
Abstract
O-glycosylation-site characterization of individual glycoproteins is a major challenge because of the heterogeneity of O-glycan core structures. In proteomic studies, O-glycosylation-site analysis is even more difficult because of the complexity of the sample. In this work, we designed a rapid and convenient workflow for characterizing the O-glycosylation sites of individual proteins and the human-plasma proteome. A mixture of exoglycosidases was used to partially remove O-glycan chains and leave an N-acetylgalacosamine (GalNAc) residue attached to the Ser or Thr residues. The O-glycosylated peptides could then be identified by using liquid chromatography–tandem mass spectrometry (LC–MS–MS) to detect the 203 Da mass increase. Jacalin was used to selectively isolate O-GalNAc glycopeptides before LC–MS–MS analysis, which is optional for individual proteins and necessary for complex human-plasma proteins. Bovine fetuin and human chorionic gonadotropin (hCG) were used to test the analytical workflow. The workflow indicated superior sensitivity by not only covering most previously known O-glycosylation sites but also discovering several novel sites. Using only one drop of blood, a total of 49 O-GalNAc-linked glycopeptides from 36 distinctive glycoproteins in human plasma were identified unambiguously. The approach described herein is simple, sensitive, and global for site analysis of core 1 through core 4 O-glycosylated proteins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glycosylation is one of the most important forms of posttranslational modification on eukaryotic proteins [1]. Two types of glycosylation, N-glycosylation at asparagine residues and O-glycosylation at threonine or serine residues, frequently occur and have important functions in many cellular processes [2, 3]. Altered glycosylation, including change of glycosylation sites and of glycan structures, has been implicated in severe diseases including cancer and Alzheimer’s disease [4–6]. Glycosylation-site analysis is critical to reveal these modifications. It provides overall insights into the number and identity of proteins which may change their glycosylation in response to specific diseases [7, 8]. Moreover, site-specific glycan structural analysis becomes more straightforward once the glycosylation sites are determined.
Methods for analysis of N-glycosylation sites are well established because the core glycan structure and potential sites on proteins are well defined. Endo-β-N-acetylglucosaminidases cleave the glycosidic bond and leave a single GlcNAc residue attached to the proteins, which provides a +203 Da mass tag to the peptides [9]. Peptide-N-glycosidase (PNGase) releases intact glycan and converts asparagine residues to aspartic-acid residues, which gives the peptide a +3 Da mass shift if the digestion is performed in H2 18O [10]. LC–MS–MS analysis can easily detect these mass differences after deglycosylation and determine the original N-glycosylation sites. In contrast, O-glycosylation-site analysis is more challenging in several aspects. Unlike N-glycosylation, which requires a conserved sequence of Asn-X-Ser/Thr (where X can be any amino acid except Pro), O-glycosylation occurs at individual Ser or Thr residues and is more difficult to predict. The core structure of O-glycan is more diverse and a universal O-glycanase has not yet been found [11, 12]. In addition, the structure of the peptide is retained and no mass tag can be incorporated during enzymatic O-deglycosylation.
Several methods have been developed for O-glycosylation-site analysis. β-elimination of the O-glycans using NH4OH incorporates one NH3 into the amino-acid residues to which the glycans are attached and yields a modified amino-acid residue with a distinct mass [13, 14]. However, the alkaline-catalyzed reaction is sometimes difficult to control and causes several side reactions on proteins [15, 16]. A mixture of exoglycosidases containing β-galactosidase, neuraminidase, and N-acetyl-β-glucosaminidase is able to cleave off side chains of O-glycans and leaves a GalNAc residue attached to the Ser or Thr residue. This strategy was used to map the glycosylation sites of proteins from Cohn IV fraction of human plasma. The glycopeptides from tryptic digestion were enriched by hydrophilic-interaction chromatography (HILIC) and partially deglycosylated with exo and endodeglycosidases. A total of 23 O-glycosylated tryptic peptides from 11 proteins were identified by LC–MS–MS analysis [17]. The number of O-glycosylated proteins detected is lower than expected for such a complex biological sample, possibly because of the limited recovery capability of HILIC, especially when short O-glycans are attached to long hydrophobic peptides. As well as the in-vitro glycan-modification approach using deglycosidases, a SimpleCell method was developed by truncating the O-glycan elongation pathway of O-glycoproteins in human cells. The O-glycoproteins interfered by zinc-finger nuclease consist of only GalNAcα or NeuAcα2-6GalNAcα O-glycans, which facilitates the downstream enrichment and LC–MS–MS analysis [18]. However, the in-vivo-modification approach is restricted to cell-culture samples and cannot be applied to human-fluid samples, for example human plasma, serum, or urine.
