
Abstract
Modified nucleosides derived from the RNA metabolism constitute an important chemical class, which are discussed as potential biomarkers in the detection of mammalian breast cancer. Not only the variability of modifications, but also the complexity of biological matrices such as urinary samples poses challenges in the analysis of modified nucleosides. In the present work, a comprehensive two-dimensional liquid chromatography mass spectrometry (2D-LC-MS) approach for the analysis of modified nucleosides in biological samples was established. For prepurification of urinary samples and cell culture supernatants, we performed a cis-diol specific affinity chromatography using boronate-derivatized polyacrylamide gel. In order to establish a 2D-LC method, we tested numerous column combinations and chromatographic conditions. In order to determine the target compounds, we coupled the 2D-LC setup to a triple quadrupole mass spectrometer performing full scans, neutral loss scans, and multiple reaction monitoring (MRM). The combination of a Zorbax Eclipse Plus C18 column with a Zorbax Bonus-RP column was found to deliver a high degree of orthogonality and adequate separation. By application of 2D-LC-MS approaches, we were able to detect 28 target compounds from RNA metabolism and crosslinked pathways in urinary samples and 26 target compounds in cell culture supernatants, respectively. This is the first demonstration of the applicability and benefit of 2D-LC-MS for the targeted metabolome analysis of modified nucleosides and compounds from crosslinked pathways in different biological matrices.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Breast cancer is the most frequently diagnosed type of cancer and the leading cause of death by cancer among females. Twenty-three percent of all cancer cases are breast cancer cases and 14 % of all deaths by cancer can be traced back to breast cancer [1].
Besides the analysis of genomic and proteomic profiles, the understanding of biochemical processes based on metabolites is picked out as a central theme in order to find characteristic biomarkers for breast cancer. Tumor markers can be produced by cancer cells or by healthy cells as a reaction to the disease. These markers can be single-protein-, RNA-, DNA-based markers as well as a molecular signature consisting of multiple compounds [2]. Regarding this, a metabolic signature of modified nucleosides has been suggested as potential biomarkers for the early diagnosis of human breast cancer [3, 4].
The altered RNA metabolism of cancer cells results in elevated excretion levels of modified nucleosides in different biological fluids. It has been reported that especially the tRNA turnover rate in tumor tissue exceeds the tRNA turnover rate in normal tissue resulting in rapid degradation and excretion of modified nucleosides [5]. As an explanation for differences of base composition in tumor tRNA, numerous reasons have been discussed, such as changes in tRNA concentration, presence of tRNA with altered sequences and aberrant modifications [6]. Regarding this phenomenon, biological matrices, e.g., urine [4, 7, 8] have been analyzed in order to find preferably specific and sensitive biomarkers for the early diagnosis of breast cancer.
In the nucleolus, RNA can be modified posttranscriptionally by several enzymes resulting in a diversity of modifications [9]. Modified RNA is degraded to modified nucleosides in the cytoplasm. The unmodified nucleosides adenosine, guanosine, uridine, and cytidine can be recycled in the salvage pathway and returned into the nucleus or can be excreted. Due to the lack of specific kinases for the synthesis of modified nucleoside triphosphates in mammalian cells, modified nucleosides do not enter the salvage pathway for RNA rebuilding and therefore they are excreted quantitatively as metabolic endproducts. Consequently, the insertion of modified nucleoside triphosphates into inappropriate positions in tRNA or rRNA is avoided [10].
Urine is one of the most commonly used and chemically complex biofluids containing typical metabolic breakdown products [11]. The application of urinary samples for the detection of a metabolic signature as biomarker for breast cancer would be beneficial because this would permit the development of a non-invasive diagnostic tool. Due to the high complexity of the biological matrices, we intended to establish a 2D-LC method being able to increase the peak capacity and therefore minimize the ion suppression effects of the matrix.
Nucleosides have been analyzed by numerous methods before, e.g., high performance liquid chromatography (HPLC) coupled to time of flight (TOF) MS via electrospray ionization (ESI) [3], capillary electrophoresis (CE) TOF MS [12], liquid chromatography (LC) ion trap (IT) MS [13], LC-ESI-MS [14, 15], ESI Fourier transform ion cyclotron resonance (FTICR) MS [4] or matrix assisted laser desorption/ionization (MALDI) TOF-MS [16, 17]. LC-ESI-MS provides a highly sensitive and selective analytical technique with a soft ionization technique, but has shown to be susceptible to matrix signal suppression effects [18].
Numerous investigations have been made leading to a better understanding of the molecular mechanism of electrospray ionization as well as matrix and analyte effects in electrospray ionization [19, 20].
Since metabolomics often deals with distinguishing marks between two different biological states there is a need for improvement regarding the analysis of target compounds in difficult biological matrices. The complexity and high dynamic range of metabolite concentrations pose the challenge of qualitative and quantitative analysis. Therefore, a highly effective chromatographic separation is mandatory. The use of two-dimensional (2D)-LC may be an effective alternative to carry out the analysis of modified nucleosides in complex samples.
There are two general criteria in multidimensional separations that have been postulated by Giddings [21]. The first criterion is that components are subjected to two or more largely independent separative dimensions. The second is that the separation must be structured such that whenever two components are adequately resolved in any one displacement step, they will generally remain resolved throughout the process.
2D-LC can be classified into two basic techniques: In comprehensive 2D-LC, the whole effluent from the first dimension is transferred onto the second dimension, using a dedicated switching valve and running ultrashort gradients. In heart-cutting 2D-LC, only one or two cuts from the first dimension effluent are transferred onto the second dimension with longer gradients than in comprehensive 2D-LC [22].
