
Abstract
We review two essential features of the intermolecular interaction energies (ΔE) computed in the context of quantum chemistry (QC): non-isotropy and non-additivity. Energy-decomposition analyses show the extent to which each comes into play in the separate ΔE contributions, namely electrostatic, short-range repulsion, polarization, charge-transfer and dispersion. Such contributions have their counterparts in anisotropic, polarizable molecular mechanics (APMM), and each of these should display the same features as in QC. We review examples to evaluate the performances of APMM in this respect. They bear on the complexes of one or several ligands with metal cations, and on multiply H-bonded complexes. We also comment on the involvement of polarization, a key contributor to non-additivity, in the issues of multipole transferability and conjugation. In the last section we provide recent examples of APMM validations by QC, which relate to interactions taking place in the recognition sites of kinases and metalloproteins. We conclude by mentioning prospects of extensive applications of APMM.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Quantum Chemistry
- Focal Adhesion Kinase
- Molecular Electrostatic Potential
- Water Network
- Polarization Energy
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1.1 Introduction
The field of molecular simulation is undergoing major strides forward. It is spearheaded by ab initio quantum chemistry (QC) , whose realm of applications is being extended to complexes of increasing size, nowadays totaling a few hundreds of atoms, and are amenable as well to increasingly long simulation times as is the case for Car-Parrinello (CP) [1, 2] or Born-Oppenheimer (BO) [3, 4] molecular dynamics (MD) approaches. Such extensions are enabled by advances both in informatics and in the QC codes themselves. The first kind of advances results from progress both in computer hardware, as exemplified by the advent of Graphics Processor Units (GPU) [5–6] and in computer software, as enabled by parallelism on Open MP/MPI software [7]. The second advances are due to promising developments in linear-scaling and divide-and-conquer approaches [8–13]. Despite such advances, simulations of very large molecular complexes and/or on very long-time scale MD or of computer-intensive Monte-Carlo (MC) simulations are likely to remain out of reach of high-level QC for a long foreseeable time in a diversity of domains. These encompass drug design, protein folding, or material science. An appealing alternative consists into the so-called QM/MM (Quantum mechanics/molecular mechanics) approaches, in which the core of the recognition site is treated quantum-mechanically while the periphery is treated by classical MM. Multiscale approaches are a generalization of QM/MM [14–22]. However, such approaches could not be used in the absence of a single ‘privileged’ recognition site, as is the case for multidomain sites in, e.g., protein-nucleic acid (NA), protein-protein or NA-NA recognition. There are also examples of enlarged or accessible recognition sites, such that long-range polarization effects could blur the distinction between QM and MM zones. This can occur with the complexes of inhibitors to the Zn-metalloenzyme phosphomannose isomerase, in which networks of polarizable water molecules can connect together the inhibitor in the recognition site to farther sites on N- and/or C-terminal sites [23]. ‘Classical’ MM/MD potentials are on the other hand fully able nowadays to handle very large molecular complexes over time-scales which even for proteins can approach the microsecond if not the millisecond time. They have rendered prominent service to the field and will most probably continue to do so for a long-time to come. Nevertheless their limitations are duly recognized, notably the absence of non-additivity and the lack of appropriate directionality. Such shortcomings will not be commented in this review. An ideal situation would consist into a QC-grounded MM potential formulated and calibrated so as to reproduce each individual QC contribution. Transferability can be compromised if the aim of a given MM potential were limited to reproduce the total QC intermolecular interaction, ΔE(QC) forfeiting that on its individual contributions. It is also clear that a term-to-term agreement with the ΔE(QC) contributions should carry out beyond the training set used in the calibration. Striving at reproducing QC results has been the object of several endeavors, past and present. These have been surveyed in several recent reviews [24–29]. Summarizing, we recall that the main efforts bore on the inclusion of polarization: dipole polarization [24–29], charge equalization [30–32], or Drude models [33, 34]. It is fitting to recall that the very first inclusion of polarization for the modeling of biological molecules were from the mid-sixties [35–37] and mid-seventies [14]. A subset of ‘polarizable’ MM/MD potentials have also targeted first-order electrostatics, resorting to distributed multipoles derived from ab QC calculations on the fragments [23, 24, 26, 29, 38–50]. This is the case of the SIBFA (Sum of Interactions Between Fragments Ab initio computed [38, 39] procedure, on which this review focuses, and which was among the prime ones in this regard. Further refinements have also borne on the two short-range contributions, repulsion in first-order and charge-transfer in second-order. This appears to restrict the subset of polarizable potentials to SIBFA, ORIENT [40, 41], and the Effective Fragment Potential (EFP) [42–45].
This review is organized as follows. We use here italics to underline the sought-for features. We will first summarize the features of the SIBFA potential, in light of the requirements for transferability. We will then present the results of validation tests regarding first anisotropy, then non-additivity. Their synergistic impact will be illustrated with the complexes of halobenzene derivatives with a guanine-cytosine base-pair in the recognition site of the HIV-1 nucleocapsid. This will be extended to a case problem in which both come into play and should enable to address in synergy two additional issues, namely multipole transferability and conjugation. We will next present results from recent studies from two of our Laboratories. The first study relates to the organization of highly structured waters in a bimetallic Zn/Cu enzyme, superoxide dismutase (SOD) . The two other relate to inhibitor or ligand binding to a tyrosine kinase, Focal Adhesion Kinase (FAK) , on the one hand, and to a Zn-metalloenzyme, phosphomannose isomerase (PMI) on the other hand. These will help to highlight the importance of the second-order contributions, and possibly non-additivity, on molecular recognition.
1.1.1 Procedure
In the SIBFA procedure, the total intermolecular interaction energy between molecules or molecular fragments is computed as a sum of five contributions:
which are the electrostatic multipolar, the short-range repulsion, the polarization, the charge-transfer, and the dispersion contributions.
EMTP is computed as a sum of multipole-multipole interactions, encompassing six terms from monopole-monopole till quadrupole-quadupole. The multipoles are located on the atoms and mid-points of the chemical bonds, and are derived from the ab initio QC molecular orbitals (MO) of the fragment. We resort to a procedure pioneered in 1970 by Claverie, Dreyfus and Pullman (CDP) at the Institut de Biologie Physico-Chimique in Paris [51–53]. It was initially applied to computations of the Molecular Electrostatic Potential (MEP) around biologically important molecules, such as the nucleic acid bases [54–57], DNA and RNA [58–60], proteins [61], phospholipids [62] and ionophores [63, 64]. The CDP method was first applied to the computations of intermolecular interaction energies in 1979 in the context of a polarizable potential [65] and, in 1980-1983, for a series of molecular recognition problems [66–71] using forerunners of the SIBFA procedure. Current applications have since resorted to a variant of the CDP procedure due to Vigné-Maeder and Claverie [72]. For completeness we mention that the derivation of distributed multipoles was also done in the context of IEHT wave-functions by Rein in 1973 [73, 74], and, regarding ab initio QC MO’s, in the early eighties by Stone et al. [75, 76], Sokalski et al. [77, 78], and Karlstrom et al. [47, 79]. The more recent OPEP procedure enables to derive both distributed multipoles and polarizabilities from such MO’s [80, 81].
Subsequent improvements to EMTP have consisted into including an explicit ‘penetration’ term, Epen. This is an overlap-dependent term: it translates the fact that at short intermolecular distances, there is a lesser shielding of the nuclear charge of a given atom by its own electronic density, due to its ‘penetration’ by the corresponding density of the incoming atom, and conversely. This results into an actual increase of the electron-nucleus attraction of the interacting pair. The first formulations of Epen were in 2000–2001 in the context of EFP [82, 83] and in 2003 in the context of SIBFA [84]. The latter has been used systematically ever since 2005 with this procedure. Other formulations have also been recently put forth [85–87]. The formulation by Piquemal et al. recently underwent a promising extension enabling to use it in conjunction with the Particle Mesh Ewald (PME) procedure [194]. This will pave the way for efficient MD simulations on very large systems in the context of the SIBFA or AMOEBA procedures.
The other contribution of predominantly electrostatic nature is polarization. It translates the gain in energy upon rearrangement of the electronic distribution of a given molecular fragment due to the electrostatic field generated on it by all the other interacting fragments. Epol on any ‘polarizable’ center is a function of the electrostatic field it undergoes and of its polarizability. The field is computed with the same distributed multipoles as EMTP and is screened by a Gaussian function, S, of the distance between the center and the interacting polarizing partner, modulated by the mean of the effective radii of the interacting pair. The polarizability can be used: -either as a scalar, the magnitude of which is derived from experimental measurements. This was done in the earlier versions of SIBFA and in most contemporary polarizable potentials which use the induced dipole approach [27–29]; -or as a QC-derived tensor. This is the case of but a handful of polarizable potentials, namely EFP, SIBFA, and ORIENT. In fact, both SIBFA and EFP resort to the same procedure to derive the polarizabilities, specifically a method published in 1989 by Garmer and Stevens (GS) [88], locating them on the centroids of the Boys localized molecular orbitals (LMO) : these are the barycenters of the chemical bonds and the ‘tips’ of the saturated lone pairs. The two first papers reporting the use of the GS polarizabilities, namely using EFP and SIBFA , were actually published in the same 1994 issue of the ACS Symposium series [89, 90].