Lectin-affinity chromatography is widely used to isolate specific types of glycan, glycopeptide, or glycoprotein on the basis of their selective binding affinity to specific carbohydrate structures [19, 20]. Jacalin is selective for binding GalNAcα that is unsubstituted at the C-6 position, for example the O-glycan core 1 structure Galβ1–3GalNAcα1-Ser/Thr and core 3 structure GlcNAcβ1–3GalNAcα-Ser/Thr [21]. In two other main types of O-glycan core structure, core 2 and core 4, the C6 position of GalNAcα attached to Ser/Thr is substituted by GlcNAc and cannot bind to jacalin. Saroha et al. extracted O-glycoproteins from plasma of rheumatoid-arthritis patients using jacalin-affinity chromatography. The proteins differentially expressed between patients and normal controls were analyzed using two-dimensional gel electrophoresis and identified by matrix-assisted laser-desorption/ionization time-of-flight (MALDI-TOF) MS. The O-glycosylation sites of 11 proteins were predicted using the Net-O-Glyc3.1 bioinformatics tool without experimental evidence [22]. Darula and Medzihradszky reported a jacalin-affinity-enrichment and exoglycosidase-deglycosylation method for characterization of bovine-serum proteins [23]. The method was restricted to core-1-type glycopeptides and a total of 26 O-glycosylation sites at 13 proteins were elucidated. The method was then improved by adding an ion-exchange step to fractionate jacalin-enriched glycoproteins or an electrostatic-repulsion-hydrophilic-interaction-chromatography step to separate tryptic glycopeptides to recover more glycosylation sites and O-glycosylated proteins from bovine serum [24]. A sialic-acid capture-and-release procedure was developed to enrich O-glycopeptides from tryptic digestion of human-urine and cerebrospinal-fluid glycoproteins, followed by nano-LC–ESI-collision-induced-dissociation (CID)-MS2–MS3 and electron-capture and electron-transfer dissociation (ECD and ETD). The glycosylation sites that the sialylated O-glycans originally attached to were characterized [25–27].
O-glycosylation-site analysis is essential for characterization of individual proteins, including recombinant-therapy proteins or diagnostic biomarkers, and for proteomic studies. However, the lack of a conserved sequence and the neutral loss of GalNAc residue during MS–MS make analysis of O-glycosylation more challenging than analysis of N-glycosylation. In this paper, we describe a simple and universal site-mapping approach for core 1 through core 4 O-glycosylated proteins. After tryptic digestion of proteins, the core-structure heterogeneity of O-glycopeptides was eliminated by endoglycosidase digestion. The O-glycosylation sites of two representative proteins, bovine fetuin [28–30] and human chorionic gonadotropin [31–33], were characterized using LC–MS–MS. Human-plasma proteins were also analyzed by adding a jacalin-affinity-chromatography step to selectively isolate O-GalNAc glycopeptides after endoglycosidase digestion. We unambiguously identified 49 glycopeptides from 36 glycoproteins in human plasma. The result covered most glycoprotein species reported elsewhere [17], and revealed 25 more O-glycosylated proteins in human plasma.
Experimental
Materials
β(1-3,4) galactosidase, the GlycoPro Enzymatic Deglycosylation Kit, and prO-LINK Extender Kit containing PNGase F, β-N-acetylglucosaminidase, sialidase A, and standard glycoprotein bovine fetuin were purchased from Prozyme (San Leandro, CA, USA). Trypsin Gold (MS grade) was purchased from Promega (Madison, WI, USA). HCG was obtained from USBIO (Swampscott, MA, USA). ProteoExtract Albumin Removal Kit was purchased from Merck KGaA (Darmstadt, Germany). Agarose-bound jacalin was purchased from Vector Laboratories (Burlingame, CA, USA). The combined plasma specimen was obtained from healthy donors under an institutional-review-board-approved procedure. All other chemicals and reagents of the best available grade were purchased from Sigma–Aldrich (St. Louis, MO, USA) or Fisher Scientific (Morris Plains, NJ, USA).
Depletion of albumin from plasma
Albumin was removed from human plasma using ProteoExtract Albumin Removal Kit according to the manufacturer’s procedure. Briefly, 20 μL combined plasma was diluted with binding buffer to a final volume of 400 μL. The sample was applied to the affinity column and then eluted with 600 μL binding buffer twice. Albumin-depleted samples were collected, concentrated, and desalted with Microcon centrifugal-filter devices (MWCO 10 kDa).
In-solution tryptic digestion
Glycoproteins (50 μg) or albumin-depleted plasma proteins (equal to 20 μL plasma) were denatured with 6 mol L−1 guanidine hydrochloride in 100 mmol L−1 ammonium bicarbonate buffer (pH 8.2). The samples were reduced by adding 1.0 mol L−1 dithiothreitol to a final concentration of 100 mmol L−1, and incubated for 1 h at 37 °C. Iodoacetamide solution (1.0 mol L−1) was then added to obtain a final concentration of 150 mmol L−1, and the mixture was incubated for 30 min at room temperature in the dark. The reaction buffer was replaced with 25 mmol L−1 ammonium bicarbonate using Microcon centrifugal-filter devices (MWCO 10 kDa). Trypsin (approximately 1–2 % w/w to the estimated protein content) was added, and the mixture digested at 37 °C overnight. The enzymatic digestion was stopped by heating at 100 °C for 2 min.
N and partial O-deglycosylation
A mixture of PNGase F and endoglycosidases including β(1-3,4) galactosidase, β-N-acetylglucosaminidase, and sialidase A (~1 μL each enzyme solution per 100 μg protein) was added to the tryptic digests and incubated for 24 h at 37 °C. The reaction was stopped by heating at 100 °C for 2 min.