Erni and Frei [23] executed a 2D-LC approach for the first time in 1978. It has been shown that online 2D-LC approaches coupled to MS can be applied for analysis of different structural classes in biological samples [24–26]. The implementation of an orthogonal second dimension separation can increase peak capacity in complex mixtures resulting in the product of peak capacities of the first and second dimension separation [27]. The concept of peak capacity was introduced by Giddings in 1967 [28]. To maximize the separation utilizing the multiplicative behavior of the peak capacities of the first and second dimension, the column separation mechanisms should be as orthogonal as possible. Orthogonality is generally understood as the independency of two separation mechanisms [22]. This challenge can be mastered either by generally different analytical techniques, e.g., standard HPLC and GC-MS or HPLC and SFC, but also by different LC systems, consisting of stationary and mobile phase. Li et al. have shown that a continuously shifted gradient can be beneficial in analysis of complex samples using comprehensive 2D-LC because of the combination of the peak compression effect of full gradient mode and the tailed gradient program in parallel gradient mode [29].
The aim of this work was to develop a 2D-LC-MS method for the detection of compounds from RNA metabolism and crosslinked pathways. In the course of this targeted metabolomics approach, different biological matrices with complex structural composition should be investigated. The peak capacity and orthogonality of the two chromatographic separations should be maximized in order to separate not only co-eluting matrix components from target substances but also target substances themselves.
Materials and methods
Chemicals
Methanol and acetonitrile (LC-MS grade, Carl Roth GmbH & CO, Karlsruhe, Germany), ultrapure water prepared from Purelab Ultra laboratory water purification system (Elga LabWater, Celle, Germany) and analytical-grade formic acid (FA) (Carl Roth GmbH & CO, Karlsruhe, Germany) for 2D-LC analysis were used. Ammonium acetate (VWR international, Darmstadt, Germany), sodium chloride (Carl Roth GmbH & CO, Karlsruhe, Germany), and boronate-derivatized polyacrylamide gel (Biorad, Richmond, USA) were used for affinity chromatography. Inosine, adenosine, N 4-acetylcytidine, uridine, 5,6-dihydrouridine, 5-aminoimidazol-4-carboxamid 1-β-d-ribofuranosid, 5-methyluridine, 1-methyladenosine, N 6-methyladenosine, 1-methylguanosine, N 2-methylguanosine, 5-desoxy-5-methylthioadenosine, xanthosine dihydrate, and nucleoside test mix were purchased from Sigma-Aldrich (Munich, Germany). Pseudouridine and isoguanosine were purchased from Carbosynth (Berkshire, UK). N 2,N 2,7-trimethylguanosine was purchased from Biolog (Bremen, Germany).
Cell culture and culture conditions
The starting culture of the breast cancer cell line MCF-7 was kindly donated by Prof. Stickeler of University Hospital Freiburg, Germany. MCF-7 was grown in RPMI 1640 (Life Technologies, Darmstadt, Germany)/10 % Newborn Calf Serum (Life Technologies, Darmstadt, Germany)/1 % penicillin/streptomycin (Life Technologies, Darmstadt, Germany)/1 % HEPES (Life Technologies, Darmstadt, Germany). The starting culture of the reference cell line MCF-10A was provided by Gillian Lehrbach and Roger Daly, Garvan Institute of Medical Research, Sydney. MCF-10A was grown in DMEM F12 (PAN-Biotech GmbH, Aidenbach, Germany)/5 % horse serum (PAA, Cölbe, Germany)/1 % glutamine (PAN-Biotech GmbH, Aidenbach, Germany)/1 % HEPES (PAN-Biotech GmbH, Aidenbach, Germany)/1 % penicillin/streptomycin (PAN-Biotech GmbH, Aidenbach, Germany)/250 μg hydrocortisone (Sigma-Aldrich, Munich, Germany)/50 μg cholera toxin (Sigma-Aldrich, Munich, Germany)/10 μg epidermal growth factor (R&D Systems, Wiesbaden-Nordenstadt, Germany)/4.858 mg insulin (Novo Nordisk Pharma GmbH, Mainz, Germany). Cells were seeded in 75 cm2 plastic flasks (Greiner Bio One GmbH, Frickenhausen, Germany) and grown at 37 °C/5 % CO2. After reaching confluence, cells were washed with 8 mL 1× PBS (Life Technologies, Darmstadt, Germany) twice and incubated with 5 mL 1× trypsin/EDTA (Life Technologies, Darmstadt, Germany) at 37 °C/5 % CO2. Trypsin triggered detachment of adherently growing cells was stopped by adding medium with serum. To check the cell viability 10 μL cell suspension was mixed with 40 μL Trypan Blue solution, consisting of 0.5 % Trypan Blue (Sigma-Aldrich, Munich, Germany) and 0.9 % NaCl in double-distilled water, and incubated for 5 min at 37 °C/5 % CO2. The number of living cells was averaged using a Neubauer counting chamber under a light microscope. Subsequently, cells were centrifuged (400g, 5 min, 4 °C) in order to remove the serum-containing medium. Afterwards, defined numbers of cells were seeded in three flasks for each cell line using the proper media containing additives without serum (sera contain nucleosides and individual components). After four days of incubation (37 °C/5 % CO2) the supernatant was collected and centrifuged (1000 g, 10 min, 4 °C) to remove cell debris. The supernatant was stored at −20 °C.
