
Abstract
Metabolomics protocols are used to comprehensively characterize the metabolite content of biological samples by exploiting cutting-edge analytical platforms, such as gas chromatography (GC) or liquid chromatography (LC) coupled to mass spectrometry (MS) assays, as well as nuclear magnetic resonance (NMR) assays. We have developed novel sample preparation procedures combined with GC-MS, LC-MS, and NMR metabolomics profiling for analyzing bronchial wash (BW) and bronchoalveolar lavage (BAL) fluid from 15 healthy volunteers following exposure to biodiesel exhaust and filtered air. Our aim was to investigate the responsiveness of metabolite profiles in the human lung to air pollution exposure derived from combustion of biofuels, such as rapeseed methyl ester biodiesel, which are increasingly being promoted as alternatives to conventional fossil fuels. Our multi-platform approach enabled us to detect the greatest number of unique metabolites yet reported in BW and BAL fluid (82 in total). All of the metabolomics assays indicated that the metabolite profiles of the BW and BAL fluids differed appreciably, with 46 metabolites showing significantly different levels in the corresponding lung compartments. Furthermore, the GC-MS assay revealed an effect of biodiesel exhaust exposure on the levels of 1-monostearylglycerol, sucrose, inosine, nonanoic acid, and ethanolamine (in BAL) and pentadecanoic acid (in BW), whereas the LC-MS assay indicated a shift in the levels of niacinamide (in BAL). The NMR assay only identified lactic acid (in BW) as being responsive to biodiesel exhaust exposure. Our findings demonstrate that the proposed multi-platform approach is useful for wide metabolomics screening of BW and BAL fluids and can facilitate elucidation of metabolites responsive to biodiesel exhaust exposure.

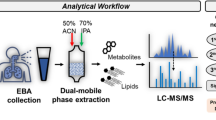
Graphical abstract illustrating the study workflow. NMR Nuclear Magnetic Resonance, LC-TOFMS Liquid chromatography-Time Of Flight Mass Spectrometry, GC Gas Chromatography-Mass spectrometry
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Metabolomics studies aim to comprehensively characterize the vast metabolite content of biological samples. Since these small molecular weight analytes are typically found at very different abundance levels and display diverse physicochemical properties, meticulous sample preparation protocols and detection techniques must be employed [1]. At present, no single metabolomics platform is able to cover the whole biochemical space of occurring metabolites. However, metabolomics assays have been developed for mass spectrometry (MS) and nuclear magnetic resonance (NMR) applications [1–6], which are largely complimentary techniques, at least for plasma analysis [7]. Hence, by using a multi-platform approach exploiting liquid and gas chromatography (LC and GC) coupled to MS as well as NMR, the largest possible portion of detectable analytes in a given sample can be covered. Subsequent classification of samples through multivariate analysis and identification of differentially expressed metabolites allow the sample class to be defined and provide information about disturbed metabolic pathways in relation to a particular stimuli or disease.
Metabolite profiling of lung lavage fluid presents a particular challenge due to extremely low metabolite levels and high salt content [8, 9]. Nevertheless, metabolomics profiling of bronchoalveolar lavage (BAL) fluid has been successfully used to assess silica exposure in an animal model for lung inflammation and in asthma models, as well as human lung injuries and pulmonary diseases such as cystic fibrosis, asthma, and respiratory distress syndrome [3, 4, 9–18]. As a result, key metabolic markers have been identified and used to enhance the understanding of the studied pathologies. To date, the analytical technique that has detected the greatest number of unique metabolites in a BAL sample (23) is LC-MS [3]. However, no study has yet used GC-MS, LC-MS, and NMR together to analyze aliquots of the same sample, which might have contributed to the limited number of metabolites identified in previous studies.
The spatial resolution of sampling for different lung compartments during metabolite profiling can be increased by performing a bronchial wash (BW) before BAL: The content of the BW reflects the metabolite profile of the central airways, while the BAL fluid reflects the metabolite profile of the lungs’ more peripheral regions. This sequential approach has been used to investigate compartment-specific fatty acid metabolite (oxylipin) profiles [19]. A similar analysis of BW and BAL fluids examining a wider range of metabolites could provide important insights into the effects of fuel exhaust exposure that would be complementary to information obtained by studying traditional markers of exposure such as cellular and soluble markers of inflammation [20–22].
The chemical and toxicological properties of particulate matter (PM) originating from fossil fuel combustion allow it to penetrate deep into the respiratory tract and cause significant damage to the lungs when inhaled [23]. The effects of exposure to such PM have been investigated in a variety of ways. For example, controlled chamber exposure studies have shown that exposure to petrodiesel exhaust induces neutrophil-dependent inflammation and adverse cardiovascular effects in healthy human subjects, and the underlying mechanisms have been thoroughly described [20–22, 24–30]. In addition, several epidemiological studies have shown that PM exposure is associated with multiple adverse health effects including increased risks of mortality, morbidity, cardiovascular events, respiratory symptoms, and distorted lung function [31–35]. Because global oil reserves are limited and there are significant concerns about the negative environmental and health effects of fossil fuel combustion, there is considerable pressure to replace fossil fuels with biofuels derived from renewable sources [36]. However, it is not yet clear how switching from fossil diesel to biofuels such as rapeseed methyl ester (RME) biodiesel will affect human health [37].
