
Abstract
Tryptophan and phenylakylamines (PAAs) are important biomolecules, which are involved in a myriad of processes. These molecules have been experimentally reported to exhibit N–H+…π interactions in their protonated form; however, this has not been theoretically investigated in detail. Generally, such interactions are observed in diverse biological systems, and their evaluation would be useful for understanding protein folding and functioning. Therefore, in this study, we have described the N–H+…π interactions of the abovementioned classes of molecules using the Quantum Theory of Atoms In Molecules (QTAIM), Natural Bond Orbital (NBO), and Non-Covalent Interaction (NCI) analyses. The results of our N–H+…π interaction energy calculations were consistent with the experimental energies derived from the redshift of the vibrational stretches. The energy values obtained using the QTAIM-based Espinosa’s approach provided a relatively better result than similar approaches previously reported in the literature. Furthermore, we observed that the N–H+…π interaction energy in tryptophan is weaker than the resonance-assisted hydrogen bond energy of the N–H+…O=C interactions of its three most stable conformers accessible at room temperature. In contrast, the strength of the N–H+…π interaction in PAAs was observed to increase with increasing alkyl lateral chain length. The increased flexibility of longer chains increases the distance between nitrogen and the phenyl ring without disturbing the N–H+…π interaction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Tryptophan (Fig. 1) is a ubiquitous biomolecule that participates in essential life processes such as cell growth and maintenance, neurotransmission, and cellular signaling [1]. Owing to its UV and fluorescence properties, this molecule has been extensively explored by spectroscopists, and there have been several studies dedicated to the assessment of its structure and its derivatives in both solution [2,3,4] and gas phases [5, 6].

Structures of protonated tryptophan and protonated phenylalkylamines (PAAs)
For instance, the infrared spectrum of the gas-phase protonated tryptophan exhibits a broad band [7,8,9] that could be attributed to the intramolecular interaction of the ammonium group and the indol ring (N–H+…π) [7, 8]. The protonated phenylalkylamines (PAAs) containing two to four carbons in their lateral chain (Fig. 1) exhibit similar behavior, and their band width and frequency depend on their lateral chain length [10, 11].
Chiavarino et al. [10] reported that the bands associated with the N–H+ stretches of phenylethylamine (PEA), phenylpropylamine (PPA), and phenylbutylamine (PBA) were redshifted in comparison with the calculated vibrational spectra, indicating that these ions also exhibit a N–H+…π interaction. However, in the most stable conformation for phenylmethylamine (PMA), the protonated amino group is at an anti orientation with respect to the phenyl group (Fig. 2b), which prevents not only the N–H+…π interaction but also the broadening as well as the redshift of the N–H stretch band.

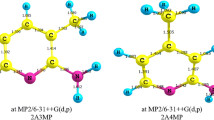
Plots of the reduced density gradient s(r) against sign(λ2)ρ and NCI isosurfaces for: a Tryp C, b PMA, c PEA, d PPA, e PBA A, obtained at the M06-2X/6-31 + G** level of theory for the lowest energy conformers; where, s(r) = 0.3 (s(r) = 0.6 for PPA) and a blue–green–red color scale from −0.04 < sign(λ2)ρ < + 0.06 a.u
The investigation of these intramolecular interactions is complicated, especially while evaluating the strength and magnitude of the interaction energies. The commonly employed methods to describe such interactions include the quantum theory of atoms in molecules (QTAIM) [12], natural bond orbital (NBO) [13] and non-covalent interaction (NCI) [14] analyses; these have been successfully employed over the years to analyze diverse systems [15,16,17,18,19,20].
While neutral tryptophan has already been modeled by QTAIM and NBO—which demonstrate the existence of a H-bond between the amine and carboxylic acid groups [2, 21], we could not find studies focusing on the intramolecular interactions of protonated tryptophan. Moreover, although there have been reported investigations of the N–H+…π interaction in PAAs using energy decomposition analysis (EDA) [22], as well as NCI calculations of PEA and its derivatives [23, 24], the PAAs and tryptophan have not been evaluated together using the set of calculations that we have used in this study.
Therefore, this overview aims to provide a detailed description of the N–H+…π interactions in these model ions, specifically protonated tryptophan; this is because such interactions in the gas and solution phases are observed to be relevant for the cation–π and other similar interactions in protein structures [25,26,27,28]. Furthermore, this characterization is also relevant for studies related to other gas-phase species because the N–H+…π interactions are often invoked to rationalize experimental spectral features; however, a formal description of such studies is rare. Therefore, we performed QTAIM, NBO, and NCI calculations in this study to assess the properties of the N–H+…π bond type, evaluate its strength, estimate the H-bond energy, and correlate the theoretical results with the gas-phase ion spectroscopy results and acceptor/donor stabilization energies.
For this purpose, the species were modeled using the Gaussian 09 (rev. D01) [29] computational package at the D3-M06–2X/6-31 + G** level of theory [30, 31].
This methodology is adequate as the choice of the basis set and method does not substantially affect the description of the weak H-bond interactions in QTAIM analysis, as reported by Jabłoński and Palusiak [32, 33]. Moreover, the use of M06-2X, or similar hybrid meta-GGA functionals for similar studies have also been reported [34,35,36,37], and are considered suitable for the systems reported herein, as their equilibrium geometries are well characterized [9, 10].