Extraction of cis-diols
Following the method from Liebich et al. [30], cis-diol-specific affinity chromatography applying boronate-derivatized polyacrylamide gel was used to purify the samples before 2D-LC analysis. Urinary samples of healthy volunteers (20 mL) were spiked with 200 μL of an internal standard solution (0.25 mM isoguanosine), alkalized to pH 8.8 with ammonia solution and put on a column (column volume, 200 mL; column diameter, 15 mm) with 500 mg boronate-derivatized polyacrylamide gel. Washing was achieved with 25 mL ammonium acetate solution (0.25 M, pH 8.8) and 6 mL methanol/water (1:1, v/v). The cis-diols were eluted with 20 mL 0.2 M formic acid in methanol/water (1:1, v/v). In case of cell culture supernatants, 50-mL samples spiked with 500 μL internal standard solution were eluted with 50 mL 0.2 M formic acid in methanol/water (1:1, v/v). The solvent was removed by evaporation and the pellet was dissolved in water. Thereby, the eluted fraction was concentrated 50-fold for urinary samples, 150-fold in case of cell culture supernatants. Injection volumes for the residues of urinary samples and cell culture supernatants were 5 and 15 μL, respectively.
Instrumentation
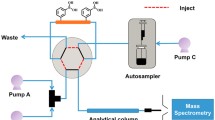
The 2D-LC analysis was performed on an Agilent 1290 Infinity 2D-LC System (Agilent, Waldbronn, Germany) consisting of a 1290 Infinity Quaternary Pump (G4204A), a 1290 Infinity Binary Pump (G4220A), a 1290 Infinity Autosampler (G4226A) with 1290 Thermostat (G1330B), coupled to a 1290 Infinity Valve Drive (G1170A) with a 2pos/4-port Duo Valve.
For the first dimension, the following columns were tested: Zorbax SB-Aq (100 × 2.1 mm, 3.5 μm), Eclipse XDB-C18 (150 × 4.6 mm, 5 μm), Zorbax HILIC Plus (150 × 2.1, 1.8 μm) and Eclipse Plus C18 (150 × 2.1 mm, 1.8 μm). The following second dimension columns were applied during method development: Zorbax Bonus-RP (50 × 3 mm, 1.8 μm), Zorbax Bonus-RP (50 × 4.6 mm, 1.8 μm), Zorbax Eclipse Plus C18 (50 × 3 mm, 1.8 μm), Poroshell 120 PFP (50 × 3 mm, 2.7 μm), Poroshell 120 Phenyl Hexyl (50 × 3 mm, 2.7 μm). For the final column combination, we used a Zorbax Eclipse Plus C18 (150 × 2.1 mm, 1.8 μm) column in the first dimension and a Zorbax Bonus-RP (50 × 4.6 mm, 1.8 μm) column in the second dimension. The effluent from the first dimension was continuously transferred to the second dimension column, achieved by filling two loops alternately using a valve switching. The setting of the valve/loop configuration is depicted in Fig. 1, the volume of both loops was 80 μL for each. In the first dimension, we used the following solvents at a flow rate of 0.07 mL/min: H2O + 0.1 % FA (A) and methanol + 0.1 % FA (B). In the second dimension, the following solvents were used at a flow rate of 2.5 mL/min: H2O + 0.1 % FA (A) and acetonitrile + 0.1 % FA (B). The idle flow was set to 0.5 mL/min. The modulation time was set to 54 s. A flow rate of 300 μL/min was directed to the ESI source of the MS system using a metallic T-piece. The applied gradients are depicted in the Electronic Supplementary Material (ESM) Fig. S1. The following gradient was used for the first dimension: 0–20 min, 2 % B; 20–45 min; 2–30 % B; 45–65 min, 30 % B; 65–75 min, 30–50 % B; 75–85 min, 50–75 % B; 85–90 min, 75 % B; 90.1 min, 2 % B. The 2D gradient is illustrated in the ESM Table S2. After each run, the columns were flushed for 5 min at 98 % B, for re-equilibration we flushed at starting conditions for 10 min in both column dimensions.

Schematic depiction of the valve/loop configuration
Mass spectra were acquired on an Agilent 6460 triple quadrupole mass spectrometer (Agilent, Waldbronn, Germany) equipped with an electrospray ionization source (Agilent Jet Stream) using the following settings: capillary voltage, 4000 V, nozzle voltage to 500 V; gas temperature, 300 °C (7 l/min); sheath gas, 350 °C (7 l/min); nebulizer pressure, 50 psi. All scan modi were carried out in the positive ionization mode with a step size of 0.1 amu, a threshold of 0 and a cell accelerator voltage of 7 V. The data storage was centroid. Nitrogen was used as collision gas.
Fragmentor voltages and collision potentials of the purchased reference standards were determined using MassHunter Optimizer Software (Agilent, Waldbronn, Germany). By application of the 1290 Infinity Binary Pump and the 1290 Infinity Autosampler 10 μl of each reference standard solution (0.1 mg/mL) was injected into the Agilent 6460 triple quadrupole mass spectrometer for 1 min with a flow rate of 0.5 mL/min. The mobile phase consisted of water and methanol at 98:2 (v/v) ratio acidified with 0.5 % formic acid. The fragmentor course range was set to 50–200 V and the collision energy range was set to 5–60 V. The low mass cut-off was set to 50 m/z and the optimization dwell time was 20 ms. Compounds that were not available as standard substance were detected in full scan, product ion scan, and MRM scan. Collision potentials were set according to product ion scans (data not shown). Therefore, the following collision potentials were tested at a fragmentor voltage of 100 V and a scan time of 300 ms: 5, 10, 20, 30, 40, 50, 60 V. The fragmentation pattern was compared to databases [31–33] and literature [4, 30]. For the full scan, the fragmentor voltage was set to 135 V. The mass range was m/z 50–700 with a scan time of 500 ms. The applied fragmentor voltages and collision potentials applied for MRM scans are summarized in Table 1. The dwell time was set to 5 ms and the cell accelerator voltage was set to 7 V. The neutral loss scans were performed at a fragmentor voltage of 100 V and a collision potential of 10 V. The scan time was set to 400 ms.