Jacalin-affinity chromatography
Agarose-bound jacalin (1.7 mL) was packed into perfluoroalkoxyalkane tubing (1 × 1900 mm) equipped with a 0.22 μm frit at its distal end and washed with 20 column volumes of wash buffer (100 mmol L−1 Tris–HCl, pH 7.4). Affinity enrichment was performed on an LC-20AT HPLC system (Shimadzu, Tokyo, Japan). After introducing the peptide sample at a flow of 100 μL min−1, the column was washed with eight column volumes of wash buffer. Bound materials were then eluted with five column volumes of elution buffer containing 0.8 mol L−1 galactose. Both wash and elution fractions were sequentially collected. Fractions were desalted on a HyperSep C18 column before LC–MS–MS analysis.
Mass spectrometry
LC–MS–MS experiments were performed on a linear ion trap–Orbitrap hybrid mass spectrometer (LTQ-Orbitrap Velos Pro, Thermo Fischer Scientific) coupled with a nano-LC system (Shimadzu, Tokyo, Japan). Sample injection and on-line desalting were performed using a C18 trap column (Chemicals Evaluation and Research Institute, Japan) at a flow of 50 μL min−1. A custom-made column (15 cm × 75 μm i.d.) packed with Reprosil-Pur C18 beads (3 μm) was used to separate peptides, eluting with a stepping gradient of 2 % solvent B (0.0–5.0 min); 2 to 15 % solvent B (5.0–25.0 min); 15 to 40 % solvent B (25.0–55.0 min); 40 to 98 % solvent B (55.0–60.0 min); 98 % solvent B (60.0–70.0 min); 98 to 2 % solvent B (70.0–75.0 min); and 2 % solvent B (75.0–90.0 min) at a flow of 300 nL min−1. Solvent A was 2.0 % ACN–water (v/v) with 0.1 % formic acid and solvent B was 98 % ACN–water (v/v) with 0.1 % formic acid. The LTQ-Orbitrap mass spectrometer was set at 60,000 isotopic resolution and m/z 400–1800 mass range during precursor scans. The mass spectrometer was operated in the data-dependent mode using the standard “top10” CID-MS–MS method. The normalized collision energy was set to 35 % and the target was set to 10,000.
Data processing
Mass-spectra data processing was performed using Mascot Distiller and searched with MASCOT (Version 2.4.0) against the SwissProt database version 2013_12. Mascot search parameters were set as follows: species, Homo sapiens (20,274 sequences); enzyme, trypsin with a maximum of two missed cleavages; fixed modification was carbamidomethylation of Cys residues; variable modification was HexNAc (203 Da) on Ser and Thr residues together with neutral loss of the same mass. Other variable modifications were Asn-to-Asp conversion (+0.9840 Da), methionine oxidation, N-terminal acetylation, and cyclization of N-terminal Gln residues. The mass accuracy was 15 ppm for precursor ions and 0.8 Da for the fragment ions. All results were filtered with expectation value (E-value). E-value less than 0.1 and Mascot ion score more than 15 were set as the acceptance criteria of glycopeptides and glycoproteins. All identified glycopeptides were further investigated by examining their MS–MS spectra manually to evaluate the acceptance criteria. Identification of a glycopeptide was accepted only when the neutral sugar-loss ion and at least four peptide-backbone fragmentation ions from the parent ion were assigned.
Results and discussion
Workflow for O-glycosylation-site analysis
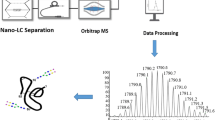
MS analysis of glycopeptides is challenging because of their glycan structural heterogeneity and low ionization efficiency. A series of exoglycosidases can partially remove O-glycans and leave a single GalNAc residue still attached to the Ser or Thr residue. The exoglycosidases include sialidase A to remove terminal α-(2-3,6,8)-linked sialic-acid residues, β(1-3,4)-galactosidase to remove β(1-3,4)-linked galactoseresidues, and β-N-acetylglucosaminidase to remove β-linked N-acetylglucosamine. PNGase F was also used to remove N-glycans, because N and O-glycosylation sites may co-exist in the same tryptic peptide. The deglycosylated samples were subjected to direct LC–MS–MS analysis for the individual glycoprotein characterization. Two representative proteins, bovine fetuin and hCG, were used to evaluate this approach. For complex proteomic samples, for example human-plasma proteins, a jacalin-enrichment step is necessary before LC–MS–MS analysis. Jacalin is a plant lectin from Artocarpus integrifolia that binds specifically to GalNAcα-peptides when the C6 position of GalNAcα is not substituted [21]. It also binds to mannose residues in N-glycans [34]. However, interference is not a problem here because the N-glycans are removed by PNGase F before jacalin enrichment. The workflow for O-glycosylation-site analysis is summarized in Fig. 1.

