
Abstract
Gas chromatography coupled to high resolution hybrid quadrupole time-of-flight mass spectrometry (GC-QTOF MS), operating in negative chemical ionization (NCI) mode and combining full scan with MSMS experiments using accurate mass analysis, has been explored for the automated determination of pesticide residues in fruit and vegetables. Seventy compounds were included in this approach where 50 % of them are not approved by the EU legislation. A global 76 % of the analytes could be identified at 1 μg kg–1. Recovery studies were developed at three concentration levels (1, 5, and 10 μg kg–1). Seventy-seven percent of the detected pesticides at the lowest level yielded recoveries within the 70 %–120 % range, whereas 94 % could be quantified at 5 μg kg–1, and the 100 % were determined at 10 μg kg–1. Good repeatability, expressed as relative standard deviation (RSD <20 %), was obtained for all compounds. The main drawback of the method was the limited dynamic range that was observed for some analytes that can be overcome either diluting the sample or lowering the injection volume. A home-made database was developed and applied to an automatic accurate mass data processing. Measured mass accuracies of the generated ions were mainly less than 5 ppm for at least one diagnostic ion. When only one ion was obtained in the single-stage NCI-MS, a representative product ion from MSMS experiments was used as identification criterion. A total of 30 real samples were analyzed and 67 % of the samples were positive for 12 different pesticides in the range 1.0–1321.3 μg kg–1.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fruit and vegetables are the primary food most consumed throughout the world. To keep the produce free from pests and plagues, feed the population, as well as to have reasonable business productivity, farmers apply pesticides through the whole production chain. But pesticides are noxious to human health and the environment, forcing food authorities to keep a strict control on the occurrence of its residues on foodstuff. This control can be only pursued if regulatory and surveillance organisms have the appropriate analytical methodologies which, in an effective and confident way, are able to check the accomplishment of the ruling maximum residue limits (MRLs) for the almost 900 pesticides in the agrochemical market. As new toxicological information is gathered, pesticide MRLs are continuously lowered [1, 2], forcing the development of more sensitive analytical methodology. Food for special groups of population like children and babies is specifically targeted [3], which bears the lowest MRLs and, in some cases, must be pesticides free, which is also the case for ecological products. For such cases, the limit of identification (LOI) and the limit of quantification (LOQ) of the analytical methods are never low enough. Sample preparation is a useful tool to reach high sensitivity but, nowadays, only coupled with the most advanced instrumental is it possible to determine trace amounts (mg kg–1) of hundreds of pesticides simultaneously, either approved or not in different matrices.
Increasing sensitivity in pesticide residue analysis is fostered by mass spectrometry developments. Twenty years ago, it has been through the use of single quadrupole (MS) in SIM mode. More recently, the advent of tandem mass spectrometry (MS-MS), which only can achieve unit mass resolution (MS) or are limited to the selected transitions (MS-MS), allowed the sensitive determination of pesticide residues in most of the food matrices. Few years ago, the use of high-resolution mass spectrometry (HRMS) has shown to be a useful tool for the determination and elucidation of known and unknown compounds [4, 5], at trace level, due to its high resolution power to discriminate between different molecular formula with the same nominal mass. As the full mass spectrum is recorded, it is possible to look for specific compounds afterwards through retrospective analysis. Nowadays, the trends in food and environmental science are the use of HRMS for pesticide residue and their metabolites determination, which can be applied in combination of different ionization modes as electron ionization (EI) [6–8] atmospheric pressure chemical ionization (APCI) [9–11], negative and positive chemical ionization (NCI and PCI, respectively) [6, 12, 13]. Particularly, HRMS and NCI are an excellent combination to provide a selective-scope, based on the difference of electronegativity between the compounds that can be ionized. Compounds bearing halogenated, NO2, or P ester groups are electron-acceptor and have enhanced response up to several orders of magnitude with NCI [14]. While EI commonly results in a more extensive fragmentation and covers a wide-scope of pesticides, NCI provides limited fragmentation and relevant M- ions in such spectra. Some reports show promising results using NCI in single stage-MS combined with μ-ECD [15] or with MSMS experiment at low resolution [16, 17], but there are few examples in the literature that report on the application of the coupling of NCI-QTOFMS, looking for accurate mass analysis with mass errors less than 5 ppm [6, 12]. The use of HRMS allows the use of libraries of exact masses for identification purposes and gives indications of possible analytes. The usefulness of this approach has been explored by our group either to identify emerging contaminants or their degradation products [18]. Furthermore, an automated mass accurate data processing method is an excellent tool to analyze huge amounts of samples in a short time, assuring the correct detection and determination of pesticide residues in ecological and pooled samples, where the absence of pesticide residues has to be assured.