We hypothesized that metabolite profiling using multi-platform metabolomics assays (GC-MS, LC-MS and NMR) would overcome current limitations in the metabolite coverage of lung lavage fluids and reveal potential effects of biodiesel exhaust exposure on the metabolome that may indicate adverse effects on pulmonary health. To enable such profiling, we developed an extraction protocol for efficient identification of a wide range of metabolites in BW and BAL fluid collected from subjects exposed to biodiesel exhaust and filtered air in a randomized fashion. Metabolites whose levels differed between the two exposure treatments were identified, providing information on the metabolic pathways affected by exposure to biodiesel exhaust. To our knowledge, this study is the first to provide comprehensive information on the abundance of compounds spanning a wide range of metabolites in BW and BAL samples based on results acquired using three different platforms/assays.
Materials and methods
Chemicals
All reagents for GC-MS analysis were of analytical grade. Methyl stearate and methoxyamine were purchased from Sigma (St. Louis, USA). N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA), 1 % trimethylchlorosilane (TMCS), and pyridine (silylation grade) were purchased from Thermo Scientific (Rockford, IL, USA), and heptane was purchased from Fischer Scientific (Loughborough, UK). Water purified with a MilliQ gradient system (Millipore, Milford, MA, USA) was used to prepare all of the aqueous solutions for the assays.
All solvents and chemicals for the LC-MS analysis were of HPLC grade. Acetonitrile (ACN) was obtained from Merck (Darmstadt, Germany). Isopropanol was from VWR PROLABO (Fontenay-sous-Bois, France).
Sodium phosphates, including Na2HPO4 and NaH2PO4, were purchased at analytical grade or higher from Scharlau S.L. (Barcelona, Spain). Deuterium oxide (D2O, 99.9 % D) and the reference species sodium 3-trimethylsilyl [2,2,3,3-2H4] propionate (TSP) were purchased from Cambridge Isotope Laboratories, Inc. (Tewksbury, MA, USA). The preservative sodium azide (NaN3) was purchased from Sigma Aldrich (Buchs, Switzerland).
Study design
Fifteen non-smoking healthy individuals (eight males, seven females) were exposed to both pure biodiesel exhaust (RME100) with an average PM10 concentration of 159 μg/m3 and filtered air in a randomized, controlled, and double-blind crossover trial. The bronchoscopy sampling (BW and BAL) was performed after each exposure resulting in a total of 60 (15 × 2 × 2) lung lavage samples. The study participants gave their written consent and were informed of the risks and purpose of the procedures, which were conducted in accordance with the Declaration of Helsinki. The Regional Ethical Review Board at Umeå University approved the study (Dnr 2013-184-31M).
Each exposure session lasted for 1 h, during which subjects alternated between 15-min intervals of exercise and rest. Exposures were carried out at least 3 weeks apart in a human exposure chamber using the urban part of the European Transient Cycle, in order to mimic urban driving conditions [25, 28]. A Volvo engine (Volvo TD40 GJE, 4.0 L, 4 cylinders) was used to generate the exhaust emissions, and the controlled environment in the chamber was continuously monitored for pollutant gases (total hydrocarbons 0.8 ppm, NO2 0.6 ppm, NO x 6.9 ppm) as well as particulate number and concentration. The subjects abstained from alcohol and caffeine 24 h prior to exposure and were requested not to take any anti-inflammatory drugs or dietary supplements during the week before the experiment. Furthermore, they were instructed to have a light, ordinary breakfast (with no ham), and to eat as similarly as possible before the two exposures.
Bronchoscopy was performed, as described previously, using a flexible video bronchoscope (Olympus BF-1T160; Olympus, Tokyo, Japan) [38]. BW (2 × 20 mL) and BAL (3 × 60 mL) samples were extracted with sterile saline solution (pH 7.3, 37 °C) after the tip of the bronchoscope was carefully inserted into the lingual or middle lobe bronchus. Aspirates recovered from the first and second 20-mL instillations of the BW and pooled BAL fluid were collected in separate siliconized containers and immediately placed on ice. After collection, the fluids were centrifuged and the cell pellet removed and stored separately. The samples were then frozen and kept at −80 °C.
There were no significant statistical differences between the recoveries of lavage fluid in either the air or biodiesel exposure experiments as calculated with Student’s t test (see Electronic Supplementary Material (ESM) Table S1). Recoveries ranged from 33 to 70 % and 50 to 86 % for BW and BAL, respectively.
Sample preparation for GC-MS analysis
BW and BAL samples were thawed at room temperature and then subjected to a modified protocol for metabolite extraction that was originally developed by A et al. [39] and is used as a standard procedure at the Swedish Metabolomics Centre (SMC) in Umeå, Sweden. Samples (1 mL) were evaporated to dryness under vacuum with a miniVac QUATTRO concentrator (Genevac LTD, Ipswich, UK) for approximately 2–3 h at room temperature. Next, 30 μL of toluene was added to each sample to enhance the drying process, which was lengthy because of the samples’ high salt contents. The dried extracts were methoxymated with 15 μL of methoxyamine solution in pyridine (15 μg/μL) by heating at 70 °C in an oven for 1 h before being left to stand at room temperature for 16 h. After methoxymation, the samples were trimethylsilylated with 15 μL of MSTFA at room temperature for 1 h, then 15 μL of heptane (containing 0.5 μg of methyl stearate as an injection standard) was added. This modified procedure yielded a 20-fold higher concentration factor than was achieved with the unmodified protocol.