Workflow of O-glycosylation-site analysis for individual proteins or human-plasma proteins
O-glycosylation-site analysis of bovine fetuin
Bovine fetuin is a widely-used model glycoprotein consisting of 359 amino-acid residues. It is N-glycosylated at N159, N156, and N176, and O-glycosylated at S271, T280, S282, and S341, according to the UniProt Database [35]. After tryptic digestion, the sample was treated with PNGase F and exoglycosidase mixture. The digests were subjected to LC–MS–MS analysis without any further enrichment. The Mascot search result gave peptide sequencing coverage as 87 % of mature bovine fetuin, and six glycopeptides containing at least one GalNAc residue were also revealed (Table 1). The first was doubly-charged 334-TPIVGQPSIPGGPVR-348 containing one GalNAc with m/z = 839.4. As shown in Fig. 2a, the dominant fragment ions corresponded with the loss of one GalNAc, because the glycan–peptide linkage bond is more fragile than peptide bonds during CID-MS–MS. There are only two possible O-glycosylation sites in this peptide, T334 and S341. On the basis of two diagnostic fragments, y9-GalNAc and y11-GalNAc, the GalNAc residue was assigned to be attached at the S341. Figure 2b shows the CID-MS–MS spectrum of 313-HTFSGVASVESSSGEAFHVGK-333 with one GalNAc. Although there are several Ser and Thr residues in this peptide, the y9-GalNAc fragment ion was clear evidence that the O-GalNAc occurred at S325. This is a recently reported O-glycosylation site [24] that has not been included in the UniProt Database. A series of glycopeptides corresponding to different numbers of O-GalNAc residues attached to the same peptide, 246-VTCTLFQTQPVIPQPQPDGAEAEAPSAVPDAAGPTPSAAGPPVASVVVGPSVVAVPLPLHR-306, was observed, with retention times ranging from 71 min to 74 min (Table 1). They eluted in reverse order to the number of O-GalNAc residues attached because the carbohydrate moieties reduced their hydrophobicity. The extracted ion chromatogram of triply-O-GalNAc-substituted peptide (m/z = 1104.9) had two peaks at 71.4 min and 72.6 min. This suggested that there were at least two forms of glycosylation, which required a minimum of four different O-glycosylation sites for three O-GalNAc residues to attach. Studies by other research groups [28–30] claimed that S271, T280, S282, and S296 within this peptide sequence were O-glycosylated. However, the determination of exact sites was not successful because most fragment ions were generated by sequential cleavages of glycan–peptide linkage bonds when multiple O-glycosylation sites were present in the parent ion. The peptide-backbone-cleavage fragment ions were able to confirm the identity of glycopeptides, but were not sufficient to locate the O-glycosylation sites (data not shown). This challenge is likely to be solved if ETD is equipped and performed simultaneously with CID. In the ETD fragmentation process, radical anions transfer an electron to the peptide backbone and induce cleavage through peptide bonds, whereas the carbohydrate moieties are minimally affected [36].

CID-MS–MS spectra of O-GalNAc glycopeptides from bovine fetuin. (a) MS–MS spectrum of the [M+2H]2+ precursor ion at m/z 839.5 corresponds to the peptide TPIVGQPSIPGGPVR carrying one O-GalNAc residue. (b) MS–MS spectrum of the [M+2H]2+ precursor ion at m/z 1162.0 corresponds to the peptide HTFSGVASVESSSGEAFHVGK carrying one O-GalNAc residue. The y and b ion series associated with neutral loss of O-GalNAc residue are indicated, and the fragment ions with O-GalNAc residue retained are annotated with a yellow square
O-glycosylation-site analysis of hCG
hCG is a glycoprotein hormone secreted by placental trophoblasts and trophoblastic tumors. It is found in the blood and urine of women during pregnancy. Its concentration may increase in patients with some types of cancer, including testicular, ovarian, liver, stomach, and lung cancer [37, 38]. HCG is composed of α and β-subunits, and glycosylations occur at both subunits. The N52 and N78 from the α-subunit and N13 and N30 from the β-subunit are N-glycosylated, whereas S121, S127, S132, and S138 from the β-subunit are attached with O-glycans [31–33, 39]. Abnormal glycosylation of hCG, namely hyperglycosylation, has been revealed to be associated with malignancy and other disorders [40].
Tryptic digestion of hCG generates complex peptides with many miscleavage sites because of the steric hindrance of the heterodimer structure and heavy glycosylation, which significantly reduces the proteolytic efficiency. After PNGase F and exoglycosidase digestion, a total of 15 peptides containing at least one HexNAc residue was detected by LC–MS–MS analysis (Table 2). Most O-glycosylation occurs near the C-terminus of the β-subunit. The largest O-glycosylated peptide observed is 115-FQDSSSSKAPPPSLPSPSRLPGPSDTPILPQ-145, which contains two miscleavage sites. Ions at m/z = 1130.9, 1198.5, 1266.2, and 1333.9 indicate that different numbers of O-HexNAc residues, ranging from one to four, are attached to this peptide. All four previously-reported O-glycosylation sites, S121, S127, S132, and S138, are within this peptide sequence. The peptide 123-APPPSLPSPSRLPGPSDTPILPQ-145, containing one tryptic miscleavage site, was also revealed to carry from one to four HexNAc residues, because the ions 1262.6, 1364.2, 1465.7, and 1567.2 had a series of 203 Da mass differences after deconvolution. Only S127, S132, and S138 within this peptide sequence are known O-glycosylation sites, suggesting there is at least one novel O-glycosylation site present in this peptide. It could be either S130 or T140. However, the neutral loss of HexNAc residues is dominant during CID fragmentation. The MS–MS spectrum of 123-APPPSLPSPSRLPGPSDTPILPQ-145 with four HexNAc residues provides insufficient peptide-backbone fragment ions to determine the exact O-glycosylation sites.