The aim of this work was to evaluate the hybrid GC-QTOF instrumentation operating in negative chemical ionization mode, for the determination and quantification of 70 common pesticides (22.8 % of these are not included in the coordinated multi-annual control program of the European Union [19]). Using Mass Hunter software, we report on the evaluation of the automated accurate mass data processing method of the diagnostic ions generated by NCI mode in fruit and vegetables. The fitness of the method was checked through the analysis for pesticide residues of 30 real samples from the Almería area.
Experimental
Reagents
Acetone obtained from J.T. Baker (Deventer, The Netherlands), ethyl acetate (EtAc) from Fluka Analytical Pestanal (Steinheim, Germany), and acetonitrile (MeCN) from Sigma-Aldrich (Steinheim, Germany) were used throughout the work. All high purity reference standards were purchased from Dr. Ehrenstofer (Augsburg, Germany), Sigma-Aldrich (Steinheim, Germany), and Riedel-de Häen (Selze, Germany), and were stored at –30 °C. Individual pesticide stock solutions (1000–2000 μg mL–1) were prepared by dissolving reference standards in the appropriate solvent and stored in amber screw-capped glass vials in the dark at –20 °C. Two working solutions at 10 μg mL–1, used for the calibration, were prepared by diluting stock solutions in EtAc. Anhydrous magnesium sulphate (MgSO4) was obtained from Panreac Quimica S.A. and sodium chloride (NaCl) was from J.T. Baker.
GC-MS/MS system and chromatographic conditions
An Agilent 7890A gas chromatograph (Agilent Technologies, Palo Alto, CA, USA), equipped with an Agilent autosampler, was coupled to an accurate-mass quadrupole time-of-flight (Q-TOF) mass spectrometer, Agilent 7200, operating in NCI mode. Agilent MassHunter software GC-MS Acquisition ver. B07.01 and MassHunter Qualitative analysis B06.00 were used to acquire and to process data obtained, respectively.
The samples were injected using a multimode injector inlet in splitless mode. The injection volume was 2 μL and was carried out at 280 °C. Helium and nitrogen, both with a purity of 99.999 %, were used as carrier and collision gases, respectively. The GC separation was performed using two fused silica HP-5MS UI capillary column of 15 m × 0.25 mm inner diameter and a film thickness of 0.25 μm (Agilent, Palo Alto, CA, USA) connected by a capillary flow technology (CFT) union. The oven temperature was programmed as follows: 60 °C for 1 min; 40 °C min–1 to 120 °C, and finally up to 310 °C at 5 °C min–1. The total run time was 40.5 min with two additional minutes for backflushing at 310 °C. Backflushing was used to shorten the analysis time and reduce system maintenance. The source and interface temperatures were set to 150 and 280 °C, respectively, and a solvent delay of 4 min was selected in order to prevent damage in the ion source filament. Retention time locked (RTL) setting was used to eliminate the need for adjusting retention times of the compounds, employing chlorpyrifos methyl as the locking compound at the retention time of 18.11 min. The instrument worked at constant flow.
Backflush system
The end of the chromatographic column is connected to the second column through a CFT union (used as purged capillary flow device), which allows system backflushing. During the run time, the flow was set at 1.0 mL min–1 in the first column and 1.2 mL min–1 in the second column (with a difference of 0.2 mL min–1 over the flow in the first column). Once the analysis is finished, there is a 2 min post-run time where a change in the flow is set: 6 mL min–1 in the second column and consequently the flow in the first column decreased to –5.7 mL min–1.