Thus given any rigid molecule or molecular fragment, we derive once and for all, in a consistent fashion both distributed multipoles and polarizabilities. These are stored in the SIBFA library as one file, along with the information on the internal geometry and connectivities and types of atoms. Each fragment-specific file can be extracted and concatenated with others whenever the fragment is needed to assemble a large, flexible molecule, or a biomolecule, such as a protein or a nucleic acid.
SIBFA embodies two overlap-dependent contributions.
Erep in first-order is formulated under the form of a sum of bond-bond, bond-lone pair, and lone pair-lone pair interactions. This was inspired by an earlier proposal by Murrell et al. [91] following which the exchange-repulsion is proportional to the square of the intermolecular overlap between localized orbitals. For each pair of interacting orbitals, it could be formulated as S2/Rn, S denoting their overlap and R the distance between their centroids, n taking the values of 1, 2 and possibly beyond. In the context of SIBFA, representations of the orbitals are the chemical bonds and the lone-pairs.
The formulation of Ect in second order is based on another work by Murrell et al. [92], in which it is formulated as the integral of an overlap transition density convoluted with the electrostatic potential it undergoes. Starting from it, Ect was derived as a function of the overlap between representations of the localized orbitals of the electron-donor fragment and of the virtual orbitals of the electron-acceptor fragment [93, 94]. On account of their greater mutual proximities, they are restricted to the sole lone-pairs of the donor, and to the sole B-H bonds of the acceptor, where B denotes any heavy atom. The different contributions are calculated for all pairs of such orbitals, and embody dependencies upon the electrostatic potentials V separately undergone by the electron-donor and by the electron acceptor in the large molecular complex. These potentials also intervene in the denominator of Ect. In it the ionization potential, I, of the electron donor is modulated by the (predominantly positive) potential it undergoes; while the electron affinity of the electron acceptor, A, is modulated by the (predominantly negative) potential it undergoes. In the whole range of energy-relevant distances, this prevents the I-A differences from being negative (and therefore Ect from becoming positive) whenever I has a smaller magnitude than A, as is the case for most di- or multivalent cations. We also note that those Vs, along with the permanent multipoles, embody the contributions due to the induced dipoles, whence an indirect coupling between the polarization and charge-transfer contributions.
The last contribution, Edisp, uses a formulation by Creuzet et al. [95]. It is computed as a sum of atom-atom terms with 1/R6, 1/R8, and 1/R10 dependencies. It is augmented by an explicit ‘exchange-dispersion’ term (Eexch-disp) and by contributions from the lone pairs at their centroid positions.
Two points could be noted at this stage. Firstly, all five SIBFA contributions, not only Erep and Ect, embody dependencies upon the overlap: Epen for EMTP; a Gaussian screening factor of the distance between each interacting pair for Epol; and Eexch-disp for Edisp. Secondly, with the exception of EMTP, all contributions embody dependencies upon explicit representations of the lone pairs.
We denote throughout by ΔE the energies in the absence of Edisp, and by E1 and E2 the summed first- and second-order contributions, namely E1 = EMTP + Erep and E2 = Epol + Ect, respectively.
On account of its very separability, critical for the refinement of the SIBFA potential is the availability of energy-decomposition analyses. The Reduced Variational Space (RVS) procedure by Stevens and Fink [96] was particularly instrumental. On the one hand, it affords an operational and dependable separation of E2 into Epol and Ect. On the other hand, it lends itself to analyses of complexes having more than just two interacting partners. This enable to compute the separate components of E1 and E2 in multi-molecular complexes, evaluate the extent of their non-additivities, and how well these could be accordingly retrieved by their SIBFA counterparts. There are also other energy-decomposition analyses, such as the Constrained Space Orbital Variation (CSOV) [97] and the Symmetry-Adapted Perturbation Theory (SAPT) [98], which can both be done at the correlated level. We have resorted to the former upon dealing with open-shell cations [99] and for correlated computations [100] and are presently resorting to the latter in analyses of the interactions involving nucleic acid bases (Gresh et al. submitted).
1.2 Aspects of Anisotropy and Non-additivity in Molecular Complexes
-
A.
Anisotropy
MM potentials should be able to account for the fine angular features of ΔE(QC ) upon performing in- and out-of-plane variations of the approach of two interacting atoms at fixed equilibrium distance: that is, the departure from the assumption of atom-centered spherical symmetry. This might not be warranted by simple point-charge electrostatics and atom-atom Lennard-Jones-like formulas for Erep and Edisp, unless neighboring atoms restricted the available space.
-
(1)
Linear water dimer. The earliest example we considered dating back from the mid-eighties is the linear water dimer at equilibrium distance (dOO = 2.95 A) [93, 94]. We monitored the evolutions, as a function of the theta angle, of ΔE(SIBFA) and its EMTP, Erep and Ect contributions (Fig. 1.1a–c). For such evolutions the H and O atoms of the incoming OH bond of the electron-acceptor monomer predominantly sense the electrostatic potential around the O atom of the electron-donor monomer and their overlaps with its sp3 lone-pairs and there is little, if any, overlap with its two OH bonds. A very close parallelism was observed not only between the total ΔE(SIBFA ) and ΔE(HF ) energies, but also between the corresponding individual contributions. This early demonstration already lends credence to the representation of electrostatics with QC multipoles and to that of the short-range contributions with lone-pairs. The behavior of Ect was instructive. Its relative shallow behavior matching that of Ect(HF) was found to result from strongly opposed trends from the two sp3 lone-pairs. The contribution from the first lone-pair increases regularly until for theta = 60° the direction of the incoming HO bond aligns with it, while that of the second lone-pair decreases in concert. Past 60°, the two trends are reversed. The overall shape of ΔE appears dictated by that of EMTP. This appears consistent with the proposal by Buckingham and Fowler [101]. Nevertheless the present example as well as several subsequent others (vide infra) clearly show instances where the other contributions do have inherently anisotropic characters of their own and do bear on the overall angular dependencies of ΔE.
Fig. 1.1 
Linear water dimer. Compared evolutions of: a ΔE(SIBFA), ΔE(HF), and EMTP (full, dashed, and dotted lines, respectively; b Eexch(HF), and Erep(SIBFA) (full and dashed lines); and c Ect(HF) and Ect(SIBFA) (full and dashed lines). The dotted and dashed-dotted lines represent the contributions to Ect(SIBFA) from each of the two individual sp3 lone pairs of the donor oxygen. Reprinted with permission from Gresh et al. [94]. Copyright 1986 Wiley
-
(2)
Cation-ligand complexes. Zn(II) is a ‘soft’ divalent cation. This translates into large values of the charge-transfer and of the dispersion contributions [102, 103], and to an enhanced propensity to bind to ‘softer’ ligands than oxygen, such as nitrogen and in particular sulfur. Its divalent charge gives rise to very large values of EMTP and Epol thus to a propensity to bind to oxygen ligands as well. In proteins and NAs , Zn binds in a versatile fashion to N, O, and S ligands, can adopt a diversity of coordination numbers ranging from 4 to 6, and can exert a structural as well as a catalytic role. It is thus most important, but it could also be particularly challenging, to correctly represent it with PMM potentials. This was addressed in 1995 [104]. By parallel QC and SIBFA computations, we probed by Zn(II) the in- and out-of-plane angularities of ΔE and its individual contributions in a diversity of complexes with N, O, and S ligands. Marked directionalities were found for both Erep and Ect for in-plane variations of the theta angle around the X–O–Zn bond upon binding to hydroxy (X = H), formate and formamide (X = C), and around the C–S–Zn bond of methanethiolate. These could complement or oppose the directionalities of EMTP. Lesser directionalities of Erep and Ect were found on the other hand for out-of-plane variations on a cone at a fixed theta angle. Epol was found to display a generally shallower behavior than the other contributions, although it is a major contributor to non-additivity.
-
(3)
Hydrogen bonding to an anionic ligand. Water acting as proton acceptor can be also used to probe the electron-rich sites of a bound ligand. An illustrative example is that of its monodentate complex with formate, upon performing in-plane variations of the C–O–H angle, at a fixed O–H distance (Fig. 1.2). Eexch(HF) has a marked angular character with a maximum at 120º, which corresponds to a maximum overlap with one sp2 oxygen lone-pair. Its behavior is paralleled by Erep(SIBFA), while by contrast a 1/R12 atom-atom repulsion expression gives rise to a shallow behavior [105].
Fig. 1.2 
Monodentate formate-water Compared evolutions as a function of the theta angle of Erep(SIBFA) Eexch(HF) and a 1/R12 atom-atom repulsion. Reprinted with permission from Piquemal et al. [105]. Copyright 2007 American Chemical Society
-
(4)
Directionality in stacked complexes. Stacking interactions are a major determinant of NA stability, but act also to stabilize a diversity of ordered structures. We have considered a representative stacked dimer of formamide and rotated step-wise the second monomer with respect to the first around the z-axis [105]. The curve of EMTP augmented with Epen is virtually superimposable over that of EC (Fig. 1.3a. The curve of Erep matches well that of Eexch with only a small indentation at the most repulsive point (Fig. 1.3b. The parallelism of ΔE(SIBFA ) with ΔE(HF ) is evidenced in Fig. 1.3c. Extensions are presently underway to NA bases, and are carried out at both uncorrelated and correlated levels, and they appear conclusive as well (Gresh et al. J Phys Chem B, 2015, 119, 9477). Such results suggest that the anisotropy of ΔE(QC ) in stacked complexes can be reliably accounted for in the context of the SIBFA procedure.