The data was acquired on a personal computer with Agilent MassHunter Workstation Data Acquisition B.04.01, which was externally started by a separate personal computer with OpenLab CDS ChemStation 2D LC A.01.05. For post-processing, Agilent MassHunter Qualitative Analysis B.04.00 (Agilent, Waldbronn, Germany) and LCxLC-HRMS Version 2.4b2 (GC Image, Lincoln, Nebraska, USA) were used. Log K OW values have been calculated by Estimation Programs Interface Suite™ v 4.11 (United States Environmental Protection Agency, Washington, DC, USA).
Results and discussion
At present, modified nucleosides are discussed as being potential biomarkers for breast cancer [3, 4, 7, 34–37]. In the present study, we established a comprehensive 2D-LC method with a triple quadrupole mass spectrometer detection, which can be applied to the analysis of nucleosides and derivatives from pathways crosslinked with the RNA metabolism. Collectively, we investigated 22 compounds from RNA metabolism, three from polyamine/methionine cycle, one from histidine metabolism, two from purine metabolism, and three from nicotinate and nicotinamide metabolism. Exemplary chemical structures of target metabolites are depicted in Fig. 2. Therefore, we analyzed urinary samples of healthy volunteers, cell culture supernatants, and standard substances. To the authors’ best knowledge, this is the first time that modified nucleosides from RNA metabolism have been analyzed by 2D-LC-MS.

Structures of some exemplary target compounds (MTA: 5-desoxy-5-methylthioadenosine, m 1G: 1-methylguanosine, Ψ: pseudouridine, t 6 A: N 6-threonylcarbamoyladenosine, 3,4-PCNR: 1-ribosyl-pyridin-4-one-3-carboxamide, AICAR: 5-aminoimidazol-4-carboxamid 1-β-D-ribofuranosid, ms 2 t 6A: 2-methylthio-N 6-threonylcarbamoyladenosine, m 2 G: N2-methylguanosine, m 3 C: 3-methylcytidine)
For prepurification, we extracted the cis-diols of urinary samples and cell culture supernatants using phenylboronic acid affinity chromatography. Because the prepurification by phenylboronic acid chromatography mainly separates low molecular weight substances with cis-diol group, such as nucleosides, catecholamines, coenzymes, and sugars, the main matrix components should be polar as well as the target compounds, too. Therefore, the two-dimensional separation should be an improvement concerning the most matrix components. According to this as the first targeted approach for the analysis of modified nucleosides by 2D-LC-MS, a stable and reproducible method had to be evaluated.
In 2D-LC, the eluent from the first dimension is continuously transferred to the second dimension column, achieved by filling two loops alternately using a valve switching. The valve (Fig. 1), which was designed exactly to be used as interface of the first and second dimension, offers two absolutely symmetrical flow paths in order to avoid any interferences when changing the positions. This cannot be achieved in this accuracy, when using a setup with a 2/10-valve, which needs one additional bridging capillary. Also, a setup with two 2/6-valves, which is sensitive to any disturbance in one of the two valve or valve drives can lead to noisy baseline characterized by binary patterns. In the described 2D-LC, 2pos/4-port valve two concentric circles with symmetrical connections eliminate these potential issues and help to ensure the best repeatability of the measurements.
Column selection
Because of the exceptionally high diversity of chemical functionalities in metabolomic samples, the coupling of two reversed-phase materials has great potential. Based on the good chemical selectivity, applicability to numerous chemical classes and reproducibility, reversed-phase chromatography is commonly used in metabolomics [27]. There are numerous reversed-phase materials available characterized by selectivity parameters [38], but the combination of two reversed-phase materials may reduce orthogonality between the columns. Nevertheless, there is a potential in 2D-LC applications using reversed phases because the separation is much more versatile than in normal phase, ion exchange chromatography (IEC), and size exclusion chromatography. The versatility and mechanisms of gradient elution in reversed-phase chromatography have been studied extensively [39–41]. Particularly in metabolomics, the samples’ chemical diversity cannot be analyzed utilizing, e.g., IEC in one of the both dimensions, making the combination of reversed-phase separations an attractive alternative [27].
When coupling two different LC systems, a flow instability phenomenon called viscous fingering can occur. It is caused by viscosity differences between the first and second dimension solvents in multidimensional chromatography [42]. To avoid viscous fingering, the solvent transfer from the first to the second dimension should be as low as possible. To minimize the injection volume to the second dimension, a low flow rate is usually used in the first dimension [24, 25].