Mass spectrometer conditions: negative chemical ionization
The ion source and quadrupole temperatures were fixed at 150 °C. Methane was used as reagent gas (purity 99.995 %) and was set at a flow of 40 % (2 mL min–1). The ionization energy was set at 70 eV and the emission current at 50 μA. Mass spectrometric grade perfluorotributylamine (PFTBA) was used daily as external calibrant in TOF mass calibration and, when a complete autotune was done, for quadrupole calibration. To ensure mass accuracy during analysis, a TOF mass calibration is carried out prior to each injection, being its eight m/z exact masses adjusted. This step can be programmed in the sequence table and when the internal mass calibration is not approved, the run is stopped automatically. Looking for the optimization of the MS parameters for each compound, all pesticides were monitored in full scan mode and TOF MS was registering at 2, 3, 4, 5, and 10 spectrum s–1, acquiring in the 50–550 m/z mass range. TOF MS resolution was about 12,000 (FWHM). For some pesticides, additional diagnostic ions can be obtained by fragmentation in the QTOF by performing acquisition in MS2 mode. For its acquisition, the optimum collision energy for each precursor ion was selected after evaluating the fragment intensities at different collision energies (ranging from 3 to 30 eV). For MS2 experiments, the quadrupole isolates the precursor ion at a medium MS resolution and the linear hexapole collision cell fragments it using nitrogen at 1.5 mL min–1.
Validation procedure
In order to validate the method, parameters as percentage of recovery, linearity, limits of identification and quantification, and repeatability were studied. For the recovery studies, five replicates at three different concentration levels were done. For this, three representative portions of 60 g of homogenized sample were spiked with the mixture standard solution. Each portion was weighed and transferred to a beaker and fortified homogeneously with 60, 300, and 600 μL of a 1000 μg L–1 standard solution in ethyl acetate in order to obtain 1, 5, and 10 μg kg–1, respectively. The mixtures were gently mixed in a beaker for 30 min and, following this, they were kept standing for another 30 min. After that, the ethyl acetate extraction procedure [20] described below was carried out. To determine linearity and detection limits, a volume of 50 μL of tomato blank extract was evaporated and reconstituted with an equal volume of the desired concentration of the mix in ethyl acetate in the range of 0.1–500 μg L–1. Finally, repeatability of the instrumental method was evaluated by analyzing five replicates at two concentration levels: 5 and 20 μg kg–1 on the same day.
Real samples
A total of 30 samples of various kinds of fruits, vegetables, and fresh herbs (apple, asparagus, aubergine, bean, chive, dill, lettuce, mango, onion, parsley, papaya, pear, pepper, pineapple, plum, potato, rosemary, spinach, tomato) were analyzed. Fruits and vegetables were purchased from different local markets in Almeria (south-eastern Spain) and fresh herbs were from a local herbal store. All samples were stored in their original packaging under the recommended conditions until use.
The fresh matrices were chopped and triturated separately. After homogenization, a 10 g portion of sample was weighed and transferred into a 50 mL PTFE centrifugal tube. Following this, 10 mL of ethyl acetate was added and then the tube was shaken vigorously for 3 s by hand. Afterwards, 8 g of anhydrous magnesium sulphate and 1.5 g of sodium chloride were added and the samples were shaken in an automatic axial extractor (AGYTAX; Cirta Lab. S.L., Madrid, Spain) for 15 min. The extract was then centrifuged (3500 rpm) for 5 min, ending up with the equivalent of 1 g of sample per mL in 100 % ethyl acetate. The obtained final extract was injected into the GC-(NCI)-QTOF MS and GC-(EI)-QqQ MS systems.
Results and discussion
Optimization of the GC-(NCI)-QTOF MS acquisition method
The mass selective detector with high resolution quadrupole time-of-flight analyzer was operated in negative chemical ionization mode. A home-made database containing the ions generated in the ionization was developed. To build this database, a 50 μg kg-1 solution of standards was injected and all ions observed were matched to its molecular formula. The retention time and the ions monitored for each pesticide, used for identification and quantification, are presented in Table 1.
The ionization energy was evaluated by two trials using 70 and 140 eV. These values were chosen for different reasons: 140 eV value was setting for default after adjusting the masses of the calibrant (the best signal for PFTBA was obtained using 140 eV), and 70 eV is a common value used in different laboratories. The results showed no difference in the molecular ionization between both potentials and showed chromatograms with the same sensitivity, so that 70 eV was chosen as ionization energy value along all the work.