Fig. 1.3 
Stacked formamide dimer Compared evolutions of a EMTP and EC(HF), b Erep(SIBFA) and Eexch(HF), ΔE(SIBFA) and ΔE(HF). Reprinted with permission from Piquemal et al. [105]. Copyright 2007 American Chemical Society
-
(5)
Directionality in halobenzene complexes. About thirty-five per cent of therapeutic drugs embody at least one halogen atom [106–108]. There is an outstanding feature of the CX bond in halobenzenes (X = F, Cl, Br, I) which was discovered thanks to quantum chemistry [109–111]. It is the presence of a ‘sigma-hole’, namely a zone of electron depletion along the bond, concomitant with a zone of electron buildup around a cone circumscribing the bond. Such features increase along the F < Cl < Br < I sequence. To account for electron depletion in the context of classical MM, a fictitious charge of δ+ has been located prolonging the CX bond, with the charge of X accordingly modified by δ -. The magnitude of δ and the δ-X distance can be optimized in order to fit the QC-computed interaction energy of an electron-rich ligand such as water or formamide approaching the CX bond through its O atom [112–114]. While this representation can be successful to translate the binding of electron-rich ligands along the CX bond, it leaves the outcome unresolved regarding approaches along the electron-rich cone. In the prospect of future simulations with APMM potentials in drug design or material science, it was thus critical to evaluate if the EMTP contribution, without any extra fitting and/or fictitious center, could reproduce the angular features of halogen bonding. We have thus resorted to a divalent cation, Mg(II), as a probe of the CX bond (X = F, Cl, Br) [115] in halobenzenes. A doubly charged species was deliberately chosen to exacerbate the impact of the sigma hole on the angularity of ΔE. The Mg-X distance, d, was first optimized for an approach at theta (C–X–Mg) of 180°. This was followed by in-plane variations of theta and by out-of-plane of the phi angle describing rotations of Mg(II) on a cone at fixed d and optimized theta (see Fig. 1.4). RVS energy-decomposition showed the EC to be the essential contribution to the angularity of ΔE(HF ). Would its representation going as far as distributed quadrupoles enable EMTP to match EC? This is evaluated in Fig. 1.5a, b. Figure 1.5 confirms this to be the case all throughout for all three halogens: for both Cl and Br, there is a pronounced maximum at 180º and two accented minima: in the 105–120° and 240–255° regions for Cl and more accented ones at 105° and 255° for Br. In marked contrast, the curve is for F shallow over the whole 135–225° region. It is also possible to unravel the origin of both the angularities of the Cl and Br curves, and of the shallowness of the F curve. Thus Fig. 1.6a–c separate each EMTP curve into its charge-charge (CC), charge-dipole (CD), and charge-quadrupole (CQ) components. The angularities of the Cl and Br curves is clearly dominated by CQ overcoming the opposed preferences of both CC and CD. Possibly just because of its featurelessness, the F curve is revealing. It shows that the flat behavior of EMTP is due to a near-exact compensation between the antagonizing angular features of CQ on the one hand and of CC and CD on the other hand. This is an important a posteriori and not taken for granted demonstration that the three components are correctly balanced within EMTP.
Fig. 1.4 
Representation of the variations undergone by an Mg(II) probe around halobenzenes a in-plane variations of d with theta = 180, b in-plane variations of theta, c out-of-plane variations of phi. Reprinted with permission from El Hage et al. [115]. Copyright 2013 Wiley
Fig. 1.5 
a–c Compared evolutions of EC(HF) and EMTP for in-plane variations of the theta angle for Mg(II) binding to the C–F (a) C–Mg (b), and C–Br (c) bonds of halobenzene, d–f in-plane theta evolutions of the charge-charge charge-dipole and charge-quadrupole components of EMTP for Mg(II) binding to the C–F (d), C–Mg (e), and C–Br (f) bonds of halobenzene. Reprinted with permission from El Hage et al. [115]. Copyright 2013 Wiley
Fig. 1.6 
Representation of the tetraligated complex of Pb(II) with six water molecules in a holo- and b hemi-directed arrangements. Reprinted with permission from Devereux et al. [119]. Copyright 2011 American Chemical Society
For the out-of-plane variations of phi, EMTP again displayed parallel features to EC. Thus for both Cl and Br, one maximum for found at 180º, and two minimum-energy regions at 60–120° and 240–300°. For F, the curve was virtually flat throughout.
We have performed a similar evaluation now with a water probe approaching through either one H atom or through it O atom. For all three halogens, and for theta as well as for phi variations, EMTP could invariably match the angular features of EC.
-
(6)
Could there be departures from spherical symmetry around some metal cations? In the polyligated complexes of metal cations, the ligand cations could justifiably be anticipated to bind around a ‘crown’ surrounding the cation. On account of the spherical symmetry of the cation, this is largely borne out experimentally and computationally in the vast majority of cases, but could there be exceptions? A notable exception is indeed provided by the Pb(II) cation, which in some instances is prone to prefer ‘hemi-directed’ arrangements, namely all ligands on one side of the cation, over ‘holo-directed’ ones, namely all ligands over the whole periphery of the cation (Fig. 1.6) [116–118]. To what an extent could be this be accounted for, if at all, by an APMM approach?






Table 1.1 reports the results of SIBFA versus HF comparisons for Pb(II) complexes with 4, 5, and 6 water ligands in the hemi- versus holo-directed arrangements [119]. The QC /RVS results indicate a small preference in favor of the hemi-directed arrangements, decreasing from 6.1 to 4.8 to 1.6 kcal/mol upon passing from 4 to 6 of out 160–200. ΔE(SIBFA) is able to match this trend, accordingly decreasing from 3.6, to 2.8, to −1.5 kcal/mol. The preferences favoring the hemi-arrangements can in SIBFA be traced back to the Pb(II) polarization energy, that is, in its absence, such arrangements would have lesser stabilities than the holo-ones. This is consistent with the RVS analyses. It translates the large dipole polarizability of Pb(II), biasing the arrangements towards the hemi-arrangements since in the holo-ones, the summed electrostatic field polarizing the cation has near-zero values. It is thus fitting to observe that while for the mono-ligated complexes of a cation such as Zn(II) Epol had a limited in-plane directional character, in the polyligated complexes of Pb(II), it is the dominant factor in favor of directionality.
-
B.
Non - additivity.
-
(1)
Sign and magnitude of δE nadd . Non-additivity is a critical feature of ΔE. That, is, in multimolecular complexes, its magnitude differs from the sum of its magnitudes in all bimolecular (pair-wise) complexes considered separately. Non-additivity could either increase or decrease the stability of the complexes, resulting into cooperativity or anticooperativity, respectively: these two cases are mostly encountered in multiply hydrogen-bonded complexes and in polycoordinated complexes of metal cations, respectively. The onset of non-additivity, δEnadd, was realized in the earlier stages of development of polarizable potentials. However, to our knowledge until the early nineties there have been surprisingly few, if any at all, attempts to quantify δEnadd on model complexes by QC computations and the separate weights of the ΔE(QC ) contributions, let alone evaluate if PMM were able to reliably match δEnadd. This could be traced back to the fact that with the sole exception of the RVS method, all available energy decomposition procedures are limited to bimolecular complexes. Thanks to this procedure, we could consider several multimolecular complexes and address such points. Such QC/SIBFA comparisons date back from the mid-to late nineties and have borne on: (a) mono-, bi-dentate, and through-water complexes of Zn(II) with formate and first- and second-shell waters [120]; polycoordinated complexes of Zn(II) with water [104], and water, hydroxy, methanethiol and/or methanethiolate ligands [121]; (b) polyligated complexes of Zn(II) with the end side-chains of proteins and some Zn-ligating groups of metalloprotein inhibitors [122]; (c) several representative water oligomers [105, 123–125] and d) the complexes of N-methylformamide in an array of linear H-bonded complexes as encountered in models of multilayered β-sheets [126]. The most important conclusions were:
-
(a)
With all ligands in the first coordination shell, polycoordinated complexes of divalent metal cations are anticooperative. Ect has a greater anticooperativity than Epol, undergoing a smaller increase in magnitude than Epol upon increasing the coordination number n. A different outcome however occurs in through-water complexes, where for both contributions, a balance between cooperativity and anticooperativity can occur. Both Epol(SIBFA ) and Ect(SIBFA) can match the behavior of their RVS counterparts upon increasing n, regarding their magnitudes, extent of non-additivities, and dependence upon the number of ligands in first second shells. As an illustration, we give in Table 1.2 a comparison between the QC and SIBFA computations for the complex of Zn(II) with six water molecules. The three considered complexes have either 0, 1, or 2 second-shell water molecules. There is a close correspondence between the SIBFA and QC contributions. As noted originally concerning the best-bound [Zn(H2O)6]2+ complex [104, 121], while the number of ligand has increased by 6, Epol and Ect with both RVS and QC have increased in magnitude by factors of only 2.6 and 2 with respect to their corresponding values in the monoligated [Zn-H2O]2+ complex at the optimized Zn-O distance of 2.10 A found in the hexamer. This, and several other related examples [120, 121, 127] give a clear indication of anticooperativity and of its control by both SIBFA Epol and Ect. This could have been expectable regarding Epol, but was not granted for Ect: it shows that the formulation of Ect embodying dependencies upon the electrostatic potential and field undergone by the electron donor as well as by the electron acceptor can reliably ensure for this critical feature.