All reported species were subjected to vibrational analysis to confirm their true minima nature. For the PAAs, the geometries reported in the literature were used as inputs [10], while the multiple conformers of tryptophan and PBA, which are accessible at room temperature (298.15 K), were evaluated (identified as conformers A, B, and C, for each ion) [9, 10]. The relative free energy values for the conformers of tryptophan and PBA were less than 1 kJ/mol, as detailed in Table S1.
The NBOs were obtained using the NBO 7.0 software [38]; the topology of the electron density in the interatomic space was obtained by QTAIM, using AIMALL v. 17.11.14 [39]; and the NCIs were obtained using NCIPLOT v. 3.0 [40]. For the graphical visualization of the ions and cubes, VMD v. 1.9.3 [41], Chemcraft v. 1.8 [42], and AIMStudio (as implemented in AIMALL), were used [39].
Although QTAIM is a useful tool for describing the H-bonds, its interpretation is not straightforward because of the non-explicit relationship between the diverse parameters obtained at the bond critical points (BCPs) and the intramolecular H-bond strength.
The increase in the H-bond strength may be evaluated either by the increase in the electronic density (ρ(r), where r stands for the BCP coordinates) and kinetic energy density (G(r)); or by a decrease in the potential energy density (V(r)) and total electronic energy density (H(r)), which are accompanied by the shortening of the distance between the hydrogen atom and the H-bond BCP (d(H…BCP)). According to Fuster and Grabowski [43] the analysis of H(r) and d(H…BCP) may be used to estimate the H-bond strength. Furthermore, Rozas et al. [44] classified the H-bond interactions according to the Laplacian of electronic density of their BCPs (i.e., ∇2ρ(r)) and H(r)): ∇2ρ(r) and H(r) > 0 indicates a weak interaction; ∇2ρ(r) > 0 and H(r) < 0 indicates a medium interaction; and ∇2ρ(r) and H(r) < 0 indicates a strong interaction. Other studies have also evaluated the relationship between H(r) and G(r) to describe H-bond strengths: as 2G(r) + V(r) and the ratio |V(r)|/G(r) [45].
Moreover, the H-bond strength can also be determined from the bond energy: Grabowski suggested that H-bonds could be classified by their H-bond energies (EHB) as weak (~ 4–17 kJ/mol), moderate (~ 17–63 kJ/mol), and strong (~ 63–168 kJ/mol) [46]. However, the estimation of the intramolecular H-bond energies (EIHB) is not as simple as that of the intermolecular H-bond energies (EHB) [22, 47]: the latter is calculated by the difference between the total energy of a complex (formed by the H-bond) and its isolated constituents; nevertheless, in case of the former, it is not possible to entirely separate the residues containing the hydrogen donors from the hydrogen acceptors. Therefore, among the existing QTAIM-based strategies for calculating EIHB, the approaches proposed by Espinosa (E ESPIHB ) [48] and by Afonin (E AFIHB ) [47, 49], which correlate EIHB with the absolute potential energy density |V(r)| at a given BCP, are prominent; these are described as follows:
In this study, we have compared the abovementioned approaches with the empirical H-bond energy that can be estimated using Iogansen’s equation [50], which is described as follows:
where ∆H represents the enthalpy of the H-bond, and ∆ν is the difference (in cm−1) between the free (ν0) and bound (ν1) D-H stretch bands (where D represents the proton donor atom). This equation is valid when the H-bond causes a redshift in the D-H stretch band and ∆ν is higher than 40 cm−1. To determine −∆H, we compared the redshifted N–H+…π stretch bands of PEA and PBA with the free N–H stretch band of PMA, which was reported by Chiavarino [10]; meanwhile, for tryptophan, we compared the N–H+…π stretch band [9] with the free N–H+ stretch band of glycine (Table S2) [51]. While some researchers would consider the average of the symmetric and asymmetric stretch bands as ν0 [52, 53], we have used the −∆H obtained by both values, thereby establishing a range for the N–H+…π interactions. Although its efficiency in estimating the intramolecular H-bonds has been demonstrated [47, 54], in reality, Eq. (III) was formulated for intermolecular H-bonds. Hence, its results must only be considered as an estimate of the of the H-bond energy range to allow a comparison with the theoretical results.
Table 1 lists the QTAIM parameters of the intramolecular interactions at their corresponding BCPs. An overall analysis of the QTAIM parameters demonstrates that all H-bonds identified in this study can be classified as weak, as they exhibit positive values of ∇2ρ(r) and H(r). This is corroborated by the E AFIHB values; however, in case of the E ESPIHB values, some interactions are considered as moderate according to Grabowski’s classification [46].