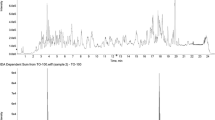
TOF MS was operated at 2, 3, 4, 5, and 10 spectrum s–1 acquiring in the 50–550 m/z mass range at 20, 50, and 100 μg kg–1, the latter being the concentration where saturation phenomena were expected. Nevertheless, the more influencing parameter in detector saturation during method development was the speed of spectra acquisition (see Fig. 1). At lower acquisition speed, higher sensitivity was observed and, consequently, a greater number of compounds saturated the detector (see Fig. 1). At the same acquisition rate and higher concentrations, the effect was more remarkable, as expected. Similar saturation phenomena for the same pesticides were observed at higher acquisition speed at 50 and 100 μg kg–1, respectively. The two concurrent phenomena registered at 2 spectrum s–1 compelled us to select a compromise acquisition speed of 3 spectrum s–1.

(a) Extracted ion chromatogram for azoxystrobin ion (m/z 371.0911) at different acquisition speed (spectrum s–1): black, blue, orange, green, and reed (2, 3, 4, 5, and 10 spectrum s–1, respectively); (b) saturation effect observed for tetradifon ion (m/z 317.9081) with the acquisition speed, comparing 2 and 3 spectrum s–1 (black and blue, respectively); (c) Saturation effect observed for vinclozolin ion (m/z 241.0066) at the selected acquisition speed (3 spectrum s–1) with various concentration levels: 5, 10, 20, and 50 μg kg–1 (red, green, blue, and black, respectively)
As we have mentioned, saturated peaks could be seen in some analytes at different concentration levels: 20, 50, 100, 200, and 500 μg kg–1. The detector saturation was evidenced especially for giving a double peak observed when the base peak (Q ion) at its exact mass was extracted with a narrow window of 10 ppm (see Fig. 1c). In some cases, no double peak was observed; nevertheless, the peaks had a plateau. In both cases, the experimental q/Q ratios (q: qualifier ion) using this saturated quantifier ion did not fit with the theoretical ones. Because of this phenomenon, the confirmation of analytes by evaluating the acceptable q/Q ion ratio could be a risk. To solve this problem, three different solutions can be useful: diluting samples, injecting less volume, and working with a higher number of spectrum s–1 lowering the sensitivity.
CID fragmentation (MS2 approach)
MS2 approach was employed as some pesticides presented only one fragment in full scan. For MSMS experiments, it was necessary to evaluate the speed of acquisition. Looking for the better conditions 20, 100, and 200 ms spectrum-1 were evaluated. From the data obtained, it could be seen that at higher acquisition speed, lower intensity was observed. Finally, 200 ms spectrum–1 was chosen to develop the method.
Precursor ion fragmentation was performed by collision induced dissociation with nitrogen, from which the best product ion was chosen. The collision gas flow was 1.5 mL min–1. The optimum collision energy for each precursor ion was selected after evaluating the intensity of each fragment ion at different collision energies (CEs). The adequate CE for each transition was assayed in the range between 3 and 30 eV. The suitable CE is the one for which the selected precursor ion produces relative abundance approximately 25 % with respect to the selected product ion. The transitions obtained with the possible molecular formula, its neutral exact mass, and CEs chosen are shown in Table 2.
Data processing method
After optimizing the acquisition parameters for each compound, a target method to process full scan MS and MS2 was developed. MassHunter Qualitative analysis was used to process data obtained from standards and samples in the analysis of target compounds. The processing method developed was linked to a home-made database of compounds to monitor the exact mass of each fragment ion using a narrow window of ±10 ppm and ±0.2 min over the established retention time. The home-made database contains two diagnostic ions from the full scan spectrum for each analyte with its molecular formula, exact neutral masses, retention times, and the names of the compounds. Detection is based both in the exact mass of each ion as in the retention time for each analyte according to the DG-SANCO [21]. The general methodology applied was based on the evaluation of the presence of the radical that was detected through its formula and searching for its accurate mass at the expected retention time. Two diagnostic ions were included for each pesticide in processing the screening method; in some cases, the two ions were from the MS spectrum and in others, one ion was from the MS spectrum and the other one was from the MS2 spectrum. For identification purposes, the mass accuracy requirement must be less than 5 ppm for at least one diagnostic ion.