Modified nucleosides are mostly polar and are usually analyzed by standard reversed-phase HPLC utilizing traditional columns with C18 stationary phases and aqueous mobile phases with increasing organic phase content. Therefore, we tested three different C18 columns (ESM Table S1) at low flow rates for the first dimension. To determine the maximum 1D flow rate, which depends on the maximum injection volume to the second dimension, each 1D column had to be investigated not only as 1D-, but also as 2D-LC application. The Eclipse XDB-C18 (150 × 4.6 mm, 5 μm) column showed insufficient separation at low flow rates, because of the wide column and particle diameter. Using the Zorbax SB-Aq (100 × 2.1 mm, 3.5 μm) column at low flow rates, the separation could be improved, but especially in the beginning of the analysis the peak capacity was unsatisfactory. By application of an additional splitter after the 1D column, we were able to increase the flow rate and therefore improved the 1D separation, but the separation was still inadequate. In a further approach, we selected a Zorbax HILIC Plus (150 × 2.1, 1.8 μm) column in the first dimension. In contrast to reversed-phase chromatography, HILIC starts with a high organic solvent composition. Combination of the Zorbax HILIC Plus column with different reversed-phase materials (ESM Table S1) led to inadequate chromatographic separation in the second dimension because of the early elution of target compounds based on the organic solvent injection from the first dimension. Reduction of the injection volume by application of a post-1D splitter could not solve this difficulty (data not shown).
Finally, the Eclipse Plus C18 (150 × 2.1 mm, 1.8 μm) column resulted in adequate separation at low flow rate (0.07 mL/min). Eclipse Plus C18 columns are valuable for reversed-phase separation of acidic, basic, and other compounds with high polarity. The packing material consists of a dimethyl-n-octadecylsilane monolayer bonded to a double-endcapped, porous silica support providing maximum deactivation of the silica surface. According to the manufacturer, the silica support reduces adsorption of basic and highly polar compounds. The performance of the first dimension separation has been evaluated by analysis of a urinary sample using the Eclipse Plus C18 column directly coupled to a triple quadrupole mass spectrometer. Therefore, the same chromatographic conditions as in the first dimension of the resulting 2D-LC setup have been applied to the 1D-LC-MS approach. The chemical complexity of the urinary sample and the incomplete separation of target compounds from matrix components can be observed by comparison of the TIC in full scan (Fig. 3a) and the TIC in MRM scan mode (Fig. 3b). The quantifier MRM transitions are displayed in Fig. 3c as overlaid spectra, showing the incomplete separation of target compounds in the 1D-LC approach.

Analysis of prepurified urinary sample using 1D-LC analysis with the Eclipse Plus C18 (150 × 2.1 mm, 1.8 μm) column coupled to a triple quadrupole mass spectrometer showing (a) TIC of full scan, (b) TIC of MRM scan, and (c) overlaid spectra of quantifier MRM transitions of target compounds (compound abbreviations are shown in Table 1)
For the second dimension, we have also investigated numerous columns (ESM Table S1), different solvents, modulation times, shifted gradients, and flow rates (data not shown). Finally, we selected the Zorbax Bonus-RP column with 4.6 mm diameter, which allowed a rapid analysis and short re-equilibration time. The diameter extension to 4.6 mm allowed higher flow rates and consequently resulted in significantly better separation in contrast to 3-mm diameter columns. The Zorbax Bonus-RP consists of diisopropyl-C14 groups, which are covalently bonded to silica by an amide functionality. Silanol interactions are reduced by the high polarity of the amide function.
2D-LC-MS with Eclipse Plus C18-Zorbax Bonus-RP column combination
The final setup consisted of the Eclipse Plus C18 (150 × 2.1 mm, 1.8 μm) column in the first dimension and a Zorbax Bonus-RP column (50 × 4.6 mm, 1.8 μm) in the second dimension. We performed a slow analysis in the first dimension (0.07 mL/min), resulting in a transfer of 63 μl to the second dimension. In the first dimension, we used methanol as eluent and in the second dimension acetonitrile, because of its lower back pressure at high flow rate and the slightly higher eluting power in reversed-phase chromatography. To reduce the coelution of matrix components with target substances, we performed a shifted gradient in the second dimension with a maximum of 28 % B. This retained numerous matrix compounds on the column. The retained matrix components were eluted between single runs by application of 98 % B. By application of a shifted gradient, we were able to separate the target compounds. The target substances have a particularly narrow polarity range. This can be underlined by the estimated log K OW values (Table 1). The hydrophobic character of a substance can be expressed by the octanol water partition constant K OW, that can be calculated or investigated experimentally [43, 44]. Therefore, a small shift of the solvent B percentage in the second dimension (ESM Figure S1, Table S2) delivered the best separation of target substances. The impact of the additional chromatographic dimension can be observed by comparison of the 1D-LC-MS (Fig. 3) and the 2D-LC-MS (Fig. 4) analysis of the same urinary sample of a healthy volunteer. Retention time differences can be explained by the different dead volume of the 1D- and 2D-LC system. The separation of numerous matrix components from target substances is depicted in Fig. 4a, showing the TIC of the full scan, and in Fig. 4b, showing the TIC of the neutral loss scan (−132 Da). The improved separation of the target substances is viewed in Fig. 4c, showing the TIC of the MRM scan. Retention times of target substances are listed in Table 2. In consideration of the repeated retention time estimation (Table 2), we have concluded that the second dimension separation shows adequate reproducibility. The additional separation of the second dimension is accentuated in Fig. 5, showing the TIC of the MRM scan as 3D plot with corresponding projections of both dimensions. The additional chromatographic separation can be recognized by comparison with the 1D-LC approach (Fig. 3).