Almost all identified BCPs obeyed the criteria for H-bonds proposed by Koch and Popelier [55] (the graphical visualization of the structure of all studied ions and their BCPs are presented in Fig. S1); the only exception is observed in conformer C of tryptophan (Tryp C), wherein a H…H attractive interaction is observed between the hydrogen atoms attached to C2 and C10 (Fig. 1). This interaction is described as a weak Van der Waals interaction, as depicted by its QTAIM parameters (e.g., low values of ρ(r) and |V(r)|). This (controversial) type of interaction has already been documented in the literature and is considered as a stabilizing interaction [56, 57] of a quantum nature (i.e., higher contribution of the exchange–correlation term) rather than of an electrostatic nature [58].
In contrast to the proposition by Fuster and Grabowski [43], the H(r) and d(H…BCP) values were not observed to be useful in estimating the H-bond strength for the systems reported herein. This could be because their approach was focused on moderate and strong H-bonds. Conversely, the ∇2ρ(r) and G(r) values exhibited a linear relationship with E ESPIHB (R2 = 0.950 and 0.984, respectively; Fig. S2). In the case of ρ(r), the data exhibited a linear trend when the N–H…O H-bond was not considered (R2 = 0.982; Fig. S2).
Similarly, d(H…A) exhibited an almost linear relationship with E ESPIHB (R2 = 0.879; Fig. S2) when the H…H and the N–H…O interactions were not considered; a similar linear trend was also observed while comparing the QTAIM parameters with the E AFIHB values (Fig. S2). Nevertheless, E AFIHB values are lower than the E ESPIHB values, as shall be discussed subsequently.
Both E AFIHB and E ESPIHB values are observed to be considerably lower than those of EEDA. Nevertheless, the empirical H–bond energies (−ΔH) for tryptophan (20–23 kJ/mol), PEA (13–17 kJ/mol), and PBA (18–22 kJ/mol) in Table 1 is in better agreement with the respective E ESPIHB values (i.e., 14–18, 13, and 16–26 kJ/mol, respectively; Table 1).
Therefore, in the context of our study, Eq. (II) yields better values than Eq. (I), and E ESPIHB is considered as the most adequate energy scheme to represent the ions of interest of this study and explain their vibrational features.
Interestingly, the E AFIHB value of N–H+…π calculated using Eq. (I) (8 kJ/mol) agrees with the much lower energy reported by Pereverzev et al. (7 kJ/mol), which they had calculated as the difference between the electron density of neutral and protonated tryptophan (Δρ = ρ(TrpH+) − ρ(Trp)) [9]. However, this approach only considers the polarization effects, as pointed out by previous studies, which may be the reason for the underestimated EIHB values.
Conformers A and B of tryptophan form a BCP between N–H+ and C11, but on opposite faces of the ring plane. However, Tryp C forms a BCP between N–H+ and C4. Furthermore, the QTAIM results suggest that N–H+…π is weaker than the N–H+…O=C interaction, which is considered to be a resonance-assisted hydrogen bond [59, 60]. Nonetheless, this behavior is less accentuated in conformer C, where the difference between the N–H+…π (E ESPIHB = 18.23 kJ/mol, Table 1) and N–H+…O (E ESPIHB = 25.52 kJ/mol, Table 1) interaction energies is lower than 7.29 kJ/mol. Thus, both interactions contribute similarly to the overall stabilization of Tryp C.
Furthermore, all PAAs, excluding PMA, exhibit BCPs corresponding to the N–H+…π interactions (Fig. S1). Conformer C of PBA (PBA C) exhibits only one N–H+…π interaction, while PBA A exhibits two N–H+…π interactions—promoted by proton H1, which interacts with C6, and proton H2, which interacts with C2 (Fig. S1). Among all PAAs, the ellipticity of the PBA H2…π interaction is closer to zero, indicating that the electron density is concentrated in a plane containing the bond path in a higher extension [61, 62], thereby increasing the bond stability. The abovementioned interaction between H1 and C6 further increases the stability of this conformation. On the other hand, PBA B exhibits only one N–H+…π interaction and a weaker C–H…π interaction.
An analysis of the PAAs indicates an increase in G(r), from 1.18 to 1.33 and 1.89 a.u., and in |V(r)|, from 0.99 to 1.21 and 1.70 a.u. with increasing side chain length, i.e., from PEA to PPA and PBA (corresponding to the total contributions of the PBA A interactions). Consequently, E ESPIHB increases from 13 to 16 and 22 kJ/mol, respectively (Table 1). Although PBA A has been considered as an example, the same trend is observed in all PBA conformers.
These interactions can be visualized using the NCI calculations and by mapping the space between the atoms to find the regions where the reduced density gradient (RDG), s(r), tends to zero. Thus, NCI can be considered to complement QTAIM, as it presents troughs in s(r) at each critical point and maps the entire 3D space in addition to the atomic interaction line, thereby allowing the identification of the electron depletion zones [14, 40, 63]. The NCI isosurfaces are represented on a color scale indicating the interaction strength (the strongly attractive, strongly repulsive, and weak interactions are represented by the blue, red, and green colors, respectively). Furthermore, the absolute value of ρ increases along with the interaction strength, while the sign of λ2 in the Hessian matrix indicates the type of interaction—i.e., negative for attractive interactions and positive for repulsive interactions.