Optimization of automated searching parameters
The target screening method was developed aiming to identify pesticides with mass accuracy errors less than 5 ppm and not detecting false positives or negatives automatically. Pursuing this objective, it was necessary to consider how the MassHunter software determines mass errors. It is important to note that in the database, neutral mass must be used, as the calculation performed by the software takes into account the mass of the electron when the ion is generated. If the mass accuracy is determined manually is necessary to add an electron mass at the theoretical neutral mass. In the software, some parameters are available to be used, such as the average scans that can be taken at different percentages of peak height. In our work, we evaluated three strategies for average scans: > 0 %, 35 %, and 50 % of peak height (see Fig. 2). Using these strategies, it was possible to see that highest errors were obtained at the beginning of the peak, and also in the tail. In light of these results, the error was estimated by calculating the average at the 50 % upper part of the peak. Also, as the error increased in the region of saturation, the criterion of excluding 10 % of the peak was set up. Other parameters for obtaining the extracted ion chromatogram (EIC), the cleaned spectrum for each analyte detected and extracting MS/MS spectrum per CE with precursor tolerance ±10 ppm can be set.

Three different approaches evaluated in the automatic search, varying the height of the peak (>10 %, >35 %, and >50 %) where the spectrum is selected to make an average while the part of the peak eliminated to avoid saturation is kept constant in 10 %
Application of automated home-made accurate mass data processing
Mass accuracy was evaluated in several injections at different concentration levels. Matrix matched calibrations were analyzed by the processing method looking for false negative detection while real samples previously analyzed in GC-QqQ MS served as a source of possible false positive detection. Retention time shifts were lower than 0.2 min for all the studied compounds and typical errors obtained from automated processed results of each chromatogram at 1, 5, and 10 μg kg–1 are shown in Table 3. For at least one ion of the total 70 analyzed pesticides, the accuracy of the masses measured was less than 5 ppm in almost 100 % at the three concentration levels. In the case of the second ion, the accuracy of the masses measured was less than 10 ppm, except for chinomethionate isotopic peak (C9H6N2S[34S]) and fenhexamid isotopic peak (C14H16[37Cl]NO2) that presented larger errors consistently. Several factors can affect the measure of mass accuracy when TOFMS is used. The principal one is due to saturation of the detector, but also variables such as temperature changes, data acquisition rate, and resolving power also play a role [6]. As five replicates at two concentration levels (5 and 20 μg kg–1) were injected to check the repeatability of the area for validation parameters, we were able to evaluate the repeatability of the automatic application of the home-made accurate mass database. No false negatives were obtained using the database and all the ions were detected in each analysis, under the parameters previously described.
Analytical parameters evaluated
Method sensitivity: limits of detection, limits of quantification, and linearity of calibration curves
Based on the calibration curves, the LOIs of the method were determined. A matrix matched calibration curve in tomato was injected, ranging from 0.1 to 500 μg kg–1. To establish the LOI for each pesticide, the two selected ions (in the case of pesticides fully identified with full scan) had to be present, at least one diagnostic ion with mass accuracy less than 5 ppm and a signal-to-noise ratio higher than 3. For pesticides in MS2, this criterion is needed for the precursor ion in full scan, whereas for the MS2 experiment only the presence of the transition is needed. MSMS experiment was developed for each case and at least one transition as the second parameter for a complete identification. The LOIs for most of compounds (75.7 %) were between 0.1 and 1 μg kg–1. However, around 23 % of the compounds had LOIs between 2 and 5 μg kg–1 and only myclobutanil had a LOI of 10 μg kg–1. The LOIs and LOQs obtained for the studied pesticides are presented in Table 4.