A novel O-glycosylation site from the α-subunit was discovered. As shown in Fig. 3, ions of m/z 678.8 (retention time 15.6 min) and m/z 779.8 (retention time 15.4 min) (both doubly-charged) were detected, representing two different glycosylation patterns of 52-NVTSESTCCVAK-63. N52 in this peptide is an N-glycosylation site. The treatment with PNGase F converted the Asn residue to an Asp residue and added +1 Da mass to the original peptide mass. The MS–MS spectrum in Fig. 3a confirms the sequence of this originally N-glycosylated peptide. The other glycosylation form of this peptide, with +203 Da mass compared with the original peptide mass, was also observed. The b3-HexNAc fragment ion at m/z 518.2 (Fig. 3b) provides evidence of O-glycosylation at the T54 residue. Interestingly, the combination of these two glycosylation patterns, which would result in +204 Da mass to the peptide 52-NVTSESTCCVAK-63, was not observed. The mechanism controlling this peptide, either N-glycosylated or O-glycosylated, may be worth further study.

Extract ion chromatograms (EICs) and CID-MS–MS spectra of two distinct glycosylation forms of peptide NVTSESTCCVAK derived from hCG α-subunit. (a) EIC of the deamidated N-glycosylated peptide DVTSESTCCVAK (m/z 678.8). (b) EIC of the peptide NVTSESTCCVAK carrying one O-GalNAc residue (m/z 789.8). (c) MS–MS spectrum of the [M+2H]2+ precursor ion at m/z 678.8 corresponds to the deamidated N-glycosylated peptide DVTSESTCCVAK. (d) MS–MS spectrum of the [M+2H]2+ precursor ion at m/z 789.8 corresponds to the peptide NVTSESTCCVAK carrying one O-GalNAc residue. The y and b ion series associated with neutral loss of O-GalNAc residue are indicated, and the fragment ions with O-GalNAc residue retained are annotated with a yellow square. The aspartic-acid residue D in red was modified by PNGase F and indicates the original N-glycosylation site
Affinity fractionation of O-GalNAc peptides
The non-glycosylated peptides in high abundance compete with O-GalNAc peptides during ionization and mass-analyzing processes in LC–MS–MS analysis. Enrichment of specific groups of peptides is necessary for complex samples, for example human-plasma proteins. Jacalin-affinity chromatography is capable of selectively enriching the O-GalNAc peptides generated by exoglycosidase treatment. The trypsin and exoglycosidase-digested hCG consists of complex peptides with many miscleavage sites and variable O-GalNAc substitutions. Digested hCG was used to evaluate the jacalin-affinity-chromatography enrichment method. According to the UV detection at 214 nm, the unbound peptides eluted at the beginning. Then a bump, corresponding to O-GalNAc peptides with relatively weak affinity, eluted during wash buffer elution. At the end the mobile phase was switched to elution buffer containing 0.8 mol L−1 galactose and the strong-binding O-GalNAc peptides were washed out. Three fractions were sequentially collected, desalted, and subjected to LC–MS–MS analysis. No O-GalNAc peptide was detected in fraction 1. Five peptide species with a single O-GalNAc residue were detected in fraction 2. The main components in fraction 3 were peptides with multiple O-GalNAc residues, by which the binding affinity to jacalin was increased. The hCG experiment revealed the satisfactory O-GalNAc-residue-binding selectivity of jacalin-affinity chromatography by recovering 14 out of 15 O-GalNAc peptides of hCG. Only the very-low-abundance peptide FQDSSSSKAPPPSLPSPSRLPGPSDTPILPQ with one HexNAc residue was missing (Table 2).
O-glycosylation-site analysis of human-plasma proteins
The analysis of O-glycosylation sites in human-plasma proteins is valuable to understanding the critical functions of this category of posttranslational modification in biological processes and diseases. Compared with tissue samples, human blood is more easily accessible for sampling. However, the proteomic method must still be sensitive, comprehensive, and high-throughput to be used in biomarker discovery and clinical studies. A small volume of 20 μL human plasma was used for O-glycosylation-site analysis, equivalent to only one drop of human blood.
The highly abundant albumin in human plasma was first depleted using an albumin-affinity column. The recovered proteins were digested with trypsin, followed by partial deglycosylation with PNGase F and exoglycosidases. Jacalin-affinity-chromatography enrichment was then performed, and the peptide complexity was significantly reduced in the subsequent LC–MS–MS analysis. Figure 4a is the affinity chromatogram of human-plasma proteins. As with the hCG enrichment, three fractions were collected and subjected to LC–MS–MS analysis. Two parallel LC–MS–MS experiments were performed for each fraction and the identified peptides were combined. Fraction 1 contained only non-glycosylated peptides. Mascot search results revealed 210 proteins from human plasma in this fraction. Fraction 2 and fraction 3 consisted of O-GalNAc peptides, eluted in the order of binding affinity to jacalin from low to high. The Mascot database search results of fraction 2 and fraction 3 were combined and a total of 58 O-GalNAc-attached peptides were detected and identified (Table 3). These glycopeptides correspond to 49 distinctive peptides from 36 human-plasma proteins carrying different numbers of O-GalNAc residues, ranging from one to six. The peptides AQDGGPVGTELFR derived from fractalkine, FIANSQEPEIR derived from protein MENT, and ALSLAPLAGAGLELQLER derived from protein HEG homolog 1 each have only a single potential O-glycosylation site. Therefore the T183 in AQDGGPVGTELFR, S143 in FIANSQEPEIR, and S43 in ALSLAPLAGAGLELQLER can be assigned unambiguously as the O-glycosylation sites. All other peptides contain more than one Ser or Thr residue, and sufficient backbone-fragmentation ions with retained GalNAc residue(s) are required to determine their exact O-glycosylation site. Because the primary fragmentation events in CID are cleavages of the glycosidic bonds, it is challenging to differentiate the O-glycan-modified Ser/Thr from unmodified Ser/Thr because of the neutral loss of GalNAc residues. Automatic database searching combined with manual examination of the tandem-MS spectra provided a few fragment ions with retained GalNAc residue. Figure 5 shows the CID-MS–MS spectrum of the doubly-charged ion of peptide 872-SPDESTPELSAEPTPK-887 carrying one GalNAc residue (m/z = 944.4), derived from proteoglycan 4. A series of y ions with the GalNAc residue attached were observed. The fragment ion m/z = 645.4, interpreted as y4 with one GalNAc residue, leads to the determination of T885 as the O-glycosylation site. A total of 13 O-glycosylation sites were assigned unambiguously and are summarized in Table 3. Compared with the UniProt Database, the human-plasma-proteins analysis was able to cover many known O-glycosylated proteins and peptides. A substantial number of O-GalNAc-modified peptides not included in the database were also discovered. Among these peptides, nine novel O-glycosylation sites were successfully revealed and confirmed by CID-MS–MS. Representative associated product-ion spectra with different scores are shown in the Electronic Supplementary Material (ESM) Figs. S1–S13.

