables by liquid chromatography-quadrupole time-of-flight high resolution mass spectrometry. Anal Methods. 2015;7(15):6385–402. doi:10.1039/C5AY01478F.
Malato O, Lozano A, Mezcua M, Agueera A, Fernandez-Alba AR. Benefits and pitfalls of the application of screening methods for the analysis of pesticide residues in fruits and vegetables. J Chromatogr A. 2011;1218(42):7615–26. doi:10.1016/j.chroma.2011.06.110.
Decaestecker TN, Vande Casteele SR, Wallemacq PE, Van Peteghem CH, Defore DL, Van Bocxlaer JF. Information-dependent acquisition-mediated LC-MS/MS screening procedure with semiquantitative potential. Anal Chem. 2004;76(21):6365–73. doi:10.1021/ac0492315.
Document No SANCO/12571/2013. Guidance document on analytical quality control and validation procedures for pesticide residues analysis in food and feed. http://www.eurlpesticides.eu/library/docs/allcrl/AqcGuidance_Sanco_2013_12571.pdf (2013). Accessed 22 Aug 2016.
Document No SANCO/12495/2011. Method validation and quality control procedures for pesticide residues analysis in food and feed. http://www.eurlpesticides.eu/library/docs/allcrl/AqcGuidance_Sanco_2011_12495.pdf (2011). Accessed 22 Aug 2016.
Lehotay SJ, Mastovska K, Amirav A, Fialkov AB, Martos PA, de Kok A, et al. Identification and confirmation of chemical residues in food by chromatography-mass spectrometry and other techniques. Trends Anal Chem. 2008;27(11):1070–90. doi:10.1016/j.trac.2008.10.004.
Acknowledgments
The authors acknowledge the financial assistance of the National Key Technology Research and Development Program (NO. 2012BAD29B01) and the Key Basic Research Program (NO. 2015FY111200) of the Ministry of Science and Technology, PRC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1584 kb)
Rights and permissions
About this article

Cite this article
Wang, Z., Cao, Y., Ge, N. et al. Wide-scope screening of pesticides in fruits and vegetables using information-dependent acquisition employing UHPLC-QTOF-MS and automated MS/MS library searching. Anal Bioanal Chem 408, 7795–7810 (2016). https://doi.org/10.1007/s00216-016-9883-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9883-3