According to the DG-SANCO [21], LOQ is the lowest level where a full validation has been carried out accomplishing acceptability criteria (mean recoveries in the 70 %–120 % range with RSD ≤20 %). Recovery studies were developed at three different concentration levels (1, 5, and 10 μg kg–1) with five replicates for each concentration. The LOQ for each compound was also obtained from these experiments. Around 57 % of the studied analytes showed a 1 μg kg–1 LOQ, whereas less than 36 % presented 5 μg kg–1 LOQ and only 7 % of pesticides had 10 μg kg–1 of LOQ (aldrin, heptachlor, myclobutanil, propiconazole, and trifloxystrobin). In the case of propiconazole and trifloxystrobin, the RSDs obtained were 22 % and 26 %, respectively, at 10 μg kg–1. The linearity was evaluated in the 1–500 μg kg–1 range for all pesticides spiked in tomato. Some pesticides showed a narrow lineal range of calibration curves because of the saturation phenomena. In Table 4 is presented the linear range for each compound and its correlation coefficient (R2) values. For chinomethionate, the linear range is delimited to 1–10 μg kg–1, whereas for tetracholvinphos and tolyfluanid their quantification is only possible in the 5–10 μg kg–1 range, owing to a combined saturation problem of the diagnostic ions employed in single-stage MS and the low sensitivity achieved for the selected transition in the MSMS experiment.
The dynamic limited range observed for some analytes is the major drawback of the GCQTOF MS system used. This is a common problem of the TOF MS systems attributable to fast saturation of the detector [22] hampering the analyte quantification at different levels depending of the sensitivity of the compound. However, this problem can be overcome by selecting characteristic ion fragments with lower abundance or istopes (e.g., Cl34, C13) because the stability of the isotopic pattern in TOF MS is very high. This approach, when necessary, does not represent a big negative impact in the workflow of the laboratory. Other approaches such as sample dilution or smaller volume injection are also applicable. But, in these last cases, duplicate analyses should be necessary.
Recoveries and repeatability
In order to assay the method trueness, recoveries trials were performed as described earlier. The precision was also evaluated and it was expressed in terms of relative standard deviation (RSD). In the lowest fortified level (1 μg kg–1), recoveries ranging from 70 % to 120 % were accomplished in around 77 % of detected pesticides, whereas at 5 μg kg–1 94 % were in the acceptable range see Table 4. At 10 μg kg–1, all pesticides were detected and the recovery rates were in the range of 70 % to 120 %. The RSDs obtained in the recovery experiments were below 20 %, except for propiconazole (26 %), trifloxystrobin (22 %) at 10 μg kg–1, and nuarimol (25 %) at 1 μg kg–1.
The repeatability of the instrumental method was also evaluated at 5 and 20 μg kg–1 and was expressed in terms of RSDs. The results demonstrated that the method was repeatable, obtaining RSDs <15 % at 5 μg kg–1 and RSDs <20 % at 20 μg kg–1.
Analysis of real samples
In order to check the positives found by GC-(NCI)-QTOF, a comparison was carried out with results obtained by GC-(EI)-QqQ MSMS using two transitions in the MRM mode. Table 5 shows the positives of some pesticides found in real samples. In GC-(NCI)-QTOF, the developed automatic method described above (±10 ppm extraction window and ±0.2 min over the established exact masses and retention times, respectively) for the application of the home-made database was employed to the evaluation of real samples. Positives were compared with the matrix matched calibration curve, both in the q/Q ratio and in the intensity, for quantification. A 30 % tolerance in the q/Q ratio was allowed. The method proposed in the present communication allowed the quantification of eight pesticides for which the concentration level was below the LOQ of the QqQ, as well as detecting 14 pesticides that the triple Quad could not detect.
A total of 30 real samples were analyzed using GC-(NCI)-QTOF, resulting in 33.3 % of the samples that could be considered free of the target pesticides and 66.7 % of the samples that contained at least one pesticide. The range of concentration was between 1–2936 μg kg–1 and no pesticide exceeded the EU MRLs. Of the positive samples, 80 % contained one or two pesticides, whereas the rest had three or more pesticides. A total of 12 different pesticides were detected in all the samples. The most common pesticides found were chlorpyrifos ethyl (in 16 samples), bupirimate (in six samples), and iprodione (in four samples). Only one pesticide not approved by the European Union was found (chlorfenapyr at ˂LOQ), which could not be detected by GC-QqQ MSMS because of the LOI for this compound.