Analysis of prepurified urinary sample using 2D-LC analysis with the Eclipse Plus C18 (150 × 2.1 mm, 1.8 μm) and the Zorbax Bonus-RP (50 × 4.6 mm, 1.8 μm) coupled to a triple quadrupole mass spectrometer in (a) full scan, (b) neutral loss scan and (c) MRM scan mode (retention times are summarized in Table 2, compound abbreviations in Table 1)

In general, all target compounds have been detected in full scan and MRM scan. By application of neutral loss scans all target substances could be detected except Ψ and MTA. Instead of the labile N-glycosidic bond, Ψ contains a stable C-glycosidic bond resulting in a differential fragmentation pattern. MTA eliminates a corresponding thioribose derivative with a constant neutral loss of 162 amu. In collision-induced dissociation experiments, the ribose moiety is usually split from the purine- or pyrimidine base residue as a neutral resulting in the constant neutral loss of 132 amu. The mechanisms for this phenomenon have been discussed before [45]. The precursor ion of t6A as well as the quantifier and qualifier ions of the MRM scan are shown in the ESM Fig. S2. The quantifier ion of t6A with an m/z value of 281.1 is a hallmark for the characteristic constant neutral loss of the ribose moiety (−132 amu) at modified nucleosides.
Exometabolome comparison of investigated cell lines
In order to transfer the developed 2D-LC-MS method to other biological matrices, we have investigated the exometabolome of different human cell lines. Therefore, we prepurified supernatants of the breast cancer cell line MCF-7 and the breast epithelial cell line MCF-10A by application of phenylboronic acid affinity chromatography and analyzed them using 2D-LC-MS. Due to the different excretion levels of the investigated target compounds, the comparison of the breast cancer cell line MCF-7 with the breast epithelial cell line MCF-10A showed significant differences (Fig. 6) resulting in a different metabolic signature. IAA-R, C, AICAR, m5C, m1I, m6A, ac4C, N6SAR, and m6t6A could solely be detected in the breast cancer cell line MCF-7 (Fig. 6a). I and A were solely detected in the breast epithelial cell line MCF-10A (Fig. 6b). The detected compounds in the investigated cell lines are summarized as a bubble chart in Fig. 6c, showing the compounds detected in both cell lines in blue and the compounds detected in a single cell line in red. Figure 6c has been created utilizing Image Investigator (GC Image, Lincoln, Nebraska, USA).

Comparison of breast cancer cell line MCF-7 with breast epithelial cell line MCF-10A utilizing 2D-LC-MS: (a) TIC of MRM scan (MCF-7); (b) TIC of MRM scan (MCF-10A); (c) Bubble chart of target substances showing relevant retention window (blue substances occurring in both cell lines; red substances occurring in one of both cell lines; 1 IAA-R; 2 C; 3 P; 4 m3C; 5 m1A; 6 acp3U; 7 AICAR; 8 U; 9 Isog; 10 A; 11 I; 12 m5U; 13 m5C; 14 3,4-PCNR; 15 MTA-SO; 16 m1I; 17 m2G; 18 TMG; 19 m6A; 20 ac4C; 21 m2 2G; 22 N6-SAR; 23 MTA; 24 t6A; 25 m6t6A; 26 ms2t6A; compound abbreviations are summarized in Table 1)
Due to the heterogeneity of breast cancer [46] and the variability of RNA modifications [31], biological samples should be investigated by 2D-LC coupled to different MS methods in relation to structural elucidation of RNA metabolites in a large scale. The potential of modified nucleosides as potential biomarker for breast cancer should be examined on the basis of a statistically meaningful number of patients’ biological fluids utilizing 2D-LC, especially in semi-quantitative or quantitative approaches. To accomplish these aims, the establishment of a 2D-LC method, as described in the present article, is beneficial in all intents and purposes.
Conclusions
In the present study, a comprehensive 2D-LC method with a triple-quadrupole mass spectrometer as detector has been developed for a targeted metabolomics approach. Target substances from RNA metabolism and crosslinked pathways have been obtained by phenylboronic acid affinity chromatography of urinary samples as well as cell culture supernatants. For 2D-LC method development, we tested numerous different column combinations with variable chromatographic conditions in the first and second dimension. The combination of a Zorbax Eclipse Plus C18 column in the first dimension and a Zorbax Bonus-RP column in the second dimension provided a high degree of orthogonality and adequate separation in the second dimension. The 2D-LC-MS approach led to the detection of 28 target compounds from RNA metabolism and crosslinked pathways in urine and 26 target compounds in cell culture supernatants, respectively. The established 2D-LC-MS approach with the combination of the Eclipse Plus C18 column and the Zorbax Bonus-RP column should be applied to future research based on the potential tumor marker class of modified nucleosides and interrelated compounds. In addition, real online comprehensive 2D-LC offers great potential for further analysis of complex biological samples. Cell culture and urinary metabolites, for example, can be compared and visualized as metabolic fingerprints in 3D plots. Consequently, a minimization of ion suppression and matrix effects caused by the high complexity of biological samples, that are posing the challenge especially in the field of metabolomics, will be accomplished.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D (2011) Global cancer statistics. CA Cancer J Clin 61:69–90