GC-MS analysis
One microliter of derivatized sample was injected splitlessly using a CTC Combi Pal autosampler (CTC Analytics AG, Zwingen, Switzerland) into an Agilent 7890A GC equipped with a 30 m × 0.25 mm i.d. fused-silica capillary column with a chemically bonded 0.25 μm DB5-MS stationary phase (J&W Scientific, Folsom, CA, USA). The injector temperature was set to 260 °C, and helium was used as a carrier gas at a constant flow rate of 1 mL/min. For routine analysis, the purge time was set to 75 s at a purge flow rate of 20 mL/min. The column temperature was initially held at 70 °C for 2 min, then increased from 70 °C to 320 °C at 20 °C/min and held at 320 °C for 12 min. The column effluent was introduced into the ion source of a Pegasus HT TOFMS (Leco Corp., St. Joseph, MI, USA), with the transfer line temperature set to 250 °C and the ion source temperature set to 200 °C. Ions were generated using a 70-eV electron beam. Masses were acquired from m/z 50 to 800 at a rate of 20 spectra per second, and the acceleration voltage was turned on after a solvent delay of 290 s. An alkane series (C10-C40) was run in conjunction with all samples. Samples (58 in total) were analyzed in randomized order to minimize the influence of instrumental drift on the results.
Processing of GC-MS chromatograms and compound identification
Sample files from the GC-MS analysis were exported in MATLAB 8.1 (R20013a) (Mathworks, Natick, MA, USA) in NetCDF format for further data processing and analysis. Custom Matlab-based scripts were used to perform alignment of chromatograms, peak detection, and identification based on retention indices and full MS spectra from the in-house mass spectra library established by the Umeå Plant Science Centre (UPSC) and SMC (Umeå, Sweden). Normalization of analyte peak areas against the peak area of the injection standard (methyl stearate) was done in Excel.
Sample preparation for LC-MS analysis
Samples were prepared as for the GC-MS analysis, except that after the drying step, samples were directly dissolved in 20 μL of a methanol/water mixture (1:1). The final procedure yielded a 20-fold higher concentration factor than was achieved with the unmodified protocol.
LC-MS analysis
Extracted samples (2 μL) were injected onto an Agilent UPLC system (Infinity 1290) equipped with a UPLC column (Acquity HSS T3, 2.1 × 50 mm, 1.8 μm C18 in combination with a 2.1 mm × 5 mm, 1.8 μm VanGuard precolumn; Waters Corporation, Milford, MA, USA). The UPLC system was coupled to an Agilent 6550 iFunnel Jet stream electrospray ion source Accurate-Mass QTOFMSMS (Agilent Technologies, Santa Clara, CA, USA). The mobile phases were MilliQ water with 0.1 % formic acid (A) and 75:25 acetonitrile/2-propanol with 0.1 % formic acid (B). The proportion of B in the eluent was increased from 0.1 to 10 % over 2 min at a flow rate of 0.5 mL/min, increased to 99 % over 5 min, held at 99 % for 2 min, and finally returned to the starting level of 0.1 % over 0.3 min. The proportion of B was then held constant for 0.5 min during which the flow rate was set to 0.8 mL/min, after which it was reduced to 0.5 mL/min for 0.1 min to reset the system for the next injection. Two aliquots of each sample were injected and analyzed per run, the first in positive ion mode and the second in negative ion mode. MS parameters were kept identical between the modes, with the exception of the capillary voltage, as described below.
A reference interface was connected to enable accurate mass measurements. The reference ions purine (4 μM) and HP-0921 (Hexakis (1H, 1H, 3H-tetrafluoropropoxy) phosphazine) at 1 μM, both purchased from Agilent Technologies (Santa Clara, CA, USA), were infused directly into the MS at a flow rate of 0.05 mL/min for internal calibration. The monitored masses for purine in positive and negative ion modes were m/z 121.05 and 119.03632, respectively; those for HP-0921 were m/z 922.0098 and 966.000725, respectively. The gas temperature was set to 150 °C, the drying gas flow to 16 L/min, and the nebulizer pressure to 35 psi. The sheath gas temp was set to 350 °C and the sheath gas flow to 11 L/min. The capillary voltage was set to 4000 V in positive ion mode and to 4500 V in negative ion mode. The nozzle voltage was 300 V. The fragmentor voltage was 380 V, the skimmer voltage was 45 V, and the OCT 1 RF Vpp was 750 V. The collision energy was set to 0 V, the m/z range was 70–1700, and data were collected in centroid mode at an acquisition rate of 4 scans/s (1977 transients/spectrum).
Processing of LC-MS chromatograms and compound identification
Targeted feature extraction of the acquired LC-MS data was performed using the Profinder™ software package, version B.06.00 (Agilent Technologies Inc., Santa Clara, CA, USA), together with a local retention time and mass spectra library containing data on 713 compounds. Feature detection was based on the following parameters: peak height ≥ 300 counts; allowed ion species in positive ionization mode + H, +Na, +K, +NH4; allowed ion species in negative ionization mode –H, +HCOO; peak spacing tolerance = 0.0025–7 ppm; isotope model, common organic molecules; charge state = 1; mass tolerance = 10 ppm; retention time tolerance = 0.1 min. After peak extraction, each compound was manually checked for mass and retention time agreement with appropriate standards from the in-house library. Peaks with poor characteristics (i.e., peaks that were overloaded, noisy, non-Gaussian, etc.) were excluded from the analysis. The initial identities assigned to peaks with a signal-to-noise ratio above 3 (relative to a blank) were verified by MS/MS analysis. To confirm compound identification, an appropriate standard was run in conjunction with two BW and BAL samples: MS/MS spectra obtained with collision energies of 10 and 40 V for samples and standards, respectively, were recorded and compared. Results for compounds with MS/MS spectra that matched those of relevant standards were included in the subsequent statistical analyses.