Conclusions
In the present work, a multiresidue acquisition procedure that can be coupled to an automatic data processing method has been implemented and validated for the simultaneous quantification of 70 pesticides in tomato using a home-made database. GC-QTOF operated in negative chemical ionization mode has proven to be a key tool for qualitative and quantitative determination of pesticides because of its high sensitivity and selectivity, providing very low LOIs (75.7 % of pesticides had a LOI ≤1 μg kg–1). The high selectivity of the NCI makes matrix interferences limited. This is related to the kinds of pesticides that are able to be analyzed in NCI mode, where most of them are halogenated ones, which usually have, by large, the lowest acute reference dose (ARfD) values. The use of QTOF was useful allowing the MSMS experiment, providing valuable fragmentation information, and high-mass accuracy of product ions for their use as second diagnostic ion in the identification step.
Automated accurate mass analysis in NCI was achieved using QTOF mass spectrometer with MassHunter. Data obtained for radical anions generated by NCI afforded mass accuracies within 5 ppm for most ions generated with excellent repeatability, with no false positives or negatives informed. In addition, 30 samples from Almería were analyzed, where chlorpyrifos, bupiramate, and iprodione were the most commonly-found pesticides with mass accuracies consistently below 5 ppm in at least one diagnostic ion. The reporting and detection limits of the GC-NCI(Q)TOF method were lower than the GC-(EI)-QqQ, a standard procedure for pesticide residue analysis, which was used for comparison as it was able to quantify and detect 22 affectional pesticides. From the results obtained, the combined use of HRMS and NCI shows the potential of a useful tool to analyze samples containing pesticide residues at very low concentrations and to ensure their absence in case of organically cultivated samples or baby foods. It is important to note that commercial libraries combining high resolution and different ionization modes are not currently available. This report provides an easy way to obtain this information when working with this type of automatic configuration.
References
Regulation (EC) No. 396/2005 of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending Council Directive 91/414/EEC (2005)
Commission Regulation (EU) No. 212/2013 of 11 March 2013 replacing Annex I to Regulation (EC) No. 396/2005 of the European Parliament and of the Council as regards additions and modifications with respect to the products covered by that Annex (2013)
Regulation (EC) No. 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control, and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009 (2013)
Hernández F, Sancho JV, Ibáñez M, Abad E, Portolés T, Mattioli L (2012) Current use of high-resolution mass spectrometry in the environmental sciences. Anal Bioanal Chem 403(5):1251–1264
Hernández F, Portolés T, Pitarch E, López FJ (2011) Gas chromatography coupled to high-resolution time-of-flight mass spectrometry to analyze trace-level organic compounds in the environment, food safety, and toxicology. TrAC Trends Anal Chem 30(2):388–400
Petucci C, Mallis L (2005) Automated accurate mass data processing using a gas chromatograph/time-of-flight mass spectrometer in drug discovery. Rapid Commun Mass Spectrom 19(11):1492–1498
Zhang F, Wang H, Zhang L, Zhang J, Fan R, Yu C, Wang W, Guo Y (2014) Suspected target pesticide screening using gas chromatography-quadrupole time-of-flight mass spectrometry with high resolution deconvolution and retention index/mass spectrum library. Talanta 128:156–163
Portolés T, Pitarch E, López FJ, Sancho JV, Hernández F (2007) Methodical approach for the use of GC-TOF MS for screening and confirmation of organic pollutants in environmental water. J Mass Spectrom 42(9):1175–1185
Cervera MI, Portolés T, López FJ, Beltrán J, Hernández F (2014) Screening and quantification of pesticide residues in fruits and vegetables making use of gas chromatography-quadrupole time-of-flight mass spectrometry with atmospheric pressure chemical ionization. Anal Bioanal Chem 406(27):6843–55
Portolés T, Mol JGJ, Sancho JV, Hernández F (2014) Use of electron ionization and atmospheric pressure chemical ionization in gas chromatography coupled to time-of-flight mass spectrometry for screening and identification of organic pollutants in waters. J Chromatogr A 1339:145–153