Jacalin-affinity-chromatography separation of PNGase F and exoglycosidase-treated human-plasma-protein tryptic peptides. (a) Fractionation of peptides with different binding affinities to jacalin, detected by UV absorption at 214 nm. (b) Total ion chromatogram (TIC) of fraction 1 collected from jacalin-affinity column, corresponding to peptides’ lack of binding affinity to jacalin. No O-GalNAc glycopeptide was detected in this fraction. (c) TIC of fraction 2 and (d) TIC of fraction 3 collected from jacalin-affinity column, containing O-GalNAc glycopeptides with binding affinity to jacalin from low to high

The CID fragment-ion spectrum corresponds to the peptide SPDESTPELSAEPTPK from proteoglycan 4 containing one GalNAc. The y and b ion series are indicated, and the fragmentations involving O-linked HexNAc residues are annotated with a yellow square
The N-glycosylation of human-plasma proteins has been investigated extensively [17, 41]. In contrast, only a couple of studies on O-glycosylation-site analysis of human-plasma proteins have been reported. Hägglund et al. identified 23 O-glycosylated peptides derived from 11 proteins in Cohn IV fraction of human plasma as a by-product while using endo-β-N-acetylglucosaminidases and exoglycosidases to investigate the core fucosylated N-glycans [17]. Six of the 11 O-glycosylated proteins reported by Hägglund et al., including coagulation factor XII, plasma protease C1 inhibitor, and kininogen, were also observed in our analysis. Durham and Regnier revealed 43 O-glycopeptides and 36 O-glycoproteins from human serum using a two-step lectin-selection-chromatography method, which included removal of N-linked glycopeptides by concanavalin A and enrichment of O-linked glycopeptides with jacalin [30]. Surprisingly, none of these glycoproteins overlapped with the results obtained by either Hägglund et al. or us. We also found several tryptic peptides were both N and O-glycosylated. For example, the peptide 53-MLFVEPILEVSSLPTTNSTTNSATK-77 from plasma protease C1 inhibitor was revealed to be triply O-glycosylated and N-glycosylated at N69. Four other O-glycosylated peptides, ceruloplasmin-derived 129-EHEGAIYPDNTTDFQR-144 (ESM, Fig. S14), HEG-homolog-1-derived 150-SHAASDAPENLTLLAETADAR-170, vitronectin-derived 85-NNATVHEQVGGPSLTSDLQAQSK-107 (ESM, Fig. S15), and peptidase-inhibitor-16-derived 397-SLPNFPNTSATANATGGR-414, were also N-glycosylated at N138, N159, N86, N403, and N409, respectively. These N and O-glycosylation co-modified peptides would be overlooked if the concanavalin A step was applied.
The determination of O-glycosylation sites in complex proteome samples is more challenging than that of N-glycosylation sites. First, binding specificity of lectin for O-glycosylation is less satisfactory than that for N-glycosylation [20]. For example, the peptide FQDSSSSKAPPPSLPSPSRLPGPSDTPILPQ with one HexNAc residue derived from hCG is missed, possibly because of its low abundance and weak binding affinity to jacalin. Use of a multilectin-affinity column, or combining lectin-based affinity and chemistry-based methods, may improve the recovery of glycopeptides with different physicochemical properties. Second, it is common to observe multiple O-glycosylation sites in one tryptic glycopeptide. The determination of these sites is extremely difficult because of the complex composition and neutral loss of sugar residues in tandem MS. Nonspecific protease digestion could cleave the glycopeptides to shorter pieces and provide more information on O-glycosylation microheterogeneity [29, 42]. However, this is limited to the less complex samples, because assigning of enormous peptides generated by nonspecific protease is impractical because of the lack of sophisticated bioinformatics tools. Because limitations are associated with different workflows of O-glycosylation-site determination, the integration of MS results from separate studies on human plasma would better reveal the O-glycosylation patterns of this important body fluid.