Sample preparation for NMR analysis
BW and BAL samples (1 mL each) were thawed at room temperature (25 °C) and evaporated to nearly dryness for 4 h. The residual material was then redissolved in a solution of 200 μL of D2O/H2O (4:1) containing 0.05 M sodium phosphate buffer, 0.5 mM of TSP, and 0.02 % of NaN3. Proton NMR spectra of the prepared samples were immediately acquired using a Bruker DRX600 spectrometer. This protocol was largely based on a previously published procedure, with minor changes relating to the reconstitution volume and protein precipitation step [40].
NMR analysis
One-dimensional proton NMR spectra were obtained using classic water suppression with a pre-saturation pulse sequence, which is a commonly used method for metabolite profiling of biofluids [41]. Spectra were acquired with 128 scans and transformed to 64 k data points.
Processing of NMR spectra
Spectral fitting was performed with the Chenomx NMR Suite professional software package (version 8.1, Chenomx Inc., Edmonton AB, Canada). All processed spectra were imported into the software under the environment of targeted profiling. The Chenomx NMR suite allows qualitative and quantitative analysis of an NMR spectrum by automatically fitting individual NMR resonances of interest using pure standard metabolite compound spectra stored in Chenomx.
Statistical methods
Read-outs from the assays under study, i.e., GC-MS (normalized peak areas), LC-MS (normalized peak areas), and NMR (quantified metabolites), were imported into the SIMCA software (version 14.0) from MKS Instruments AB (Umeå, Sweden) for multivariate analysis. All data were mean centered and scaled to unit variance. Principal component analysis (PCA) was used to check for trends and outliers in the data. Orthogonal partial least squares discriminant analysis (OPLS-DA) was used to compare metabolite profiles of different sample classes; 1 + 1 component models were used to avoid the risk of over-fitting [42]. Sevenfold cross-validation was used in the calculation of all models [43]. The significance of a metabolite for classification in the OPLS-DA models was specified by calculating the 95 % confidence interval for the loadings using jackknife [44].
Univariate analysis was performed with GraphPad Prism 6 (San Diego, CA, USA) to check the normality distribution for each metabolite and detect any significant differences between air and biodiesel exhaust exposure, as well as between BW and BAL sample types. Significant differences were assessed using Student’s t test and Wilcoxon’s t test for normal and non-normal distributions at p < 0.05, respectively. Receiver operating characteristic (ROC) curves [45] obtained from the OPLS-DA models were calculated in Matlab (R2014a, Mathworks; MA, USA) using predicted values for the fitted Y values for observations in the dataset computed with the cross-validation procedure.
Results and discussion
The modified extraction protocols were optimized with respect to sample concentration because of the expected low metabolite levels in lung lavage fluids. Omitting the protein precipitation step meant that sample dilution could be avoided. The derivatization step for the GC-MS analysis was performed as in the previous protocol [39] except that the volumes of the methoxyamine, MSTFA, and injection standard solutions were halved. Because the BW and BAL samples contained high quantities of salts, it was necessary to extend the drying time above 2 h and add toluene to speed up the drying process. The optimized modified procedure yielded a concentration factor that was 20 times greater than that achieved with the original protocol for the GC-MS and LC-MS analyses, and 5 times greater for NMR.
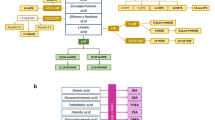
The GC-MS chromatograms (ESM Fig. S1A) were deconvoluted using an in-house script based on a local GC-MS library featuring data on 713 compounds, resulting in the detection of 76 distinct metabolites. Peaks with a signal-to-noise ratio above three (relative to blanks) were considered to have been satisfactorily identified, giving a total of 53 identified metabolites (Table 1). Fourteen metabolites with signal-to-noise ratios above three were identified in the LC-MS assays (Table 2) based on chromatograms in positive (ESM Fig. S1B) and negative (ESM Fig. S1C) mode, while 23 were identified based on NMR spectra (ESM Fig. S1D) using the Chenomx environment, excluding interference with the anesthetic applied prior to the lavage procedure (Table 3). While the detected metabolites collectively spanned a wide swath of biochemical space (including fatty acids, sugars, amino acids, and small organic acids), none were common to all of the analytical platforms. However, seven compounds were detected in both the GC-MS and NMR assays (glucose, glutamate, glycine, lactic acid, pyruvic acid, taurine, and valine), and one compound detected in the LC-MS assay was also detected by NMR (creatinine). The studied assays thus exhibited a high degree of orthogonality, with higher mass and less polar compounds (e.g., fatty acids) being detected by GC-MS and LC-MS while lower mass and more polar species (e.g., acetic acid and acetone) were detected by NMR (Fig. 1). It is thus advisable to perform all three assays in order to achieve the widest possible coverage of metabolites in BW and BAL samples. Our study is the first to provide comprehensive information on the abundance of compounds spanning a wide range of molecular weights in BW and BAL samples based on results acquired using three different platforms/assays. In previous studies, several metabolites were detected in human BAL fluid by GC-MS [15, 16]; however, not many were identified. Similarly, in previous NMR studies of human BAL samples, 19 metabolites were detected and quantified [10, 14, 15, 17]. In addition, 23 metabolites were identified and analyzed in earlier studies of BAL fluid using LC-MS [3, 46].