Nácher-Mestre J, Serrano R, Portolés T, Berntssen MHG, Pérez-Sánchez J, Hernández F (2014) Screening of pesticides and polycyclic aromatic hydrocarbons in feeds and fish tissues by gas chromatography coupled to high-resolution mass spectrometry using atmospheric pressure chemical ionization. J Agric Food Chem 62(10):2165–2174
Kazda R, Hajšlová J, Poustka J, Čajka T (2004) Determination of polybrominated diphenyl ethers in human milk samples in the Czech Republic: comparative study of negative chemical ionisation mass spectrometry and time-of-flight high-resolution mass spectrometry. Anal Chim Acta 520(1/2):237–243
Moniruzzaman M, Rodríguez I, Ramil M, Cela R, Sulaiman SA, Gan SH (2014) Assessment of gas chromatography time-of-flight accurate mass spectrometry for identification of volatile and semi-volatile compounds in honey. Talanta 129:505–515
Belmonte N, Uclés S, Gamón M, Ferrer C, Mezcua M, R. Fernández-Alba A (2014) Improving sensitivity and selectivity in pesticide analysis with GC-MS and GC-MS-MS. LCxGC Europe Advances in GC-MS analysis of pesticide. LCxGC Europe. http://www.chromatographyonline.com. Accessed 20 Sept 2014
Martínez-Uroz MA, Mezcua M, Valles NB, Fernández-Alba AR (2012) Determination of selected pesticides by GC with simultaneous detection by MS (NCI) and μ-ECD in fruit and vegetable matrices. Anal Bioanal Chem 402(3):1365–1372
Belmonte Valles N, Retamal M, Mezcua M, Fernández-Alba AR (2012) A sensitive and selective method for the determination of selected pesticides in fruit by gas chromatography/mass spectrometry with negative chemical ionization. J Chromatogr A 1264:110–116
Zhu P, Miao H, Du J, Zou JH, Zhang GW, Zhao YF, Wu YN (2014) Organochlorine pesticides and pyrethroids in Chinese tea by screening and confirmatory detection using GC-NCI-MS and GC-MS/MS. J Agric Food Chem 62(29):7092–7100
Gómez-Ramos MDM, Pérez-Parada A, García-Reyes JF, Fernández-Alba AR, Agüera A (2011) Use of an accurate-mass database for the systematic identification of transformation products of organic contaminants in wastewater effluents. J Chromatogr A 1218(44):8002–8012
Regulation (EC) No. 400/2014 of the European Parliament and of the Council of 22 April 2014 concerning a coordinated multiannual control programme of the Union for 2015, 2016 and 2017 to ensure compliance with maximum residue levels of pesticides and to assess the consumer exposure to pesticide residues in and on food of plant and animal origin (2014)
Uclés S, Belmonte N, Mezcua M, Martínez AB, Martinez-Bueno MJ, Gamón M, Fernández-Alba AR (2014) Validation of a multiclass multiresidue method and monitoring results for 210 pesticides in fruits and vegetables by gas chromatography-triple quadrupole mass spectrometry. J Environ Sci Health B 49(8):557–568
Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed. Document SANCO/12571/2013 (2013)
Thurman EM, Ferrer I, Fernández-Alba AR (2005) Matching unknown empirical formulas to chemical structure using LC/MS TOF accurate mass and database searching: example of unknown pesticides on tomato skins. J Chromatogr A 1067(1/2):127–134
Acknowledgments
The authors acknowledge funding support from the European Commission, DG SANCO, Specific Agreement No. 5 of Framework Partnership Agreement No. SANCO/2005/FOOD SAFETY/0025 Pesticides in Fruit and Vegetables. N.B. acknowledges the fellowship from ANII (Agencia Nacional de Investigación e Innovación), PEDECIBA (Programa de Desarrollo de las Ciencias Básicas), AUIP (Asociación Universitaria Iberoamericana de Postgrado), and CSIC, Uruguay (Comisión Sectorial de Investigación Científica) for economic support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection High-Resolution Mass Spectrometry in Food and Environmental Analysis with guest editor Aldo Laganà.
Rights and permissions
About this article

Cite this article
Besil, N., Uclés, S., Mezcúa, M. et al. Negative chemical ionization gas chromatography coupled to hybrid quadrupole time-of-flight mass spectrometry and automated accurate mass data processing for determination of pesticides in fruit and vegetables. Anal Bioanal Chem 407, 6327–6343 (2015). https://doi.org/10.1007/s00216-015-8514-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8514-8