Table 1.2 Polycoordinated complex of Zn(II) with six water molecules Values (kcal/mol) of the QC and SIBFA intermolecular interaction energies and of their individual contributions -
(b)
Multiply H-bonded complexes are generally cooperative. Epol is the dominant, but not unique, contributor to δEnadd. Cooperativity is optimized in cyclic structures if each molecule can act simultaneously as an H-bond donor with one neighbor and as an H-bond acceptor with the other. Anticooperativity was also noted with one cyclic water tetramer [124], in which one water molecule acts as an H-bond acceptor from both neighbors. Again, both Epol and Ect(SIBFA ) could closely match their QC counterparts. In large oligomers (n = 12 and beyond) having an ice cube-like structure, cooperativity results into a significant compression of the structures, several H-bonded distances shortening by up to 0.2 Å. The dominant magnitude of EMTP becomes strongly opposed by Erep, and thus E1 attains a much smaller magnitude than Epol and in some cases even than Ect. The fact that in such complexes E2 could have a significantly larger weight than E1 does not appear to have its pendant with any other competing PMM potential, yet is fully supported by RVS computations [105, 123].
-
(c)
It is also possible to evaluate, within Epol, the contribution to δEnadd of the iterative calculations of the induced dipoles. The polarizing field is a vector quantity and the polarization energy of a given center is proportional in first approximation to the square of the field it undergoes. Non-additivity thus appears already when the field is computed with the permanent SIBFA multipoles and its magnitude is close to the corresponding magnitude of Epol(RVS). This is consistent with the fact that in the RVS approach each monomer is considered in turn and relaxed in the presence in the frozen MO’s of all the others. At the outcome of the iterative calculation of Epol taking into account the additional contributions of the induced dipoles, Epol has values that are now close to those of Epol(QC ) calculated in a fully variational manner. We have found that in both cooperative and anticooperative complexes at equilibrium, the contribution to δEnadd of the iterative procedure was generally close to 30 %, and consistently enforced the preexisting cooperativity or anticooperativity.
-
(a)
-
(2)
On the need for off-centered polarizabilities. The importance of having off-centered polarizabilities, specifically on the tips of saturated lone-pairs, was shown in a study that bore on three model water oligomers [125]. They were denoted as bifurcated, transverse H-bonded, and longitudinal chains of helical shape (Fig. 1.7). For all three complexes, the SIBFA calculations enabled to reliably reproduce both the magnitude of Epol(QC ) and the values of the total dipole moments. This is due to the fact that in such complexes polarizable centers on the lone-pairs can be closer to an incoming molecule than the bearer atom, and thus sense a more intense field than it. This constitutes an illustrative example of the impact of non-anisotropy on non-additivity. This is a reverse situation to the one encountered in the Pb(II) oligohydrates, for which it was the non-additive behavior of Pb(II) polarization that resulted into non-isotropy.
Fig. 1.7 
Representation of three water oligomers in transverse bifurcated and linear H-bonding arrangements. Reprinted with permission from Piquemal et al. [125]. Copyright 2007 American Chemical Society
-
(3)
Impact of quadrupolar polarizabilities. Epol stems predominantly from the dipolar polarizabilities. There are cases, however, in which the polarization energy of some atoms can be increased due to their quadrupolar polarizabilities, whereby quadrupoles can be induced by the gradient of the field. This was observed with the Cu(I) cation [128]. We have compared several Cu(I) di- and oligo- planar ligated complexes in which the ligands are at equal distances from Cu(I) and on opposite sites. In such arrangements the field at the cation position is null, and so would Epol(Cu) if it were limited to the sole Cu(I) dipole polarizability. On the other hand the field gradients are non-null, resulting into a significant Cu(I) quadrupolar polarization, the inclusion of which enabled an improved agreement with ΔE(QC ). Only few attempts to include quadrupolar polarizabilities in PMM potentials have been reported so far [129].
-
(4)
Handling intramolecular polarization and the issue of multipole transferability. A large flexible molecule is assembled from its molecular fragments as follows: X and Y denoting two heavy atoms, two successive fragments, Fa and Fb, connect at the level of their X–H and H–Y bonds to create a junctional bond X–Y. The multipoles on the two H atoms and at the centers of the two X–H and H–Y bonds disappear and give rise to multipoles on X, Y, and the mid-point of the X–Y bond, according to proportionality rules related to their distances from these three centers. This preserves the net charge of the assembled molecule, but not that of the individual fragments. This is of no consequence for the calculation of EMTP, since the interactions of the charges along the junction bond connecting Fa and Fb are not computed anyway in SIBFA between the two fragments, although they are indeed with all the other fragments: the ‘missing’ interactions have no impact upon performing torsions around the junction bond, since they are located along this very bond. A different situation arises for the computation of Epol, because Fa would be polarized by the field of Fb now bearing a non-net charge (0, −1, 1 for neutral, anionic or cationic), and conversely. This would again be immaterial were it not for the presence of other fragments in a larger molecule. The summed field polarizing Fa would be that of a non-net charge before being squared, so that due to non-additivity the residual non-net charge could lead to uncontrolled over- or underestimations of Epol. These would be further amplified by the fact that Fa would be polarized by a centroid at a very close distance from it, namely half the length of the XY bond. Therefore we resorted to an alternative representation. Both HX and HY bonds are shrunk, each H atom being superimposed over its ‘bearer’ X or Y atom. All polarizing centers thus retain their net initial charge and their shortest distances from the neighboring fragments have lengths never smaller than those of XY bonds. Such a procedure should be more immune to short-distance artifacts. It has been evaluated by several comparisons with QC in a diversity of cases. We have thus compared the conformational energies, δEconf, of ten alanine tetrapeptides put forth by Beachy et al. [130] to evaluate the accuracies of MM force-fields. In our published study [131] the ten conformers differed by their sole torsion angles, i.e. with rigid constitutive fragments, pending completion of the SIBFA stretching and bending harmonic potentials. Inclusion of Epol enabled the relative SIBFA δEconf values to closely reproduce the corresponding QC values for all ten conformers. Refinements presently underway bear on the representation of the π lone pairs of the prime constituent of the peptide backbone, the N-methylformamide moiety, to further improve the accuracy of the intra-molecular short-range contributions Erep and Ect; this leads back to the issue of non-isotropy. We have also investigated the conformational dependency of: Na+ binding to conformers of glycine and the glycine zwitterion [132]; Zn(II) binding to mercaptocarboxamides [133], which constitute the Zn-binding moiety of some Zn-metalloenzyme inhibitors; and Zn(II) binding to tetra-anionic triphosphate, which is the backbone of ATP [134]. The comparisons with the QC results constituted revealing tests of the procedure, requesting the simultaneous and consistent computation of both intra- and intermolecular polarization and charge-transfer effects. They enabled to address the issue of multipole transferability [135]. Namely, a limitation of the use of distributed multipoles in intramolecular studies would reside in the variation of their intensities, thus lack of transferability, following conformational changes. The results of our above-mentioned studies on the other hand showed that, exactly as in intermolecular interactions between fragments unconnected by covalent bonds, Epol enables to account for the consequences of conformational changes on both δEconf and the intermolecular interactions with additional molecules. This occurs provided that: -the permanent multipoles of the constitutive unconnected fragments are the ones used to compute first-order electrostatics; -and the H atoms are carried back on their ‘bearer’ X and Y atoms of the XY junction bond. Very recent tests have borne on the solvation energies of four inhibitors of the PMI Zn-metalloenzyme, which embody a dianionic phosphate and a monoanionic Zn-binding hydroxamate. Shells with 64 waters were considered and up to 26 complexes in different conformations. Despite the large magnitudes of the interaction energies, the large number of interacting partners, and the diversity of binding modes, close agreements between SIBFA and QC calculations at both HF and correlated levels were demonstrated (Gresh et al. to be submitted).
-
(5)
Could the first-order exchange repulsion contribute to non-additivity? The short-range exchange-repulsion between two molecules is due to reorthogonalization of their MO’s upon complex formation. This leads to a destabilization of the total energy of the complex in first-order counteracting the Coulomb contribution. In a multimolecular complex, the energy cost due to simultaneous reorthogonalization can be expected to differ from that due to the summed separate pair-wise reorthogonalizations. The resulting non-additivity of Eexch was found to be very small, except for the polyligated complexes of some metal cations. In the context of SIBFA, it was formulated under the form of a three-center exponential involving the cation and all pairs of ligating heteroatoms, calibrated beforehand on model diligated cations, and then validated by QC comparisons on tetra- and hexaligated complexes [136]. The impact of the non-additivity of Eexch/Erep is illustrated in the case of one metal cation for which it is the most pronounced, a heavy toxic metal, Hg(II). We have thus considered the tetrahydrated [Hg(H2O)4]2+ complexes in competing square-planar versus tetrahedral arrangements (Fig. 1.8). The results are reported in Table 1.3. The three-body repulsion ΔEthree-body is 3 kcal/mol larger in the square planar arrangement than in the tetrahedral one. For both arrangements its values computed by SIBFA were close to the QC ones. Its explicit inclusion in SIBFA enabled to account for the preference of Hg(II) in favor of a tetrahedral arrangement. The energy difference between the two arrangements might appear small, but could nevertheless be sufficient to bear on the structural preferences found at the outcome of large-scale MD or MC simulations.