Schematic overview of the number of unique and commonly identified metabolites by the gas chromatography (GC) coupled to mass spectrometry (MS) assay, the liquid chromatography (LC) coupled to MS assay, and the nuclear magnetic resonance (NMR) assay; none were common to all three platforms
To determine whether the detected metabolites could be employed in a biological context, we performed pathway analysis with MetaboAnalyst 3.0 [47] on the GC-MS, LC-MS, and NMR metabolic sets. Data on the significance and impact of the represented pathways are presented in Tables S8-S10 (see ESM). Many canonical pathways were represented by the detected metabolites, although only a few were significant based on the calculated p values. The metabolites and metabolic pathways identified in this work related mainly to amino acid and fatty acid metabolism. For example, glycerolipid metabolism was the most significantly represented pathway in the GC-MS and NMR datasets based on both p values (which reflect the number of metabolites from the pathway that are detected) and pathway impact (which measures the degree to which each metabolite in the given pathway is connected to the other metabolites of that pathway). Fewer metabolic pathways were covered in the LC-MS dataset. This was probably because fewer metabolites were detected in the LC-MS analyses and those that were detected were less common than compounds such as amino and fatty acids, meaning that they were connected to fewer metabolic pathways. The wide coverage of biochemical pathways represented by the metabolites detected using the multi-platform approach shows that metabolomics analysis of BW and BAL samples has great potential for providing insights into processes occurring in the airways.
Differences in the metabolite profiles of BW and BAL fluid samples
OPLS-DA modeling indicated that the metabolite profiles determined for BW and BAL fluid samples obtained after filtered air exposure differed significantly from one another for all three of the assays used in this work, although LC-MS and NMR produced stronger models compared to GC-MS (Fig. 2). Merging data from all three platforms corroborated these results (ESM Fig. S2). We focused on filtered air exposure data when comparing BW and BAL samples to exclude potential variability due to the biodiesel exhaust exposure. The GC-MS data for the filtered air exposures indicated that 20 metabolites were significantly more abundant in the BW samples than in the BAL fluid (Table 1). The LC-MS data indicated that three compounds—chenodeoxycholic acid glycine conjugate, oxidized glutathione, and docosahexaenoic acid (22:6)—were present at significantly higher levels in BW than BAL fluid after filtered air exposure, while the reverse was true for another two compounds (caffeine and cortisone; see Table 2). Finally, the NMR data indicated that acetone, glutamine, glycine, and taurine were present at significantly higher levels in the BW samples, while alanine and dimethyl sulfone were significantly more highly concentrated in the BAL samples. For the seven metabolites detected by both NMR and GC-MS, the differences in the measured levels for the BW and BAL fluids by the two analytical methods were comparable in all cases except those of glucose and valine. The inconsistencies observed for glucose and valine may be due to the presence of other substances that influenced their detection by GC-MS and/or NMR and thus affected the deconvolution of the metabolite signal. The only compound detected by both LC-MS and NMR was creatinine, for which the levels determined by the two platforms were very similar in all cases.

OPLS-DA (1 + 1 + 0) score plots of metabolite profiles after filtered air exposure in BAL (open circles) and BW (closed circles) fluids. (A) GC-MS (R2X = 0.595, Q2(cum) = 0.290, CV-ANOVA p-value = 0.060), (B) LC-MS (R2X = 0.283, Q2(cum) = 0.586, CV-ANOVA p value = 0.00014), and (C) NMR (R2X = 0.574, Q2(cum) = 0.613, CV-ANOVA p value = 0.00023)
Relative standard deviations (RSDs) for each metabolite detected with GC-MS and LC-MS approaches are listed in ESM Tables S5 and S6, respectively. These values were higher than those typically observed for metabolomics assays in plasma, which can be as low as a few percent [48, 49]. Contributing factors for the higher variability could be the low concentrations of the metabolites (close to the noise level) and sub-optimal sampling and extraction protocols. The RSD values determined for the NMR data were in general lower than those for the GC-MS and LC-MS assays (ESM Table S7).
Biodiesel exhaust exposure alters the metabolite profiles of BW and BAL
The metabolite profiles were generally compartment-specific for both exposure regimes. However, some individual metabolites responded differently to biodiesel exhaust exposure in BW and BAL fluid. According to the GC-MS assay, levels of pentadecanoic acid in the BW samples increased significantly after biodiesel exhaust exposure, while in the BAL samples the levels of ethanolamine, inosine, and nonanoic acid increased significantly and those of 1-monostearoylglycerol decreased significantly (Table 4 and ESM Fig. S3). The trend towards decreased levels of fatty acid glycerol esters like 1-monostearoyglycerol and hexadecanoic acid methyl ester in response to biodiesel exhaust exposure in BAL samples and the increased levels of ethanolamine, phosphate, glycerol-3-phosphate, and unsaturated fatty acids suggest that biodiesel exposure enhances the degradation of cell membrane lipids. However, this conclusion is tentative and requires further validation.