To characterize the protonated tryptophan, Fig. 2 shows the NCI isosurfaces of this species, the protonated PPA series for comparison, as well as the plots of s(r) against sign(λ2)ρ; conformers Tryp C and PBA A were chosen to represent the respective ions. Table S3 lists the troughs in s(r) for all species, while the NCI isosurfaces of the remaining conformers of tryptophan and PBA are shown in Fig. S4 and Fig. S5, respectively.
As expected, PMA does not exhibit any trough from the intramolecular HB interactions. The repulsive region (sign(λ2)ρ = + 0.021; Fig. 2b) corresponds to a steric repulsion of the non-bonded overlap between the carbon atoms of the phenyl ring, which is observed in all studied compounds, including PMA.
The NCI analysis of PEA (Fig. 2c) is consistent with the previous analysis described in the literature [24]. The N–H+…π interaction is depicted by the blue region centered at the BCP (sign(λ2)ρ = − 0.015) and an orange region centered at the ring critical point (RCP) (sign(λ2)ρ = + 0.015) of the pseudo-five-membered ring, generated by the lateral chain and C1.
This repulsive region centered at the RCP is observed in all PAAs and is weakened by the increase in the distance between the nitrogen atom and C1, as observed for PPA (sign(λ2)ρ = + 0.011; Fig. 2d) and PBA A (sign(λ2)ρ = + 0.010; Fig. 2e).
For example, from PEA to PPA, the addition of one alkyl carbon to the side chain brings the proton closer to C1, thereby reducing their distance from 2.37 Å, in the former ion, to 2.28 Å in the latter; meanwhile, the distance for nitrogen to C1 increases from 2.90 to 2.95 Å. On the other hand, in PBA A, this distance is 3.07 Å, while H1 is further from the π system (2.5 Å from C6) than H2 (2.3 Å from C2).
Furthermore, for PBA A (Fig. 2e), two troughs at approximately sign(λ2)ρ = − 0.017 and sign(λ2)ρ = − 0.011 can be observed and assigned to two N–H+…π interactions, as previously discussed. The other troughs at the positive values of sign(λ2)ρ correspond to the repulsive interactions from a pseudo-seven-membered ring formation as well as in PPA where there is a pseudo-six-membered ring formation. However, owing to the relatively lower torsional stress in the former ring than the latter, the repulsive interactions in PBA are weaker than those in PPA. Moreover, unlike in QTAIM, the NCI analysis also shows the H…H interactions in PPA and all PBA conformers that were previously observed in the QTAIM analysis of tryptophan. The troughs of these interactions are not discernible in the case of PPA (Fig. 2d), although only PBA A exhibits an attractive (sign(λ2)ρ = − 0.007) and a repulsive (sign(λ2)ρ = − 0.006) character for this interaction (Fig. 2e).
The protonated tryptophan (Fig. 2a) can be compared to the characterized protonated PAAs—for Tryp C, two isosurfaces are observed: the first represents the attractive and repulsive regions, corresponding to the stronger N–H+…O=C intramolecular H-bond (sign(λ2)ρ = − 0.024 and sign(λ2)ρ = + 0.024); and the second represents the N–H+…π interaction (sign(λ2)ρ = − 0.021 and sign(λ2)ρ = + 0.024). Both isosurfaces exhibit an asymmetric volume with a distinct blue region centered at the BCP and a distinct orange region centered at the RCP of the pseudo-rings. Furthermore, the H…H interaction exhibits an attractive (sign(λ2)ρ = − 0.006) and a repulsive (sign(λ2)ρ = + 0.006) region, as observed in PPA and PBA. The trough at sign(λ2)ρ = + 0.051 corresponds to the repulsive interactions between the carbons of the pyrrolic ring. The other conformers also exhibited similar results, as shown in Fig. S3.
The NBO second-order stabilization energy (E2) is generally used to investigate delocalization and charge-transfer effects based on the interaction between occupied Lewis-like orbitals (donors) and unoccupied non-Lewis orbitals (acceptors) [64]. The results from the NBO population analysis of the analyzed species indicate the same trends observed by the QTAIM and NCI analyses. Table S4 summarizes all orbital interactions, their hybridization, and calculated E2 values: Tryp C exhibits a N–H+…π interaction that has a major contribution from the π orbital donation (C4–C5 π-bond) to the N–H+ anti bond orbital (π → σ*), and a less significant backdonation from the NH group to the π system. The sum of the E2 values of these two contributions for the N–H+…π in protonated tryptophan is −28 kJ/mol, which is slightly higher than the total E2 energy for the N–H+…O=C interaction (−25 kJ/mol). As the EIHB value of the N–H+…π interaction is lower, the higher E2 value could indicate a higher charge-transfer character for this interaction than the N–H+…O=C interaction. The other conformers of tryptophan accepts donations from the π orbitals of the C11–C6 and C10–C9 bonds to N–H+ σ*, and their E2 values (i.e., 11 kJ/mol for both) are lower than those of the N–H+…O=C interactions (Tryp A = 18 kJ/mol and Tryp B = 22 kJ/mol).