McShane LM, Hayes DF (2012) Publication of tumor marker research results: the necessity for complete and transparent reporting. J Clin Oncol 30:4223–4232
Bullinger D, Frickenschmidt A, Pelzing M, Zey T, Zurek G, Laufer S, Kammerer B (2005) identification of urinary nucleosides by ESI-TOF-MS. LC-GC Europe 16–17
Bullinger D, Fux R, Nicholson G, Plontke S, Belka C, Laufer S, Gleiter CH, Kammerer B (2008) Identification of urinary modified nucleosides and ribosylated metabolites in humans via combined ESI-FTICR MS and ESI-IT MS analysis. J Am Soc Mass Spectrom 19:1500–1513
Borek E, Baliga BS, Gehrke CW, Kuo CW, Belman S, Troll W, Waalkes TP (1977) High turnover rate of transfer RNA in tumor tissue. Cancer Res 37:3362–3366
Randerath E, Chia LL, Morris HP, Randerath K (1974) Transfer RNA base composition studies in Morris hepatomas and rat liver. Cancer Res 34:643–653
Liebich HM, Muller-Hagedorn S, Klaus F, Meziane K, Kim KR, Frickenschmidt A, Kammerer B (2005) Chromatographic, capillary electrophoretic and matrix-assisted laser desorption ionization time-of-flight mass spectrometry analysis of urinary modified nucleosides as tumor markers. J Chromatogr A 1071:271–275
Colonna A, Russo T, Esposito F, Salvatore F, Cimino F (1983) Determination of pseudouridine and other nucleosides in human blood serum by high-performance liquid chromatography. Anal Biochem 130:19–26
Boschi-Muller S, Motorin Y (2013) Chemistry enters nucleic acids biology: enzymatic mechanisms of RNA modification. Biochemistry (Mosc) 78:1392–1404
Mandel LR, Srinivasan PR, Borek E (1966) Origin of urinary methylated purines. Nature 209:586–588
Bouatra S, Aziat F, Mandal R, Guo AC, Wilson MR, Knox C, Bjorndahl TC, Krishnamurthy R, Saleem F, Liu P, Dame ZT, Poelzer J, Huynh J, Yallou FS, Psychogios N, Dong E, Bogumil R, Roehring C, Wishart DS (2013) The human urine metabolome. PLoS One 8:e73076
Urakami K, Zangiacomi V, Yamaguchi K, Kusuhara M (2010) Quantitative metabolome profiling of Illicium anisatum by capillary electrophoresis time-of-flight mass spectrometry. Biomed Res 31:161–163
Frickenschmidt A, Frohlich H, Bullinger D, Zell A, Laufer S, Gleiter CH, Liebich H, Kammerer B (2008) Metabonomics in cancer diagnosis: mass spectrometry-based profiling of urinary nucleosides from breast cancer patients. Biomarkers 13:435–449
Lee SH, Jung BH, Kim SY, Chung BC (2004) A rapid and sensitive method for quantitation of nucleosides in human urine using liquid chromatography/mass spectrometry with direct urine injection. Rapid Commun Mass Spectrom 18:973–977
Dudley E, Lemiere F, Van Dongen W, Tuytten R, El-Sharkawi S, Brenton AG, Esmans EL, Newton RP (2004) Analysis of urinary nucleosides. IV. Identification of urinary purine nucleosides by liquid chromatography/electrospray mass spectrometry. Rapid Commun Mass Spectrom 18:2730–2738
Kammerer B, Frickenschmidt A, Gleiter CH, Laufer S, Liebich H (2005) MALDI-TOF MS analysis of urinary nucleosides. J Am Soc Mass Spectrom 16:940–947
Dudley E, Tuytten R, Bond A, Lemière F, Brenton AG, Esmans EL, Newton RP (2005) Study of the mass spectrometric fragmentation of pseudouridine: comparison of fragmentation data obtained by matrix-assisted laser desorption/ionisation post-source decay, electrospray ion trap multistage mass spectrometry, and by a method utilising electrospray quadrupole time-of-flight tandem mass spectrometry and in-source fragmentation. Rapid Commun Mass Spectrom 19:3075–3085
Choi BK, Hercules DM, Gusev AI (2001) LC-MS/MS signal suppression effects in the analysis of pesticides in complex environmental matrices. Fresenius J Anal Chem 369:370–377
Enke CG (1997) A predictive model for matrix and analyte effects in electrospray ionization of singly-charged ionic analytes. Anal Chem 69:4885–4893
Kebarle P, Verkerk UH (2009) Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom Rev 28:898–917
Giddings JC (2005) Concepts and comparisons in multidimensional separation. J High Resol Chromatogr 10:319–323
François I, Sandra K, Sandra P (2009) Comprehensive liquid chromatography: fundamental aspects and practical considerations—a review. Anal Chim Acta 641:14–31
Erni F, RWF (1978) Two-dimensional column liquid chromatographic technique for resolution of complex mixtures. J Chromatogr A 149:561–569
Elsner V, Laun S, Melchior D, Kohler M, Schmitz OJ (2012) Analysis of fatty alcohol derivatives with comprehensive two-dimensional liquid chromatography coupled with mass spectrometry. J Chromatogr A 1268:22–28
Kittlaus S, Schimanke J, Kempe G, Speer K (2013) Development and validation of an efficient automated method for the analysis of 300 pesticides in foods using two-dimensional liquid chromatography-tandem mass spectrometry. J Chromatogr A 1283:98–109
Montero L, Herrero M, Ibanez E, Cifuentes A (2014) Separation and characterization of phlorotannins from brown algae Cystoseira abies-marina by comprehensive two-dimensional liquid chromatography. Electrophoresis 35:1644–1651