Fig. 1.8 
Representation of the [Hg(H2O)4]2+ complex in it’s a square planar and b tetrahedral arrangements. Reprinted with permission from Chaudret et al. [136]. Copyright 2011 Wiley
Table 1.3 Complexes of Hg(II) with four water molecules in square-planar and tetrahedral arrangements Comparisons of the QC and SIBFA intermolecular interaction energies (in kcal/mol) and their contributions


1.3 Applications
-
(1)
Conformational study of a polyconjugated drug. Interplay of cooperativity, anisotropy, multipole transferability, and conjugation.

Representation of a the molecular structure of Lig-47 and b of the structure of its central carboxythiourea (CTU) moiety. Reprinted from Goldwaser et al. [141]
Several drugs which target proteins involved in disease embody conjugated or aromatic groups connected together by conjugated or partly conjugated bonds. This is the case of an inhibitor of neuropilin-1 (NRP1) , a protein which when overexpressed is involved in diseases such as cancer or macular degeneracy [137]. A ligand, denoted as Lig-47, was identified in one of our Laboratories after experimental high-throughput screening, and shown to be endowed with a submicromolar activity on several cell lines [138]. Its structure is represented in Fig. 1.9. It is made out of the following chemical groups: benzimidazole, methyl benzene, carboxythiourea (CTU) , and benzene-substituted dioxane. It is a highly conjugated drug, and the connections between its four groups all involve unsatured atoms having sp or sp2 hybridization. The three-dimensional structure of NRP1 is known from high-resolution X-ray crystallography [139, 140], but not that of its complex with Lig-47. As a prerequisite to APMM MD or MC studies on the NRP1-Lig-47 complex, it is essential to control the ligand conformational flexibility, which is mostly governed by torsions around conjugated bonds. Overestimating it would result into too ‘floppy’ a ligand and the onset of a manifold of unlikely candidate protein-binding poses. Conversely, underestimating it would give rise to an unrealistically stiff ligand, unlikely to favorably bind its receptor. It was thus imperative to ensure if the SIBFA δEconf calculations were reliable when compared to the QC ones. The work published in [141], focused essentially on the central CTU unity, and it proceeded along several successive steps:
-
(a)
The distributed multipoles and the polarizabilities were derived from the MO’s of the constitutive fragments of Lig-47, namely benzimidazole, methyl-benzene, CTU in an extended conformation, and benzene-connected dioxane. The location of the sp2 lone-pairs of CTU was determined on the basis of ELF [142] analyses using the TopMod package [143], and is shown in Fig. 1.10;
Fig. 1.10 
Representation of the ELF contours around the CTU locating the positions of the sp2 lone-pairs of O and S atoms. Reprinted from Goldwaser et al. J. Mol. Mod. 2014, 20, 2472
-
(b)
For all four constitutive fragments, we calibrated the increments or decrements of the effective van der Waals (vdW) radii of the atoms along their lone-pair directions by probing them with an incoming water probe approaching through one of its H atoms, in order for Erep(SIBFA ) to match Eexch(RVS ) over the range of energy-relevant distances;
-
(c)
Having set the increment of the vdW radii, CTU was split into four pseudo-fragments. The first and the third one are an sp2 amine, the second one is thioaldehyde, and the fourth one is aldehyde. In this process fictitious connecting H atoms are created which are given null multipoles, the centroids of each of the two connecting bonds making up a junction are superimposed at the mid-point of the bond and given half the multipoles and polarizabilities of the original bond centroid prior to splitting. CTU thus retains its net initial charge of 0. 15° step-wise variations are performed around the four junction bonds, and the values of one- and two-fold rotational barriers V0 are calibrated so that δEconf(SIBFA) reproduces δEconf(QC ). CTU is then integrated in the entire Lig-47 ligand. The torsional variations are redone to evaluate the transferability of V0, for which only limited changes were found necessary. The values of V0 for the rotations around the junctional bonds connecting CTU to methylbenzene and dioxane, and methylbenzene to benzimidazole were similarly calibrated with respect to QC . In all cases, the curves of δEconf(SIBFA ) were found to reproduce very satisfactorily the corresponding QC ones.

Steps a-c are virtually exclusively fitting steps, but which accuracy is to be expected upon passing to a real validation step? This was carried out as follows. We selected all conformations which, for rotations around all junction bonds, corresponded to local minima or to the global one. For each of them, we performed energy-minimization with the ‘Merlin’ minimizer [144] on all torsional angles simultaneously. We were thus able to characterize up to 20 distinct conformations. The four most relevant ones are represented in Fig. 1.11. The most stable one, denoted 1b, has an intramolecular H-bond between the NH bond of the first thioamide group of CTU and the carbonyl bond of its formamide group. In Fig. 1.12 are plotted the SIBFA and QC δEconf curves. The following analyses will be limited to the HF level. Calculation at the correlated level and with Edisp contribution in the SIBFA calculations led to the same conclusions as those to follow. While 1b is confirmed by the QC calculations to be the lowest-energy minimum, its relative stability is clearly underestimated by δEconf(SIBFA) compared to δEconf(QC). The overall R2 coefficient is 0.88. Could this be improved? CTU had been used as one single building block in an extended conformation enabling to account for the impact of conjugation on the multipoles and polarizabilities. This disables, however, the computation of Epol between the four pseudo-junctions. Firstly, the intramolecular polarization of CTU, which took place during the variational HF procedure, is inherently embodied. Secondly, since the sub-fragments bear non-net charges, and for the reasons mentioned above, attempts to include Epol can be easily anticipated to severely overestimate it. We thus sought for an alternative representation, in which CTU is built from two separate fragments, thioamide and formamide. This enables for their mutual polarization simultaneously with the other Lig-47 fragments, although at the cost of the loss of conjugation between them. The two fragments were again split as before into two pseudo sp2 amine, one pseudo thioaldehyde and a pseudo aldehyde fragment. The one- and two-fold V0 torsional barriers were refit as above, and the values of δEconf for the twenty minima were recomputed. The corresponding δEconf(SIBFA ) curve is plotted in Fig. 1.13a along with the QC one, now showing a very close superposition and a R2 coefficient of 0.96. Figure 1.13b, c represent the corresponding evolutions of δE1 and δE2. It is clearly seen that the evolution of δEconf(SIBFA) is governed by E2 and not by E1. While Epol can be shown to be a leading determinant in several cases of molecular recognition, the finding that it plays the leading role in shaping the conformational energies of some conjugated molecules has no precedent. Several papers have mentioned [145, 146] that in order to treat polyconjugated compounds, introduction of couplings between successive torsion angles was necessary. Those studies were done using non-polarizable potentials. Such results might have to be reconsidered since such couplings might have been accounted for by polarization effects which are non-additive and long-range.

Three-dimensional structures of four representative energy-minimized conformations of Lig-47. Reprinted from Goldwaser et al. J. Mol. Mod. 2014, 20, 2472

Compared evolutions of the relative QC/HF and SIBFA conformational energies as a function of the conformer number Representation ‘a’ is used to construct CTU. Reprinted from Goldwaser et al. J. Mol. Mod. 2014, 20, 2472

a Compared evolutions of the relative QC/HF and SIBFA conformational energies as a function of the conformer number Representation ‘b’ is used to construct CTU, b corresponding evolutions of δE1, c corresponding evolutions of δEpol. Reprinted from Goldwaser et al. J. Mol. Mod. 2014, 20, 2472
-
(2)
Binding of halobenzene derivatives to a recognition subsite of the HIV-1 integrase Joint involvement of anisotropy and non-additivity
The HIV-1 integrase (INT) is a viral enzyme responsible for the integration of the viral DNA genome into the genome of the host cell. It is the target for the development of novel antiviral drugs. Three of these are currently used in therapy, and all three have a halobenzene ring: raltegravir, dolutegravir, and elvitegravir [147–150]. The high-resolution structure of their complexes with the DNA-bound integrase of Moloney foamy virus has been solved by X-ray crystallography [151]. This INT has very close homologies to the HIV-1 one, enabling inferences from structure-activity relationships for drug design. We have focused first on elvitegravir (EVG) , the chlorofluorobenzene ring of which interacts which a guanine-cytosine (GC) base-pair in an INT recognition subsite (Fig. 1.14a, b). Figure 1.14b shows that on the one hand, the electron-deficient prolongation of the C-Cl bond points towards the electron-rich ring of guanine, and that on the other hand, an area of its electron-rich cone can interact favorably with an electron-deficient region around the extracyclic N4H2 zone of cytosine. The double-faceted, ‘Janus-like’ property of the CX bond in halobenzene could be leveraged to enhance its affinity for each of these two regions separately, upon resorting to electron-withdrawing and to electron-donating groups, respectively [152]. A simultaneous enhancement for both sites could even be sought for. Figure 1.15 gives the structures of several substituted halobenzene candidates. Their interaction energies with the G-C base-pair were optimized by QC calculations at the B97-D level, and relative energy balances were done which took into account their desolvation energies. Several derivatives were found endowed with more favorable energy balances than the parent fluorochlorobenzene ring of EVG , including one, compound H, having both an electron-donating and an electron-withdrawing ring. While the search for improved first-order electrostatics due to anisotropy was indeed justified by the energy decomposition analyses as well by QC-derived contours of Molecular Electrostatic Potential (MEP) [152], an unanticipated result concerned the role of non-additivity now coming into play on two counts: (a) the intermolecular interaction energies in the trimeric complexes could in some cases significantly differ from the sum of the pairwise interactions in the three dimeric complexes. While this has been documented in previous work on multiply H-bonded complexes, it was unprecedented in the case of stacking interactions; (b) the energy gains resulting from di- and polysubstitutions could differ from the summed gains resulting from monosubstitutions. To what an extent could APMM account for such QC results [153]? EMTP was shown above to account faithfully for the anisotropy of EC in halobenzene binding, but is non-additivity still in control for the considered trimeric complexes, which could either involve cooperativity and anticooperativity? The values of the nonadditivities are compared in Table 1.4 for the QC and SIBFA computations. δEnadd(SIBFA) reproduces correctly the trends of δEnadd(QC), whether cooperative or anticooperative. There is a close agreement between Epol(RVS ) and Epol*(SIBFA) prior to iterating on the induced dipoles, as also noted from previous publications. Epol(KM) resulting from the Kitaura-Morokuma procedure [154] has greater δEnadd values than found from SIBFA after iterations on the induced dipoles, but this might be caused in part by the non-orthogonalization of the MO’s by this procedure. We have considered a total of 18 complexes of compounds selected from Fig. 1.15 with the G–C pair as well as with G and C separately, for which we monitored the evolutions of E1, E2, and their ΔE sums in Fig. 1.16a, b for the SIBFA and QC computations, respectively. The three SIBFA energies closely match the corresponding QC ones. This is also the case for the individual EMTP, Erep, Epol, Ect and Edisp contributions [153] and for ΔEtot as compared to ΔE(QC/B97-D) (Fig. 1.16c). These figures clearly show that both E1 and E2 are needed to confer its shape to ΔE. Moreover, we found that for all 18 complexes, whether ternary or binary, the stabilization due to E2 is larger in magnitude than that due to E1. This is reminiscent of the situation with the water oligomers in ice-like arrangements. Here again, the dominant magnitude of EMTP and EC are counteracted by those of Erep and Eexch. The present results indicate that ab initio QC -derived multipoles and polarizabilities can be necessary to control both non-isotropy and non-additivity. The latter feature also concerns the gains or losses in binding energies from polysubstitutions relative to the summed monosubstitutions, the impact of which on the MO’s can be reflected by the distributed QC multipoles and polarizabilities whence they are derived.