The E2 value for the PEA, PPA, and PBA conformers increases as the number of alkyl carbons increases. The E2 correlated with the donation of the C1-C2 π orbital to the N–H σ* orbital of PEA is −18 kJ/mol (Table S4), while the N–H σ* orbital of PPA receives charge from two π orbitals of the C1–C2 and C3–C4 bonds, resulting in a total E2 of −29 kJ/mol.
In PBA A, the N–H1 σ* orbital accepts electrons from two π orbitals (from C1–C6 and C4–C5 π-bonds), contributing to an E2 value of −9 kJ/mol, while the C2–C3 π-bond donation to the N–H2 σ* orbital results in an E2 value of −24 kJ/mol, which represents a total stabilization energy of −33 kJ/mol—i.e., almost twice as much as the total stabilization for PEA (Table S4). The E2 values of PBA B and C are still higher—at 37 and 46 kJ/mol, respectively: in the former, the proton interacts with the σ orbital of the C1–C6 bond and with the π orbitals of the C1–C2 and C5–C6 bonds. Furthermore, there is a donation from the σ orbital of the C1-C2 bond to the σ* orbital of the C10–H bond.
In PBA C, the proton interacts with all atoms participating in the π system. This observed increase in the E2 value of the PAAs can be associated with an increase in the degrees of freedom in the longer chains, which allows them to fold freely without compromising the N–H+…π interaction.
Thus, in summary, the N–H+…π interactions in tryptophan and PAAs (with one to four alkyl carbons) were modeled using QTAIM, NCI, and NBO analyses, and the following observations were made: For PAA, the calculations indicated an increase in the interaction as the alkyl side chain length was increased, which can be explained by the more flexible chain allowing a better proton interaction with the π system. Moreover, longer chains increase the distance between nitrogen and the phenyl ring without disturbing the N–H+…π interaction. For tryptophan, the H…π interaction plays a significant role in the conformation state of the ions: In conformers A and B, the N–H+…π bond is considerably weaker than the H…O bond, while in Tryp C, the difference is not so large, which indicates that both interactions contribute to the overall stabilization of this conformer. Moreover, a weak H…H Van der Waals interaction was also identified.
Furthermore, the QTAIM-based EIHB values obtained using Espinosa’s approach (E ESPIHB ) were consistent with the experimental values based on the redshift of the vibrational stretches. By formally describing these N–H+…π interactions, the results corroborate the observed experimental features and provide theoretical support for the assumption that N–H+…π is a cause for the band redshift.
The NBO calculations further corroborate the QTAIM results: The N–H+…π interaction exhibits a significant charge-transfer nature, which is more accentuated in PAAs than in tryptophan.
Availability of data and material
The datasets generated and/or analyzed during the current study shall be provided by the corresponding author upon reasonable request.
References
Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA (2019) Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov 18:379–401. https://doi.org/10.1038/s41573-019-0016-5
Duarte CJ, Cormanich RA, Ducati LC, Rittner R (2013) 1H NMR and theoretical studies on the conformational equilibrium of tryptophan methyl ester. J Mol Struct 1050:174–179. https://doi.org/10.1016/j.molstruc.2013.07.024
Anderson JS, Bowitch GS, Brewster RL (1983) Influence of conformation on the fluorescence of tryptophan-containing peptides. Biopolymers 22:2459–2476. https://doi.org/10.1002/bip.360221111
Kowalska-Baron A (2015) Theoretical study of the complexes of tyrosine and tryptophan with biologically important metal cations in aqueous solutions. Comput Theor Chem 1057:7–14. https://doi.org/10.1016/j.comptc.2015.01.010
Lioe H, O’Hair RAJ, Reid GE (2004) Gas-phase reactions of protonated tryptophan. J Am Soc Mass Spectrom 15:65–76. https://doi.org/10.1016/j.jasms.2003.09.011
Rizzo TR, Park YD, Levy DH (1986) Dispersed fluorescence of jet-cooled tryptophan: excited state conformers and intramolecular exciplex formation. J Chem Phys 85:6945–6951. https://doi.org/10.1063/1.451381
Rodrigues-Oliveira AF, Ribeiro FWM, Cervi G, Correra TC (2018) Evaluation of common theoretical methods for predicting infrared multiphotonic dissociation vibrational spectra of intramolecular hydrogen-bonded ions. ACS Omega 3:9075–9085. https://doi.org/10.1021/acsomega.8b00815
Mino WK, Gulyuz K, Wang D, Stedwell CN, Polfer NC (2011) Gas-phase structure and dissociation chemistry of protonated tryptophan elucidated by infrared multiple-photon dissociation spectroscopy. J Phys Chem Lett 2:299–304. https://doi.org/10.1021/jz1017174