The LC-MS assay indicated that biodiesel exhaust exposure led to reduced levels of chenodeoxycholic acid glycine conjugate in BW fluid and elevated levels of niacinamide in BAL fluid (ESM Fig. S4). Niacinamide is a component of the coenzyme NAD and is thus connected to the redox state of the cell.
The only metabolite found to respond significantly to biodiesel exhaust exposure in the NMR assay was lactic acid, whose concentration fell in the BW fluid (ESM Fig. S5). The GC-MS data also suggested that the levels of lactic acid in the BW and BAL samples fell after exhaust exposure, although the differences were not significant; this may have been due to the greater variability in the measured concentrations of this compound by GC-MS compared to NMR. Changes in lactic acid levels may be connected to glycolysis and/or gluconeogenesis. Lactic acid production in aerobic organisms increases when the supply of oxygen is not sufficient to meet the demand for its consumption, so it can serve as an early marker of imbalances in oxygen supply and demand.
We used OPLS-DA modeling to obtain an overview of how the metabolite patterns changed following biodiesel exposure. No significant model was produced using combined data from all three platforms. Only the GC-MS assay alone was able to detect significant shifts in the overall metabolite profile in response to biodiesel exhaust exposure (Fig. 3, data for individual metabolites are shown in Table 4). OPLS-DA models were constructed for the BW and BAL samples separately. Both models were valid according to cross-validation, meaning that we were able to observe a metabolic effect of biodiesel exhaust exposure in both BW and BAL samples. As demonstrated by the OPLS-DA cross-validated score values (Fig. 3), most of the subjects showed the same direction of change (metabolic response) between biodiesel exhaust and filtered air exposure, albeit with different magnitudes. However, a few subjects, namely the individuals labeled 4, 5, and 13 (BW) and 2, 4, and 6 (BAL), displayed deviating behavior. The fact that the magnitude of the metabolic response to biodiesel exposure differed between subjects is not surprising because it is already known that individuals respond differently to external stimuli. Thus, the results demonstrate that the presented methodology can be used to monitor exposure effects on an individual basis [41, 50].

Normalized cross-validated scores for the metabolite profiles of individual participants (t(cv)) derived from the OPLS-DA models for exposure to biodiesel exhaust and air based on GC-MS assays; normalized score vector for each individual was obtained by subtracting OPLS-DA cross-validated score for filtered air sample from the cross validated OPLS-DA score for biodiesel exposed sample, thus generating a measurement of the shift in profile; (A) bronchoalveolar lavage fluid (BAL) and (B) bronchial wash (BW). The results for most participants (other than subjects’ 2, 4, and 6 BAL samples and 4, 5, and 13 BW samples) indicate a positively directed shift in the composition of the metabolome after biodiesel exhaust exposure
The strongest OPLS-DA model (CV-ANOVA p value = 0.0009) relating biodiesel and filtered air exposure was obtained for the BAL samples (Fig. 4). The most significant metabolites for the OPLS-DA exposure model of BAL samples according to jack-knife intervals were 1-monostearylglycerol (negative correlation with biodiesel exposure) and sucrose, inosine, nonanoic acid, and ethanolamine (positive correlation with biodiesel exposure), in agreement with the univariate analysis. In the corresponding BW model, only pentadecanoic acid (positive correlation with biodiesel) was significant, again in agreement with the univariate analysis (Table 4).

Predictive correlated loading values (p(corr)) for different metabolites from the OPLS-DA model for the effects of biodiesel and air exposure in BAL samples measured by GC-MS. Metabolites are colored according to their p(corr) values and grouped according to chemical similarity; p(corr) values indicate how each metabolite is affected by biodiesel exposure
These results imply that the more peripheral regions of the lung were more responsive to biodiesel exhaust exposure with regard to metabolite levels. Another explanation could be that exposure effects were more visible in the BAL samples because of lower within-group variation compared to the BW samples for the majority of the detected metabolites (ESM Table S5), which may be reflected in the lower recovery of BW compared to BAL fluid (ESM Table S1). It is important to remember that metabolomics involves studying very small changes in metabolite levels that can be easily obscured by analytical and noise factors.
In addition, we tested the performance of the OPLS-DA models by creating receiver operating characteristic (ROC) curves based on predictions obtained from the cross-validated procedure (ESM Fig. S6). ROC curves [51] are widely used to evaluate the performance of diagnostic parameters in clinical studies. The accuracy of each model was estimated as the area under the curve (AUC), which was above 0.75 for all studied models. These results show that the approach based on OPLS-DA modeling of metabolic profiles and verification by constructing ROC curves can be used to accurately classify the effects of biodiesel exhaust exposure.
Conclusions
We have shown for the first time that extensive multi-platform metabolite profiling can be performed on BW and BAL fluid samples, providing important and novel information on the metabolome of the respiratory tract. Eighty-two different small molecule compounds were detected in BW and BAL samples from healthy individuals after air and biodiesel exhaust exposure. This largest to date number of unique metabolites detected in such samples proves that application of several orthogonal analytical platforms dramatically enhances the number of detected metabolites. Enhanced metabolite coverage is directly connected to the amount of covered biochemical pathways, which was shown by the extended number of covered pathways obtained after pathway enrichment analysis on all detected metabolites compared to the ones detected only by one platform. A larger number of biochemical pathways represented by the detected metabolites increase the chances for obtaining a valid biological interpretation of the studied system; thereby, it is a crucial issue in any metabolomics investigation.