a Representation of the three-dimensional structure of the Foamy virus integrase/DNA complex with elvitegravir, b close-up on the interaction of the halobenzene ring with the G-C subsite. Reprinted with permission from El Hage et al. [152]. Copyright 2011 Wiley

Molecular structures of halobenzene derivatives substituted with electron-donors or electron attractors or with both. Reprinted with permission from El Hage et al. [153]. Copyright 2014 American Chemical Society

a Evolutions of the SIBFA E1 E2 and E1 + E2 intermolecular interaction energies along a series of halobenzene derivatives, b Evolutions of the QC E1 E2 and E1 + E2 intermolecular interaction energies along a series of halobenzene derivatives, c Compared evolutions of ΔEtot(SIBFA) and ΔE(DFT/QC) along a series of halobenzene derivatives. Reprinted with permission from El Hage et al. 115. Copyright 2014 American Chemical Society
-
(3)
Structural waters in and around protein recognition sites. Impact of the second-order contributions. Discrete water molecules, whether individually or in arrays, are considered to be an integral part of protein or NA structures. We summarize below recent findings aiming to unravel the impact of polarization and charge-transfer on the stabilization energies they confer. These studies bore on superoxide dismutase (SOD) , a bimetallic Zn/Cu metalloenzyme, and on the complexes of inhibitors with the FAK tyrosine kinase and with the PMI Zn-metalloenzyme.
-
(a)
Superoxide dismutase. SOD is a bimetallic enzyme catalyzing the dismutation of O2− into dioxygen and hydrogen peroxide [155]. It is essential for the survival of cells, but even more so for the cancer cells. SOD is thus an emerging target for the design of novel anticancer strategies [156, 157] including photodynamic therapy which can involve Ru(II)-based compounds [158]. Its high-resolution structure has been solved by X-ray crystallography [159, 160] showing the presence of several structural waters in close vicinity to the 85/Zn/Cu binding site. This is an incentive to evaluate whether an APMM approach could single out some privileged water network(s), and the possible extent of its/their overlap with the one found experimentally. As a first step toward such an evaluation [161], we started from the X-ray structure retaining eleven well-defined structural waters, and completed the solvation with up to 296 waters. We then performed short-duration MD runs at temperatures in the 10–300 K range with a simplified SIBFA potential and constrained the waters in a 22 Å sphere centered around Cu(I) using a quadratic potential. We selected six snapshots, denoted a-f. For each we then retained the 64 waters closest to Cu(I), and performed energy-minimization (EM) on their positions, on the side-chain conformations of the SOD residues making up, or neighboring, the Zn/Cu binding site, and on the positions of the two cations. Model binding sites were then extracted at the outcome of EM for validation by parallel SIBFA and QC computations. These encompassed selected main-chains and/or the side chains of 25 residues, the two metal cations and the 28 closest waters, totaling 301 atoms. Single-point computations were done for validation by parallel QC and SIBFA computations.
Figure 1.17a gives a representation of the most stable of the six energy-minimized structure, namely c. c is the structure for which the water networks have the greatest overlap with the one determined by X-ray crystallography used as a starting structure, despite the ‘scrambling’ it underwent by the initial MD steps. Figure 1.17b shows a close-up on the water network. There is a dense array of waters connecting two ionic residues, Glu131 and Arg141, which are located beneath the bimetallic site and are at about 10 Å distance from one another. These residues are connected, in fact, by several intermeshed water networks, which are also channeled in other vicinal regions, such as between the side-chains of residues Thr56 and Asn137. These two residues are linked together by a five-water near-linear array, the first and the last of which interact with the dense Glu131-Arg141 connecting water network. Six waters have dipole moments (μ) greater than 2.70 Debye, which is the value found for water oligomers in ice using SIBFA [123]. High values of the dipole moment were also found for discrete waters in the recognition site of FAK kinase which similarly mediated the interactions between ionic sites. This is mentioned below. Parallel SIBFA and QC computations were performed on complexes a-f in the presence and in the absence of the water networks. This enabled to compute the stabilization brought by the water networks. Figure 1.18a displays the evolutions of ΔE(SIBFA ) and ΔE(HF ) in the six complexes. The evolution of ΔE(HF) is very closely matched by that of ΔE(SIBFA), the relative errors being <2 %. As was the case for substituted halobenzene binding to the G–C base pair of HIV-1 integrase , both E1 and E2 terms are necessary to confer its proper shape to ΔE. The superimposition of ΔE(QC/B97D) and ΔEtot(SIBFA) curves is even better at the correlated level (Fig. 1.18b). One means to validate the high values of the dipole moments found for water is to compare the total values of μ in the six 28-water clusters extracted from complexes a-f (retaining a net charge of 0 ensures that the dipole moment is translation-independent). Figure 1.19 compares the evolutions of μ as derived from both QC and SIBFA in the six clusters, with a close and clear correspondence. The R2 factor is of 0.99. It is also seen that the most stable complex, c, is the one for which μ has the highest value, attesting to the importance of polarization and non-additivity in its preferential stabilization.
Fig. 1.17 
a Organization of the most stable network of 28 waters around the bimetallic site of SOD, b representation of the water networks and values of the highest dipole moments of structural waters. Reprinted with permission from Gresh et al., J. Comput Chem., 2014, 35, 2096. Copyright 2011 Wiley
Fig. 1.18 
Stabilization brought by the 28-water networks in six distinct arrangements a Evolutions of ΔE(QC) and of ΔE(SIBFA) and its separate E1 and E2 terms b Evolutions of ΔE(QC/B97D) and ΔEtot(SIBFA). Reprinted with permission from Gresh et al., J. Comput Chem., 2014, 35, 2096. Copyright 2011 Wiley
Fig. 1.19 
Compared variations of the QC and SIBFA values of the total dipole moments in six distinct 28-water networks. Reprinted with permission from Gresh et al., J. Comput Chem., 2014, 35, 2096. Copyright 2011 Wiley
We stress again that the close agreements found for SIBFA with ΔE(QC ) at both HF and correlated levels could only be possible thanks to the separable nature of the potential, a proper balance of first- and second-order contributions, and control of non-additivity in large complexes. They are encouraging in the perspective of long-time MD simulations, enabled by very important recent advances in the development of a highly efficient and scalable code by one of our Laboratories [162]. We thus plan to monitor the lifetimes of the individual waters in the networks, the possible existence of other, competing networks, the possibility of their mutual interconversions, and the channeling of solutes toward the bimetallic site.