Pereverzev AY, Cheng X, Nagornova NS, Reese DL, Steele RP, Boyarkin OV (2016) Vibrational signatures of conformer-specific intramolecular interactions in protonated tryptophan. J Phys Chem A 120:5598–5608. https://doi.org/10.1021/acs.jpca.6b05605
Chiavarino B, Crestoni ME, Schütz M, Bouchet A, Piccirillo S, Steinmetz V, Dopfer O, Fornarini S (2014) Cation − π interactions in protonated phenylalkylamines. J Phys Chem A 118:7130–7138. https://doi.org/10.1021/jp505037n
Bouchet A, Schütz M, Chiavarino B, Elisa Crestoni M, Fornarini S, Dopfer O (2015) IR spectrum of the protonated neurotransmitter 2-phenylethylamine: dispersion and anharmonicity of the NH3 + –π interaction. Phys Chem Chem Phys 17:25742–25754. https://doi.org/10.1039/C5CP00221D
Bader RFW (1991) A quantum theory of molecular structure and its applications. Chem Rev 91:893–928. https://doi.org/10.1021/cr00005a013
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem Rev 88:899–926. https://doi.org/10.1021/cr00088a005
Johnson ER, Keinan S, Mori-Sánchez P, Contreras-García J, Cohen AJ, Yang W (2010) Revealing Noncovalent Interactions. J Am Chem Soc 132:6498–6506. https://doi.org/10.1021/ja100936w
Correra TC, Fernandes AS, Reginato MM, Ducati LC, Berden G, Oomens J (2017) Probing the geometry reorganization from solution to gas-phase in putrescine derivatives by IRMPD, 1H-NMR and theoretical calculations. Phys Chem Chem Phys 19:24330–24340. https://doi.org/10.1039/c7cp04617k
Masson MAC, Karpfenstein R, Oliveira-silva D, Teuler J, Archirel P, Mâitre P, Correra TC (2018) Evaluation of Ca 2 + binding sites in tacrolimus by infrared multiple photon dissociation spectroscopy. https://doi.org/10.1021/acs.jpcb.8b06523
Salehi S, Mashmool Moghaddam SM, Tarin M, Shokooh Saljooghi A (2020) Pharmaceutical nickel(II) chelation properties of 3-hydroxyflaven, deferiprone and maltol metal chelators: A density functional study. Phys Chem Res 8:91–110. https://doi.org/10.22036/pcr.2019.202156.1677
Potla KM, Poojith N, Osório FAP, Valverde C, Chinnam S, Suchetan PA, Vankayalapati S (2020) An analysis of spectroscopic, computational and biological activity studies of L-shaped sulfamoylbenzoic acid derivatives: a third order nonlinear optical material. J Mol Struct 1210:128070. https://doi.org/10.1016/j.molstruc.2020.128070
Allal H, Belhocine Y, Rahali S, Damous M, Ammouchi N (2020) Structural, electronic, and energetic investigations of acrolein adsorption on B36 borophene nanosheet: a dispersion-corrected DFT insight. J Mol Model 26:128. https://doi.org/10.1007/s00894-020-04388-3
Likhitha U, Narayana B, Sarojini BK, Madan Kumar S, Lobo AG, Karthick T (2020) A study on interwoven hydrogen bonding interactions in new zidovudine-picric acid (1:1) cocrystal through single crystal XRD, spectral and computational methods. J Mol Struct 1211:128052. https://doi.org/10.1016/j.molstruc.2020.128052
Niu X, Huang Z, Ma L, Shen T, Guo L (2013) Density functional theory, natural bond orbital and quantum theory of atoms in molecule analyses on the hydrogen bonding interactions in tryptophan-water complexes. J Chem Sci 125:949–958. https://doi.org/10.1007/s12039-013-0445-3
Su P, Chen Z, Wu W (2015) An energy decomposition analysis study for intramolecular non-covalent interaction. Chem Phys Lett 635:250–256. https://doi.org/10.1016/j.cplett.2015.06.078
Schütz M, Bouchet A, Dopfer O (2016) Infrared spectrum of the cold ortho-fluorinated protonated neurotransmitter 2-phenylethylamine: competition between NH + …π and NH + …F interactions. Phys Chem Chem Phys 18:26980–26989. https://doi.org/10.1039/C6CP05915E
Bouchet A, Schütz M, Dopfer O (2016) Competing insertion and external binding motifs in hydrated neurotransmitters: infrared spectra of protonated phenylethylamine monohydrate. ChemPhysChem 17:232–243. https://doi.org/10.1002/cphc.201500939
Kumar M, Balaji PV (2014) C-H…pi interactions in proteins: prevalence, pattern of occurrence, residue propensities, location, and contribution to protein stability. J Mol Model 20:2136. https://doi.org/10.1007/s00894-014-2136-5
Gallivan JP, Dougherty DA (1999) Cation-pi interactions in structural biology. Proc Natl Acad Sci 96:9459–9464. https://doi.org/10.1073/pnas.96.17.9459
Mahadevi AS, Sastry GN (2013) Cation − π interaction: its role and relevance in chemistry, biology, and material science. Chem Rev 113:2100–2138. https://doi.org/10.1021/cr300222d
Yamada S (2020) Cation–π interactions in organic crystals. Coord Chem Rev 415:213301. https://doi.org/10.1016/j.ccr.2020.213301