Furthermore, we have shown that there are significant differences in the levels of certain metabolites found in BW and BAL samples. This proves that the sampling procedure presented herein, which was designed to enable separate sampling of the central and peripheral airways, provides new molecular information on the metabolome of the lungs and, by increasing the spatial resolution of airway sampling, can enhance our understanding of processes occurring in this organ.
In summary, we conclude that the BW and BAL samples contained significantly different metabolite profiles, demonstrating the importance of sampling them independently. Moreover, different kinds of biochemical information can be obtained by using samples from both of the corresponding compartments in lung studies, which is particularly important from a clinical perspective. For instance, we observed shifts in the metabolite profiles of the lungs following exposure to biodiesel exhaust when compared to filtered air, particularly in the BAL samples, and identified individual metabolites whose levels shifted significantly in response to the biodiesel exhaust. Because metabolite levels in BAL and BW fluids presumably reflect the molecular species produced, at least in part, by the lung epithelium, we conclude that exposure to air pollution derived from biodiesel exhaust can alter human lung metabolism. Our methodology represents a promising tool for future metabolomics studies on the lungs and further investigations into the effects of air pollution on human health.
Abbreviations
- AUC:
-
Area under the curve
- CV-ANOVA:
-
Cross-validation-analysis of variance
- GC-MS:
-
Gas chromatography-mass spectrometry
- LC-MS:
-
Liquid chromatography-mass spectrometry
- MVA:
-
Multivariate analysis
- NMR:
-
Nucleic magnetic resonance
- OPLS:
-
Orthogonal projections to latent structures
- OPLS-DA:
-
Orthogonal projections to latent structures-discriminant analysis
- PCA:
-
Principal component analysis
- RME:
-
Rapeseed methyl ester
- ROC:
-
Receiver operating characteristic
- RSD:
-
Relative standard deviations
- SMC:
-
Swedish Metabolomics Centre
- UPSC:
-
Umeå Plant Science Centre
References
Dettmer K, Aronov PA, Hammock BD. Mass spectrometry-based metabolomics. Mass Spectrom Rev. 2007;26(1):51–78.
Ellis DI et al. Metabolic fingerprinting as a diagnostic tool. Pharmacogenomics. 2007;8(9):1243–66.
Evans CR et al. Untargeted LC–MS Metabolomics of bronchoalveolar lavage fluid differentiates acute respiratory distress syndrome from health. J Proteome Res. 2014;13(2):640–9.
Ho WE et al. Metabolomics reveals altered metabolic pathways in experimental asthma. Am J Respir Cell Mol Biol. 2013;48(2):204–11.
Xu Y-J et al. Recent developments and applications of metabolomics in microbiological investigations. TrAC Trends Anal Chem. 2014;56:37–48.
Larive CK, Barding GA, Dinges MM. NMR Spectroscopy for metabolomics and metabolic profiling. Anal Chem. 2015;87(1):133–46.
Karimpour M et al. Postprandial metabolomics: a pilot mass spectrometry and NMR study of the human plasma metabolome in response to a challenge meal. Anal Chim Acta. 2016;908:121–31.
Walters EH, Ward C, Li X. Bronchoalveolar lavage in asthma research. Respirology. 1996;1(4):233–45.
Peng J et al. Metabolomic profiling of bronchoalveolar lavage fluids by isotope labeling liquid chromatography mass spectrometry: a promising approach to studying experimental asthma. Metabolomics. 2014;10(6):1305–17.
Singh C et al. Mini-bronchoalveolar lavage fluid can be used for biomarker identification in patients with lung injury by employing (1)H NMR spectroscopy. Crit Care. 2013;17(2):430.
Connett GJ. Bronchoalveolar lavage. Paediatr Respir Rev. 2000;1(1):52–6.
Hu JZ et al. Metabolomics in lung inflammation: a high resolution (1)h NMR study of mice exposed to silica dust. Toxicol Mech Methods. 2008;18(5):385–98.
Ho WE et al. Anti-malarial drug artesunate restores metabolic changes in experimental allergic asthma. Metabolomics. 2014;11(2):380–90.
Wolak JE, Esther CR, O’Connell TM. Metabolomic analysis of bronchoalveolar lavage fluid from cystic fibrosis patients. Biomarkers. 2009;14(1):55–60.
Fabiano A et al. Metabolomic analysis of bronchoalveolar lavage fluid in preterm infants complicated by respiratory distress syndrome: preliminary results. J Matern Fetal Neonatal Med. 2011;24(S2):55–8.
Julák J et al. Bronchoalveolar lavage examined by solid phase microextraction, gas chromatography–mass spectrometry and selected ion flow tube mass spectrometry. J Microbiol Methods. 2006;65(1):76–86.
Rai R et al. Metabolic profiling in human lung injuries by high-resolution nuclear magnetic resonance spectroscopy of bronchoalveolar lavage fluid (BALF). Metabolomics. 2013;9(3):667–76.
Ho WE et al. Metabolomics reveals inflammatory-linked pulmonary metabolic alterations in a murine model of house dust mite-induced allergic asthma. J Proteome Res. 2014;13(8):3771–82.
Larsson N et al. Lipid mediator profiles differ between lung compartments in asthmatic and healthy humans. Eur Respir J. 2014;43(2):453–63.