-
(b)
Focal Adhesion Kinase (FAK) . Kinases presently account for about 40 % of the targets for drug design [163]. They are a class of enzymes which draft a phosphate group from ATP to hydroxylated amino-acids, namely tyrosine, serine and threonine. This results into cellular activation but also if overexpressed into pathologies such as cancer, arthrosis and neurodegenerative diseases [164–166]. FAK is a tyrosine kinase which subsequent to autophosphorylation, can trigger a cascade of protein-protein interactions resulting into signal transmission to the cellular nucleus to trigger cell division and motility. Its three-dimensional structure has been resolved by X-ray diffraction showing the ATP -binding site in a hinge between the N- and C-terminal lobes (Fig. 1.20) [167]. Five inhibitors in the pyrrolopyrimidine were designed, synthesized and tested by the Novartis company [168]. They all have in common a benzene substituting the five-membered ring nitrogen. The benzene is substituted by a carboxylate group at the ortho or meta position. In the latter, there are 0, 1, or 2 methylene groups interposed. They are represented in Fig. 1.21, along with their IC50 values in μM and using the notations of the original paper. Compound 16i with an ortho carboxylate substituent is the least active (1.6 μM). Compounds 17 g, 17 h, 17i all have a meta carboxylate substituent and have the same submicromolar affinity (0.04 μM), i.e. a two-order-of-magnitude enhancement in affinity. A further gain results from the replacement of benzene of 17i by pyridine, with compound 32 now nanomolar (IC50 = 0.004 μM). Thus, apparently modest structural changes can result into very large (thousand-fold) changes in the binding affinities. Could APMM procedures shed light on the factors governing such large changes [169]? Energy-minimizations were performed in which the solvation free energy was computed by a Continuum reaction field procedure, which was designed by Langlet, Claverie and their coworkers [170]. We denote this contribution as ΔGsolv(LC). The solvent is represented by a ‘bulk’ which responds to the electrostatic potential generated by the solute on its van der Waals by creating fictitious charges, which interact with the solute potential to give rise to the electrostatic contribution of ΔGsolv(LC ). The energy balances take into account: (a) on the one hand: the intermolecular ligand-protein interaction energy and the solvation energy of the complex; and (b) on the other hand, the conformational energy costs of the ligand and of the protein upon passing from their uncomplexed, solvated states to the complex, along with their corresponding desolvation energies. An overlay of the five complexes in the recognition site after energy-minimization is given in Fig. 1.22, showing an extensive overlap except at the position of the ligand carboxylates. The energy balances are given in Table 1.5 under the form of differences between a) and b) for each contribution. They show no correlation at all with the experimental results. Thus, e.g., there is 7 kcal/mol energy difference disfavoring compound 32, which is nanomolar, with respect to 17 h. Yet 17 h has a ten-fold lesser experimental affinity than 32; and there is an 11 kcal/mol energy difference between 17 g and 17 h which in fact have similar experimental affinities. Separate QC tests on the FAK recognition site having confirmed the accuracy of the SIBFA procedure, we were led to evaluate the extent to which a limited number of ‘discrete’ waters could impact the relative energy balances. For that purpose, five, six, or seven waters were located in the complexes between each ligand and a reduced model of FAK limited to the recognition site shown in Fig. 1.22. They were initially located thanks to a procedure [171] which minimizes with a simplified energy function a limited number of discrete waters around the accessible hydrophilic sites of the solute. These positions were reoptimized by a Generalized Simulated Annealing [172, 173] procedure with the SIBFA potential, then minimized with Merlin. The final resulting positions were then ported to the entire FAK and EM redone again in the presence of ΔGsolv(LC ). Figure 1.23 gives a simplified representation limited to the complex of the end carboxylate of 32, the ionic sites of FAK, and five discrete waters (denoted as complexes ‘cw’ in Ref. [169]). These waters can clearly snugly fit in the structure. They can either mediate the interactions between the ligand and PMI as occurs with residues Glu471, Arg550 and Asp564, or complement them, as occurs with residue Lys454. Several waters have much stronger dipole moments than ice, so that the polarization energy contribution could be expected to be a key contributor to ΔE. This led us to perform a parallel SIBFA/QC(RVS ) analysis of the intermolecular interactions on complexes c and cw, without and with the discrete waters. The analyses done for the two extreme compounds, nanomolar 32, and micromolar 16i, are reported in Table 1.6. In the absence of the waters, E1 favors by 17 kcal/mol 16i over 32, while E2 favors by 3–4 kcal/mol 32 over 16i. A remarkable reversal in the magnitudes of the preferences takes place in the presence of the discrete waters. E1 now favors by only 4–5 kcal/mol 16i over 32, but E2 favors 32 over 16i by a very significantly augmented preference, namely 17 kcal/mol. The resulting preference of 13–14 kcal/mol in terms of ΔE(SIBFA) and ΔE(RVS) is thus the one imposed by E2. The persistent agreements between all SIBFA and RVS individual contributions is noteworthy for all four complexes. The final energy balances in the presence of the five structural waters are given in Table 1.7. All five ligands are now ranked at least qualitatively along the correct sequence ranking first the nanomolar compound 32, then the three submicromolar compounds 17 g, 17 h, 17i, and then the micromolar compound 16i. The same conclusions hold with 6 and 7 discrete waters. The critical role of Epol in such balances is noteworthy. The need for Epol for a correct ranking of affinities had been previously shown in a study with the AMOEBA potential that bore on the complexes of trypsin with benzamidine derivatives [174], although its role was possibly not as extreme as in the present study. As an extension of the present work, we plan to resort to a massively parallel version of the TINKER software on which the SIBFA potential is ported to perform long-duration MD . These should enable us to monitor the life-times of the discrete waters and their rates of exchange with the solvent, and possibly the path for interconversion between ’DFG-in’ and‘DFG-out’ conformations [175].
Fig. 1.20 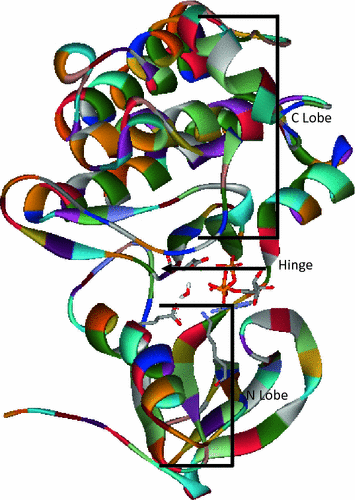
Representation of the three-dimensional structure of FAK kinase. Reprinted with permission from de Courcy et al. J. Am. Chem. Soc. 2010, 132, 3312. Copyright 2010 American Chemical Society
Fig. 1.21 
Representation of the molecular structures of the five pyrolopyrimidine inhibitors. . Reprinted with permission from de Courcy et al. J. Am. Chem. Soc. 2010, 132, 3312. Copyright 2010 American Chemical Society
Fig. 1.22 
Overlay of the inhibitors in the FAK recognition site. Reprinted with permission from de Courcy et al. J. Am. Chem. Soc. 2010, 132, 3312. Copyright 2010 American Chemical Society
Table 1.5 Relative energy balances for the binding of the five pyrrolopyrimidine inhibitors to FAK in the absence of the structural waters Fig. 1.23 
Representation of the complexes between the carboxylate group of the inhibitor and the ionic groups of the recognition site of FAK together with the discrete waters. Reprinted with permission from de Courcy et al. J. Am. Chem. Soc. 2010, 132, 3312. Copyright 2010 American Chemical Society
Table 1.6 Energy decomposition for the binding of the carboxylate of the micro- and the nanomolar compounds to the ionic sites of FAK without and with the structural waters Table 1.7 Relative energy balances for the binding of the five pyrrolopyrimidine inhibitors to FAK in the presence of the structural waters -
(c).
Phosphomannose isomerase (PMI) . PMI is a Zn-metalloenzyme which catalyzes the reversible isomerization of fructose-phosphate into mannose-phosphate [176]. It is responsible for several infectious and parasitic diseases but there is no clinically-useful inhibitor against it [177–179]. A hydroxamate inhibitor, denoted 5PAH was designed, synthesized and tested in the Laboratory of Bioorganic and Bioinorganic Chemistry at Orsay, France, and shown to display a submicromolar inhibitory potency. By contrast, an analogue with formate replacing hydroxamate was devoid of potency [180]. Based on the X-ray crystal structure of Zn-bound PMI [181, Fig. 1.24], we were able to account for these experimental results and derived a structural model locating the hydroxamate in the Zn-binding pocket and the phosphate at the entrance of the cavity where it binds simultaneously to two cationic residues, Arg304 and Lys310 [182]. In a next step, four ligands in the sugar family were considered (Fig. 1.25) [23]. The first three ones (1–3) have a dianionic phosphate. The first (1) is β-D-mannopyranose 6-phosphate (β-6-MP1). The fourth (4) is an analog of β-6-MP1 but has a malonate with two monoanionic carboxylates replacing the phosphate. Only compound 1 in the phosphate series displayed a measurable PMI binding affinity. The malonate derivative 4 displayed a ten-fold larger binding affinity than it. This could constitute a step toward the design of therapeutically relevant drugs, because malonate is more resistant to enzymatic hydrolysis than phosphate, and is also more easily transported. The SIBFA calculated have enabled to account for the greater affinity fort PMI of 1 than 2 and 3. However the energy balances with the sole ΔGsolv(LC ) terms failed to account for the greater affinity of the malonate derivative. This led us, as in the case of FAK , to solvate their complexes with discrete waters. Nine waters were optimized in the PMI-1 and PMI-4 complexes. The structure of the latter is shown in Fig. 1.26. Three networks are found. The first network bridges one O of the most accessible carboxylate of the ligand with Trp18 and Glu48 residues. These interactions are extended by ionic interactions with residues Lys100 and Glu294. The second network bridges the other anionic O with Asp17. The third network bridges the sole accessible anionic O of the second carboxylate with Arg304 and Asp300. Residues Asp17, Glu48, and Asp300 are at about 18 Å distance from one another. We thus observe an extension of the recognition site to PMI residues that do not interact directly with the ligand, and such an extension is mediated by the polarizable water molecules.