Frisch MJ, Trucks GW, Schlegel HB GES, Robb MA, Cheeseman JR, Scalmani G, Barone VBM, Petersson GA, Nakatsuji H, Caricato M, Li XHPH, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JLMH, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida MTN, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JAJ, Peralta JE, Ogliaro F, Bearpark M, Heyd JJEB, Kudin KN, Staroverov VN, Keith T, Kobayashi RJN, Raghavachari K, Rendell A, Burant JC, Iyengar SSJT, Cossi M, Rega N, Millam JM, Klene M, Knox JEJBC, Bakken V, Adamo C, Jaramillo J, Gomperts RRES, Yazyev O, Austin AJ, Cammi R, Pomelli CJWO, Martin RL, Morokuma K, Zakrzewski VGGAV, Salvador P, Dannenberg JJ, Dapprich SADD, Farkas O, Foresman JB, Ortiz JVJC, Fox DJ (2013) Gaussian 09, Revision D.01
Grimme S (2004) Accurate description of van der Waals complexes by density functional theory including empirical corrections. J Comput Chem 25:1463–1473. https://doi.org/10.1002/jcc.20078
Zhao Y, Truhlar D (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor Chem Acc 120:215–241. https://doi.org/10.1007/s00214-007-0310-x
Jabłoński M, Palusiak M (2010) Basis set and method dependence in Quantum Theory of Atoms in Molecules calculations for covalent bonds. J Phys Chem A 114:12498–12505. https://doi.org/10.1021/jp106740e
Jabłoński M, Palusiak M (2010) Basis set and method dependence in atoms in molecules calculations. J Phys Chem A 114:2240–2244. https://doi.org/10.1021/jp911047s
Forni A, Pieraccini S, Franchini D, Sironi M (2016) Assessment of DFT functionals for QTAIM topological analysis of halogen bonds with benzene. J Phys Chem A 120:9071–9080. https://doi.org/10.1021/acs.jpca.6b07578
Esrafili MD (2012) Investigation of H-bonding and halogen-bonding effects in dichloroacetic acid: DFT calculations of NQR parameters and QTAIM analysis. J Mol Model 18:5005–5016. https://doi.org/10.1007/s00894-012-1496-y
Rincón DA, Cordeiro MNDS, Mosquera RA (2016) On the effects of the basis set superposition error on the change of QTAIM charges in adduct formation. Application to complexes between morphine and cocaine and their main metabolites. RSC Adv 6:110642–110655. https://doi.org/10.1039/C6RA22736H
Shameera Ahamed TK, Rajan VK, Sabira K, Muraleedharan K (2019) DFT and QTAIM based investigation on the structure and antioxidant behavior of lichen substances Atranorin, Evernic acid and Diffractaic acid. Comput Biol Chem 80:66–78. https://doi.org/10.1016/j.compbiolchem.2019.03.009
Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Karafiloglou P, Landis CR, Weinhold F (2018) NBO 7.0
Keith TA (2017) AIMAll (Version 17.11.14)
Contreras-García J, Johnson ER, Keinan S, Chaudret R, Piquemal J-P, Beratan DN, Yang W (2011) NCIPLOT: a program for plotting noncovalent interaction regions. J Chem Theory Comput 7:625–632. https://doi.org/10.1021/ct100641a
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. https://doi.org/10.1016/0263-7855(96)00018-5
Zhurko GA Chemcraft—graphical program for visualization of quantum chemistry computations-v.1.8. https://chemcraftprog.com
Fuster F, Grabowski SJ (2011) Intramolecular hydrogen bonds: the QTAIM and ELF characteristics. J Phys Chem A 115:10078–10086. https://doi.org/10.1021/jp2056859
Rozas I, Alkorta I, Elguero J (2000) Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J Am Chem Soc 122:11154–11161. https://doi.org/10.1021/ja0017864
Sarkar R, Kundu TK (2019) Nonbonding interaction analyses on PVDF/[BMIM][BF4] complex system in gas and solution phase. J Mol Model 25:131. https://doi.org/10.1007/s00894-019-4020-9
Grabowski SJ (2006) Theoretical studies of strong hydrogen bonds. Annu Reports Sect “C” (Physical Chem 102:131. https://doi.org/10.1039/b417200k
Afonin AV, Vashchenko AV, Sigalov MV (2016) Estimating the energy of intramolecular hydrogen bonds from 1 H NMR and QTAIM calculations. Org Biomol Chem 14:11199–11211. https://doi.org/10.1039/C6OB01604A
Espinosa E, Molins E, Lecomte C (1998) Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem Phys Lett 285:170–173. https://doi.org/10.1016/S0009-2614(98)00036-0
Batista PR, Karas LJ, Viesser RV, De Oliveira CC, Gonçalves MB, Tormena CF, Rittner R, Ducati LC, De Oliveira PR (2019) Dealing with hydrogen bonding on the conformational preference of 1,3-aminopropanols: experimental and molecular dynamics approaches. J Phys Chem A 123:8583–8594. https://doi.org/10.1021/acs.jpca.9b05619
Iogansen AV (1999) Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν(XH) vibration in infrared spectra. Spectrochim Acta Part A Mol Biomol Spectrosc 55:1585–1612. https://doi.org/10.1016/S1386-1425(98)00348-5