Conclusion
An exoglycosidase treatment and jacalin enrichment two-step sample-preparation strategy in addition to tryptic digestion is a new method for O-glycosylation-site analysis of individual proteins and the human-plasma proteome. The approach described herein is simple, sensitive, and comprehensive. It requires minimal sample (as little as one drop of blood) to map the O-glycosylation sites of human-plasma proteins. By applying exoglycosidase digestion first, the heterogeneity of O-glycan core structures is diminished and the jacalin enrichment becomes global for core 1 through core 4 O-glycosylated peptides and single-O-GalNAc-modified peptides. Adding PNGase F to the exoglycosidase digestion converts N-glycosylated Asn residues to Asp residues. Therefore the N-glycosylation-site information can also be obtained in one LC–MS–MS run. It would be interesting in the future to use this feature to study the relationship of adjacent N and O-glycosylation sites within a protein; for example, the co-existing N and O-glycosylation sites in plasma-protease-C1-inhibitor-derived peptide 55-MLFVEPILEVSSLPTTNSTTNSATK-77 versus the mutually exclusive N and O-glycosylations in the hCG-derived peptide 52-NVTSESTCCVAK-63. The O-glycosylation-site-analysis result is reliable because many previously-known O-glycosylated peptides and proteins were detected. Additionally, many novel O-glycosylation locations in human-plasma proteins were discovered, with nine exact O-glycosylation sites being determined by CID-MS–MS. Incorporation of ETD-MS–MS techniques in future studies will provide more specific and comprehensive O-glycosylation-site information for human-plasma proteins and other critical glycoproteomes.
References
Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA (2001) Glycosylation and the immune system. Science 291:2370–2376
Helenius A, Aebi M (2001) Intracellular functions of N-linked glycans. Science 291:2364–2369
Konopka JB (2012) N-acetylglucosamine (GlcNAc) functions in cell signaling. Scientifica (Cairo)
Fardini Y, Dehennaut V, Lefebvre T, Issad T (2013) O-GlcNAcylation: a new cancer hallmark? Front Endocrinol (Lausanne) 4:99
Whitmore TE, Peterson A, Holzman T, Eastham A, Amon L, McIntosh M, Ozinsky A, Nelson PS, Martin DB (2012) Integrative analysis of N-linked human glycoproteomic data sets reveals PTPRF ectodomain as a novel plasma biomarker candidate for prostate cancer. J Proteome Res 11:2653–2665
Yao PJ, Coleman PD (1998) Reduction of O-linked N-acetylglucosamine-modified assembly protein-3 in Alzheimer’s disease. J Neurosci 18:2399–2411
Huang Y, Wu H, Xue R, Liu T, Dong L, Yao J, Zhang Y, Shen X (2013) Identification of N-glycosylation in hepatocellular carcinoma patients' serum with a comparative proteomic approach. PLoS One 8:e77161
Dunfee RL, Thomas ER, Wang J, Kunstman K, Wolinsky SM, Gabuzda D (2007) Loss of the N-linked glycosylation site at position 386 in the HIV envelope V4 region enhances macrophage tropism and is associated with dementia. Virology 367:222–234
Yamamoto K (1994) Microbial endoglycosidases for analyses of oligosaccharide chains in glycoproteins. J Biochem 116:229–235
Narimatsu H, Sawaki H, Kuno A, Kaji H, Ito H, Ikehara Y (2010) A strategy for discovery of cancer glyco-biomarkers in serum using newly developed technologies for glycoproteomics. FEBS J 277:95–105
Davies MJ, Smith KD, Hounsell EF (1994) In: Walker M (ed) Methods in molecular biology: basic protein and peptide protocols, 2nd edn. Humana Press, Totowa
Chiesa C, O’Neill RA, Horváth CG, Oefner PJ (1996) In: Righetti PG (ed) Capillary electrophoresis in analytical biotechnology. CRC Press, Boca Raton
Rademaker GJ, Pergantis SA, Blok-Tip L, Langridge JI, Kleen A, Thomas-Oates JE (1998) Mass spectrometric determination of the sites of O-glycan attachment with low picomolar sensitivity. Anal Biochem 257:149–160
Zheng Y, Guo Z, Cai Z (2009) Combination of beta-elimination and liquid chromatography/quadrupole time-of-flight mass spectrometry for the determination of O-glycosylation sites. Talanta 78:358–363
Hanisch FG, Teitz S, Schwientek T, Müller S (2009) Chemical de-O-glycosylation of glycoproteins for application in LC-based proteomics. Proteomics 9:710–719
Whitaker JR, Feeney RE (1977) Behavior of O-glycosyl and O-phosphoryl proteins in alkaline solution. Adv Exp Med Biol 86:155–175
Hägglund P, Matthiesen R, Elortza F, Højrup P, Roepstorff P, Jensen ON, Bunkenborg J (2007) An enzymatic deglycosylation scheme enabling identification of core fucosylated N-glycans and O-glycosylation site mapping of human plasma proteins. J Proteome Res 6:3021–3031
Steentoft C, Vakhrushev SY, Vester-Christensen MB, Schjoldager KT, Kong Y, Bennett EP, Mandel U, Wandall H, Levery SB, Clausen H (2011) Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat Methods 8:977–982
Ongay S, Boichenko A, Govorukhina N, Bischoff R (2012) Glycopeptide enrichment and separation for protein glycosylation analysis. J Sep Sci 35:2341–2372