Brook RD et al. Insights into the mechanisms and mediators of the effects of air pollution exposure on blood pressure and vascular function in healthy humans. Hypertension. 2009;54(3):659–67.
Lucking AJ et al. Diesel exhaust inhalation increases thrombus formation in man†. Eur Heart J. 2008;29(24):3043–51.
Törnqvist H et al. Persistent endothelial dysfunction in humans after diesel exhaust inhalation. Am J Respir Crit Care Med. 2007;176(4):395–400.
Löndahl J et al. Measurement techniques for respiratory tract deposition of airborne nanoparticles: a critical review. J Aerosol Med Pulm Drug Deliv. 2013;27(4):229–54.
Sivagangabalan G et al. The effect of air pollution on spatial dispersion of myocardial repolarization in healthy human volunteers. J Am Coll Cardiol. 2011;57(2):198–206.
Mills NL et al. Diesel exhaust inhalation causes vascular dysfunction and impaired endogenous fibrinolysis. Circulation. 2005;112(25):3930–6.
Mills NL et al. Ischemic and thrombotic effects of dilute diesel-exhaust inhalation in men with coronary heart disease. N Engl J Med. 2007;357(11):1075–82.
Lundbäck M et al. Experimental exposure to diesel exhaust increases arterial stiffness in man. Part Fibre Toxicol. 2009;6:7–7.
Barath S et al. Impaired vascular function after exposure to diesel exhaust generated at urban transient running conditions. Part Fibre Toxicol. 2010;7(1):1–11.
Lucking AJ et al. Particle traps prevent adverse vascular and prothrombotic effects of diesel engine exhaust inhalation in men. Circulation. 2011;123(16):1721–8.
Mills NL et al. Combustion-derived nanoparticulate induces the adverse vascular effects of diesel exhaust inhalation. Eur Heart J. 2011;32(21):2660–71.
Hoek G et al. Long-term air pollution exposure and cardio- respiratory mortality: a review. Environ Health. 2013;12(1):43.
Pope CA, Ezzati M, Dockery DW. Fine particulate air pollution and US county life expectancies. N Engl J Med. 2009;360(4):376–86.
Brook RD et al. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121(21):2331–78.
Thibodeau LA et al. Air pollution and human health: a review and reanalysis. Environ Health Perspect. 1980;34:165–83.
Langrish JP et al. Cardiovascular effects of particulate air pollution exposure: time course and underlying mechanisms. J Intern Med. 2012;272(3):224–39.
Robbins M. Policy: fuelling politics. Nature. 2011;474(7352):S22–4.
Bünger J et al. Potential hazards associated with combustion of bio-derived versus petroleum-derived diesel fuel. Crit Rev Toxicol. 2012;42(9):732–50.
Salvi S et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159(3):702–9.
A J et al. Extraction and GC/MS analysis of the human blood plasma metabolome. Anal Chem. 2005;77(24):8086–94.
Hong J-H et al. Characterization of the biochemical effects of naphthalene on the mouse respiratory system using NMR-based metabolomics. J Appl Toxicol. 2014;34(12):1379–88.
Beckonert O et al. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat Protoc. 2007;2(11):2692–703.
Trygg J, Wold S. Orthogonal projections to latent structures (O-PLS). J Chemom. 2002;16(3):119–28.
Eriksson, L., Multi- and Megavariate Data Analysis. 2006: Umetrics AB.
Efron B, Gong G. A leisurely look at the bootstrap, the jackknife, and cross-validation. Am Stat. 1983;37(1):36–48.
Egan JP. Signal detection theory and ROC analysis. Series in Cognition and Perception; New York: Academic; 1975.
Cribbs SK et al. Metabolomics of bronchoalveolar lavage differentiate healthy HIV-1-infected subjects from controls. AIDS Res Hum Retrovir. 2014;30(6):579–85.
Xia J et al. MetaboAnalyst 2.0—a comprehensive server for metabolomic data analysis. Nucleic Acids Res. 2012;40(Web Server issue):W127–33.
Want EJ et al. Solvent-dependent metabolite distribution, clustering, and protein extraction for serum profiling with mass spectrometry. Anal Chem. 2006;78(3):743–52.
Crews B et al. Variability analysis of human plasma and cerebral spinal fluid reveals statistical significance of changes in mass spectrometry-based metabolomics data. Anal Chem. 2009;81(20):8538–44.
Masson P et al. Optimization and evaluation of metabolite extraction protocols for untargeted metabolic profiling of liver samples by UPLC-MS. Anal Chem. 2010;82(18):7779–86.
Egan JP. Signal detection theory and roc-analysis. New York: Academic press; 1975.
Acknowledgments
Financial support from The Swedish Research Council Formas, the Swedish Heart Lung Foundation, Västerbotten County Council (Spjutspetsmedel), AFA-Insurance, and the Kempe Foundation is gratefully acknowledged. The Swedish Metabolomics Centre (www.swedishmetabolomicscentre.se) is acknowledged for outstanding assistance with the analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Izabella Surowiec and Masoumeh Karimpour contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 2377 kb)
Rights and permissions
About this article

Cite this article
Surowiec, I., Karimpour, M., Gouveia-Figueira, S. et al. Multi-platform metabolomics assays for human lung lavage fluids in an air pollution exposure study. Anal Bioanal Chem 408, 4751–4764 (2016). https://doi.org/10.1007/s00216-016-9566-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9566-0