Fischer KC, Sherman SL, Voss JM, Zhou J, Garand E (2019) Microsolvation structures of protonated glycine and L-alanine. J Phys Chem A 123:3355–3366. https://doi.org/10.1021/acs.jpca.9b01578
Voss JM, Fischer KC, Garand E (2018) Revealing the structure of isolated peptides: IR–IR predissociation spectroscopy of protonated triglycine isomers. J Mol Spectrosc 347:28–34. https://doi.org/10.1016/j.jms.2018.03.006
Yurenko YP, Zhurakivsky RO, Ghomi M, Samijlenko SP, Hovorun DM (2007) How many conformers determine the thymidine low-temperature matrix infrared spectrum? DFT and MP2 quantum chemical study. J Phys Chem B 111:9655–9663. https://doi.org/10.1021/jp073203j
Afonin AV, Sterkhova IV, Vashchenko AV, Sigalov MV (2018) Estimating the energy of intramolecular bifurcated (three-centered) hydrogen bond by X-ray, IR and 1 H NMR spectroscopy, and QTAIM calculations. J Mol Struct 1163:185–196. https://doi.org/10.1016/j.molstruc.2018.02.106
Koch U, Popelier PLA (1995) Characterization of C–H–O hydrogen bonds on the basis of the charge density. J Phys Chem 99:9747–9754. https://doi.org/10.1021/j100024a016
El-Emam AA, Saveeth Kumar E, Janani K, Al-Wahaibi LH, Blacque O, El-Awady MI, Al-Shaalan NH, Percino MJ, Thamotharan S (2020) Quantitative assessment of the nature of noncovalent interactions in N -substituted-5-(adamantan-1-yl)-1,3,4-thiadiazole-2-amines: insights from crystallographic and QTAIM analysis. RSC Adv 10:9840–9853. https://doi.org/10.1039/D0RA00733A
Cukrowski I, Matta CF (2010) Hydrogen–hydrogen bonding: a stabilizing interaction in strained chelating rings of metal complexes in aqueous phase. Chem Phys Lett 499:66–69. https://doi.org/10.1016/j.cplett.2010.09.013
Miranda MO, Duarte DJR, Alkorta I (2020) Anion-anion complexes established between aspartate dimers. ChemPhysChem. https://doi.org/10.1002/cphc.201901200
Gilli G, Gilli P (2000) Towards an unified hydrogen-bond theory. J Mol Struct 552:1–15. https://doi.org/10.1016/S0022-2860(00)00454-3
Gilli G (2009) The nature of the hydrogen bond: outline of a comprehensive hydrogen bond theory. Oxford University Press, Oxford
Halls MD, Velkovski J, Schlegel HB (2001) Harmonic frequency scaling factors for Hartree-Fock, S-VWN, B-LYP, B3-LYP, B3-PW91 and MP2 with the Sadlej pVTZ electric property basis set. Theor Chem Acc 105:413–421. https://doi.org/10.1007/s002140000204
Hilal R, Aziz SG, Alyoubi AO, Elroby S (2015) Quantum topology of the charge density of chemical bonds. QTAIM analysis of the C-Br and O-Br bonds. Procedia Comput Sci 51:1872–1877. https://doi.org/10.1016/j.procs.2015.05.423
Cukrowski I, de Lange JH, Adeyinka AS, Mangondo P (2015) Evaluating common QTAIM and NCI interpretations of the electron density concentration through IQA interaction energies and 1D cross-sections of the electron and deformation density distributions. Comput Theor Chem 1053:60–76. https://doi.org/10.1016/j.comptc.2014.10.005
Astani E, Heshmati E, Chen C-J, Hadipour NL, Shekarsaraei S (2016) A study of hydrogen bond effects on the oxygen, nitrogen, and hydrogen electric field gradient tensors in the active site of human dehydroepiandrosterone sulphotransferase: a density-functional theory based treatment. Chem Phys Lett 653:78–84. https://doi.org/10.1016/j.cplett.2016.04.001
Acknowledgements
The authors would like to thank the support received from São Paulo Research Foundation (FAPESP Grants 2014/15962-5, 2015/08539-1, 2017/17750-3, 2018/07308–4, and 2019/25634-9) and Coordination of Superior Level Staff Improvement (CAPES finance code 001 and Grant 23038.006960/2014-65); A.F.R. also acknowledges the support of The National Council for Scientific and Technological Development (CNPq 142342/2016-5). We also acknowledge the National Laboratory for Scientific Computing (LNCC/MCTI, Brazil) for providing the high performance computing resources of the SDumont supercomputer.
Funding
FAPESP: Grants 2014/15962-5, 2015/08539-1, 2017/17750-3, 2018/07308-4, and 2019/25634-9 CAPES: finance code 001 and Grant 23038.006960/2014-65 CNPq: Grant 142342/2016-5.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
“Festschrift in honor of Prof. Fernando R. Ornellas” Guest Edited by Adélia Justino Aguiar Aquino, Antonio Gustavo Sampaio de Oliveira Filho & Francisco Bolivar Correto Machado.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rodrigues-Oliveira, A.F., Batista, P.R., Ducati, L.C. et al. Analyzing the N–H+…π interactions of protonated tryptophan and phenylalkylamines using QTAIM, NCI, and NBO. Theor Chem Acc 139, 130 (2020). https://doi.org/10.1007/s00214-020-02643-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-02643-7