Pan S, Chen R, Aebersold R, Brentnall TA (2011) Mass spectrometry based glycoproteomics–from a proteomics perspective. Mol Cell Proteomics 10:R110.003251
Tachibana K, Nakamura S, Wang H, Iwasaki H, Tachibana K, Maebara K, Cheng L, Hirabayashi J, Narimatsu H (2006) Elucidation of binding specificity of Jacalin toward O-glycosylated peptides: quantitative analysis by frontal affinity chromatography. Glycobiology 16:46–53
Saroha A, Kumar S, Chatterjee BP, Das HR (2012) Jacalin bound plasma O-glycoproteome and reduced sialylation of alpha 2-HS glycoprotein (A2HSG) in rheumatoid arthritis patients. PLoS One 7:e46374
Darula Z, Medzihradszky KF (2009) Affinity enrichment and characterization of mucin core-1 type glycopeptides from bovine serum. Mol Cell Proteomics 8:2515–2526
Darula Z, Sherman J, Medzihradszky KF (2012) How to dig deeper? Improved enrichment methods for mucin core-1 type glycopeptides. Mol Cell Proteomics 11:O111.016774
Nilsson J, Rüetschi U, Halim A, Hesse C, Carlsohn E, Brinkmalm G, Larson G (2009) Enrichment of glycopeptides for glycan structure and attachment site identification. Nat Methods 6:809–811
Halim A, Nilsson J, Rüetschi U, Hesse C, Larson G (2012) Human urinary glycoproteomics; attachment site specific analysis of N- and O-linked glycosylations by CID and ECD. Mol Cell Proteomics 11:M111.013649
Halim A, Rüetschi U, Larson G, Nilsson J (2013) LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res 12:573–584
Zauner G, Koeleman CA, Deelder AM, Wuhrer M (2010) Protein glycosylation analysis by HILIC-LC-MS of proteinase K-generated N- and O-glycopeptides. J Sep Sci 33:903–910
Nwosu CC, Seipert RR, Strum JS, Hua SS, An HJ, Zivkovic AM, German BJ, Lebrilla CB (2011) Simultaneous and extensive site-specific N- and O-glycosylation analysis in protein mixtures. J Proteome Res 10:2612–2624
Durham M, Regnier FE (2006) Targeted glycoproteomics: serial lectin affinity chromatography in the selection of O-glycosylation sites on proteins from the human blood proteome. J Chromatogr A 1132:165–173
Kessler MJ, Mise T, Ghai RD, Bahl OP (1979) Structure and location of the O-glycosidic carbohydrate units of human chorionic gonadotropin. J Biol Chem 254:7909–7914
Morgan FJ, Birken S, Canfield RE (1975) The amino acid sequence of human chorionic gonadotropin. The alpha subunit and beta subunit. J Biol Chem 250:5247–5258
Valmu L, Alfthan H, Hotakainen K, Birken S, Stenman UH (2006) Site-specific glycan analysis of human chorionic gonadotropin beta-subunit from malignancies and pregnancy by liquid chromatography–electrospray mass spectrometry. Glycobiology 16:1207–1218
Bourne Y, Astoul CH, Zamboni V, Peumans WJ, Menu-Bouaouiche L, Van Damme EJ, Barre A, Rougé P (2002) Structural basis for the unusual carbohydrate-binding specificity of jacalin towards galactose and mannose. Biochem J 364:173–180
Pisano A, Jardine DR, Packer NH, Farnsworth V, Carson W, Cartier P, Redmond JW, Williams KL, Gooley AA (1996) In: Townsend RR, Hotchkiss AT Jr (eds) Techniques in glycobiology. Marcel Dekker, New York
Wiesner J, Premsler T, Sickmann A (2008) Application of electron transfer dissociation (ETD) for the analysis of posttranslational modifications. Proteomics 8:4466–4483
Cole LA, Butler S (2012) Hyperglycosylated hCG, hCGβ and hyperglycosylated hCGβ: interchangeable cancer promoters. Mol Cell Endocrinol 349:232–238
Cole LA, Laidler LL, Muller CY (2010) USA hCG reference service, 10-year report. Clin Biochem 43:1013–1022
Cole LA (2009) Human chorionic gonadotropin and associated molecules. Expert Rev Mol Diagn 9:51–73
Cole LA (2010) Hyperglycosylated hCG, a review. Placenta 31:653–664
Zhao X, Ma C, Han H, Jiang J, Tian F, Wang J, Ying W, Qian X (2013) Comparison and optimization of strategies for a more profound profiling of the sialylated N-glycoproteomics in human plasma using metal oxide enrichment. Anal Bioanal Chem 405:5519–5529
Zauner G, Hoffmann M, Rapp E, Koeleman CA, Dragan I, Deelder AM, Wuhrer M, Hensbergen PJ (2012) Glycoproteomic analysis of human fibrinogen reveals novel regions of O-glycosylation. J Proteome Res 11:5804–5814
Acknowledgments
This work was supported by grants from the National Basic Research Program of China (973 Program) (2012CB822102), the National High Technology Research and Development Program of China (863 Program) (2012AA021504), the National Natural Science Foundation of China (21472115, 81102783, 31000367), and Natural Science Foundation of Shandong Province, China (ZR2011HQ038).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 601 kb)
Rights and permissions
About this article

Cite this article
Bai, X., Li, D., Zhu, J. et al. From individual proteins to proteomic samples: characterization of O-glycosylation sites in human chorionic gonadotropin and human-plasma proteins. Anal Bioanal Chem 407, 1857–1869 (2015). https://doi.org/10.1007/s00216-014-8439-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-014-8439-7