Stoll DR, Li X, Wang X, Carr PW, Porter SEG, Rutan SC (2007) Fast, comprehensive two-dimensional liquid chromatography. J Chromatogr A 1168:3–43
Giddings JC (1967) Maximum number of components resolvable by gel filtration and other elution chromatographic methods. Anal Chem 39:1027–1028
Li D, Schmitz OJ (2013) Use of shift gradient in the second dimension to improve the separation space in comprehensive two-dimensional liquid chromatography. Anal Bioanal Chem 405:6511–6517
Liebich HM, Di Stefano C, Wixforth A, Schmid HR (1997) Quantitation of urinary nucleosides by high-performance liquid chromatography. J Chromatogr A 763:193–197
Cantara WA, Crain PF, Rozenski J, McCloskey JA, Harris KA, Zhang X, Vendeix FA, Fabris D, Agris PF (2011) The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res 39:D195–D201
Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, Oda Y, Kakazu Y, Kusano M, Tohge T, Matsuda F, Sawada Y, Hirai MY, Nakanishi H, Ikeda K, Akimoto N, Maoka T, Takahashi H, Ara T, Sakurai N, Suzuki H, Shibata D, Neumann S, Iida T, Funatsu K, Matsuura F, Soga T, Taguchi R, Saito K, Nishioka T (2010) MassBank: a public repository for sharing mass spectral data for life sciences. J Mass Spectrom 45:703–714
Smith CA, O’Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, Siuzdak G (2005) METLIN: a metabolite mass spectral database. Ther Drug Monit 27:747–751
Henneges C, Bullinger D, Fux R, Friese N, Seeger H, Neubauer H, Laufer S, Gleiter CH, Schwab M, Zell A, Kammerer B (2009) Prediction of breast cancer by profiling of urinary RNA metabolites using support vector machine-based feature selection. BMC Cancer 9:104
Hsu WY, Lin WD, Tsai Y, Lin CT, Wang HC, Jeng LB, Lee CC, Lin YC, Lai CC, Tsai FJ (2011) Analysis of urinary nucleosides as potential tumor markers in human breast cancer by high performance liquid chromatography/electrospray ionization tandem mass spectrometry. Clin Chim Acta 412:1861–1866
Sasco AJ, Rey F, Reynaud C, Bobin JY, Clavel M, Niveleau A (1996) Breast cancer prognostic significance of some modified urinary nucleosides. Cancer Lett 108:157–162
Zheng YF, Kong HW, Xiong JH, Lv S, Xu GW (2005) Clinical significance and prognostic value of urinary nucleosides in breast cancer patients. Clin Biochem 38:24–30
Snyder LR, Dolan JW, Carr PW (2004) The hydrophobic-subtraction model of reversed-phase column selectivity. J Chromatogr A 1060:77–116
Snyder LR (1965) Principles of gradient elution. Chromatogr Rev 7:1–51
Galante RN (1986) Gradient elution in column liquid chromatography theory and practice. Journal of Chromatography Library—Vol 31. Amsterdam, The Netherlands
Jandera P (2005) Gradient elution in liquid column chromatography-prediction of retention and optimization of separation. Adv Chromatogr 43:1–108
Mayfield KJ, Shalliker RA, Catchpoole HJ, Sweeney AP, Wong V, Guiochon G (2005) Viscous fingering induced flow instability in multidimensional liquid chromatography. J Chromatogr A 1080:124–131
Dearden JC, Bentley D (2002) The components of the “critical quartet” log Kow values assessed by four commercial software packages. SAR QSAR Environ Res 13:185–197
Finizio A, Vighi M, Sandroni D (1997) Determination of n-octanol/water partition coefficient (Kow) of pesticide critical review and comparison of methods. Chemosphere 34:131–161
Jihong Liu SC (2009) A theoretical and mass spectrometry study of the novel mechanism of N-glycosidic bond cleavage in nucleoside. Int J Mass Spectrom 282:1–5
Eccles SA, Aboagye EO, Ali S, Anderson AS, Armes J, Berditchevski F, Blaydes JP, Brennan K, Brown NJ, Bryant HE, Bundred NJ, Burchell JM, Campbell AM, Carroll JS, Clarke RB, Coles CE, Cook GJ, Cox A, Curtin NJ, Dekker LV, Silva Idos S, Duffy SW, Easton DF, Eccles DM, Edwards DR, Edwards J, Evans D, Fenlon DF, Flanagan JM, Foster C, Gallagher WM, Garcia-Closas M, Gee JM, Gescher AJ, Goh V, Groves AM, Harvey AJ, Harvie M, Hennessy BT, Hiscox S, Holen I, Howell SJ, Howell A, Hubbard G, Hulbert-Williams N, Hunter MS, Jasani B, Jones LJ, Key TJ, Kirwan CC, Kong A, Kunkler IH, Langdon SP, Leach MO, Mann DJ, Marshall JF, Martin L, Martin SG, Macdougall JE, Miles DW, Miller WR, Morris JR, Moss SM, Mullan P, Natrajan R, O’Connor JP, O’Connor R, Palmieri C, Pharoah PD, Rakha EA, Reed E, Robinson SP, Sahai E, Saxton JM, Schmid P, Smalley MJ, Speirs V, Stein R, Stingl J, Streuli CH, Tutt AN, Velikova G, Walker RA, Watson CJ, Williams KJ, Young LS, Thompson AM (2013) Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer. Breast Cancer Res 15:R92
Acknowledgments
We acknowledge Ralf Falter (Agilent Technologies, Waldbronn, Germany) for the support and fruitful discussions. We would like to thank Tilman Brummer (Albert-Ludwigs-University Freiburg, Germany) for providing the breast epithelial cell line MCF-10A and the technical support.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 84 kb)
Rights and permissions
About this article

Cite this article
Willmann, L., Erbes, T., Krieger, S. et al. Metabolome analysis via comprehensive two-dimensional liquid chromatography: identification of modified nucleosides from RNA metabolism. Anal Bioanal Chem 407, 3555–3566 (2015). https://doi.org/10.1007/s00216-015-8516-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-015-8516-6