Fig. 1.24 
Three-dimensional structure of Zn(II)-bound PMI. Reprinted with permission from Gresh et al. J. Phys. Chem. B. 2011, 115, 8304. Copyright 2011 American Chemical Society
Fig. 1.25 
Representation of the structures of four PMI ligands. Reprinted with permission from Gresh et al. J. Phys. Chem. B. 2011, 115, 8304. Copyright 2011 American Chemical Society
Fig. 1.26 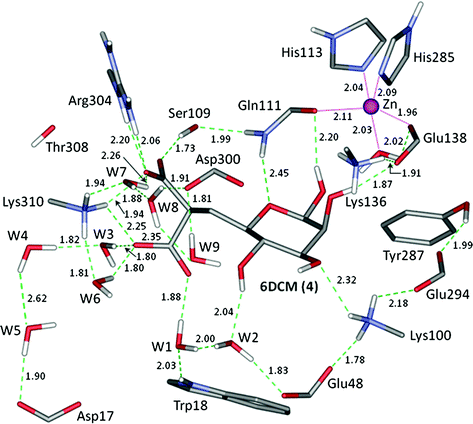
Representation of the optimized complex of the malonate derivative with PMI and nine water molecules. Reprinted with permission from Gresh et al. J. Phys. Chem. B. 2011, 115, 8304. Copyright 2011 American Chemical Society










In the complex of I, the phosphate fits more snugly than malonate between Arg304 and Lys310. This on the one hand gives rise to stronger electrostatic interactions with these two residues, but at the price of a lesser accessibility to the structural waters. The differential stability due to the nine waters can be illustrated by comparing the interaction energies of the two ligands in the recognition site. Each site has been extracted from the energy-minimized ligand-PMI complex, and single-point computations were done with and without the nine structural waters. The SIBFA interaction energies are reported in Table 1.8 and compared to the QC ones. For each contribution and for each ligand, we also report the values of δ(a-b), namely the gain due to the nine waters. The gain in E1 is by only −1.6 kcal/mol more favorable for the malonate than for the phosphate ligand. Such relative gains increase very significantly with the second-order contributions, Epol and Ect, passing to −7.7 and −5.2 kcal/mol, respectively. Thus the networks totaling nine waters would stabilize by −14.7 kcal/mole the malonate ligand over the phosphate one. This value is very close to the corresponding QC(HF ) value of −15.7 kcal/mol. Inclusion of dispersion/correlation does not alter the outcome. The comparative energy balances done on the complete ligand-PMI gave rise to the same conclusion: there is a distinct preference in favor of the malonate over the phosphate derivative, but it is only enabled by the networks of structural waters. However, as noted in [23], more exhaustive sampling of the energy surface, along with long-time MD and accounting for entropy effects, are needed for a more quantitative evaluation. This will be further discussed in the last section of this review.
1.4 Conclusions and Perspectives
There are numerous fields of application of computational chemistry where next-generation QC -derived anisotropic polarizable molecular mechanics/dynamics could very significantly extent the realm of ab initio quantum chemistry. These encompass, e.g., drug design, material science, and supramolecular chemistry. APMM should be able to handle systems with sizes larger by at least four orders of magnitude than QC, and/or enable simulations times also larger by similar orders. But a prerequisite to very large scale applications is an objective evaluation of its expectable accuracy. In this respect, a distinctive asset of the SIBFA procedure, which appears to this day to be shared by very few other potentials [42–45], is the separability of ΔE into five distinct contributions, each of which is formulated and calibrated on the basis of its ab initio QC counterpart, and subsequently extensively tested against it.
We have reviewed in this paper the inherent non-isotropy and non-additivity features of several of these contributions, and their impact on overall structure and energetics. The SIBFA procedure has lent itself to numerous confrontations against QC, more so than any other competing method. It could be adequate to conclude to shortly develop on three points: its refinements and enrichments, its integration into highly optimized softwares, and the realm of its applications.
-
(a)
Refinements.
-
Regarding electrostatics. The distributed multipoles and polarizabilies used to construct the SIBFA library of fragments were derived from QC fragment calculations using the CEP 4-31G(2d) basis set by Stevens et al. [183, 184]. Accordingly, most validation studies resorted to QC computations using this basis as well. Upon comparing the evolutions of intermolecular interaction energies for a series of different complexes, we found invariably the values of DE(HF ) with this base to very closely parallel those with larger basis sets [185, 186], the most extended one being aug-cc-pVTZ (-f) (Gresh et al. to be submitted). This attests to the high reliability of this basis set, and justifies its use to construct the SIBFA library of fragments. There are several cases, however, where it could be preferable to resort to very extended basis for calibration and validation purposes. Accordingly, we are assembling a new library of fragments with multipoles and polarizabilities now derived from uncorrelated as well as correlated aug-cc-pVTZ(-f) calculations. The parametrization phase can be automatized thanks to the I-NoLLS algorithm [187, 188]. It was recently reported in the context of SIBFA [189]. Once the ‘general’ parameters are set, the calibration of individual atom types or the introduction of new atoms, such as metal cations, becomes straightforward. This was done recently in the Li+ – Cs+ alkali cation series [100].
-
Regarding short-range. Erep, Ect, and Edisp-exch have dependencies upon the location of the lone-pair tips. Such locations can be derived from QC analyses such as Boys’ localization procedure [190] or ELF [142], as analyzed for a series of ligands by Chaudret et al. [191], whence an additional filiation of SIBFA to QC .
-
-
(b)
Enrichments. Multi-scale approaches such as QM/MM , pioneered in 1976 [14] constitute nowadays an emerging field of computational chemistry. Steps toward merging SIBFA and the Gaussian Electrostatic Model (GEM) [192] have been completed [193], and a complete integration of the two approaches is underway. Owing to their polarizable nature, APMM approaches are well suited to a merging with QM approaches. This was completed recently concerning AMOEBA (Piquemal et al. submitted) and could be pursued with SIBFA.
-
(c)
Integration into massively parallel codes. There could be very important perspectives for the use of the SIBFA potential on a much larger scale than before. It is presently being integrated in the newly developed Tinker-HP software. It will therefore benefit from novel algorithmic developments to speed up polarizable molecular dynamics . For example, the bottleneck of the polarization energy and associated derivative evaluation on parallel computers has been overcome by the use of new iterative techniques such as the Jacobi/DIIS approach offering good scaling on hundreds and even thousands processors with gains in time going up to three orders of magnitude upon using advanced MD predictor-corrector algorithms [162]. SIBFA should also benefit from the high performance Smooth Particle Mesh periodic boundary condition implementation for electrostatics, including the short-range penetration correction [194] and for the polarization energy that uses newly introduced solvers and benefits from a Nlog(N) scalability [195]. Overall, all derivatives and torques have been coded and production simulation runs could be anticipated to start this year. Moreover, another asset will be the availability of the newly developed domain decomposition Cosmo (dd-Cosmo) continuum solvation model that is now available in direct connection with polarizable molecular dynamics [196]. To conclude on the technical part, new parametrization strategies have been defined with automatic parametrization using the INOLLS software [187, 188]. Such an approach should greatly reduce the time effort required for the definition of new parameter sets [189]. A previous version of the SIBFA software had been earlier (1999–2005) deposited at the Computational Chemistry List (CCL). A version of the Tinker-HP code integrating the SIBFA potential and its gradients is destined to a release in the forthcoming year.
-
(d)
Prospective applications.
-
Ligand-macromolecule complexes. One of the most attractive fields of APMM applications is ligand-protein complexes. There have been published applications regarding kinases [169] and metalloproteins [23, 161, 197–201]. It could be rewarding to adapt Free Energy Perturbation (FEP) methods [202] or non-equilibrium MD [203] to such targets, and particularly metalloproteins, on account of the demonstrated reliable handling of metal cation-ligand interactions. Along these lines, we note that an AMOEBA application was recently coauthored by one of us, which bore on the binding to MMP-13 of four dicarboxamide inhibitors [202]. This was the first ever reported FEP study on a metalloprotein using polarizable potentials. Although the ligand structural changes bore on sites distinct from the Zn-binding site, it is expectable that changes directly affecting Zn(II)-binding are amenable to prospective SIBFA FEP calculations.
-
Supramolecular chemistry and material science. As reviewed above, SIBFA has been adapted to a diversity of metal cations. These encompass the following: alkali Li(I)-Cs(I) [100], alkaline-earth Mg(II) and Ca(II) [103], transition metals Cu(I) [128], Cu(II) [99], Zn(II) and Cd(II) [103, 104, 127], heavy metals Pb(II) [119] and Hg(II) [136], and lanthanides and actinides [204]. On the one hand, this should enable to investigate their binding, stationary or transitory, to a diversity of proteins and NA’s . On the other hand, this should enable to address the issue of preferential entrapment of one cation over that of others by a supramolecular host, and possibly the design of cation-selective hosts for extraction or detoxification. The computation of free energies of binding could be based on FEP, non-equilibrium MD , or possibly by computing the contribution of vibrational entropy [205, 206]. Finally, there is a little charted domain of application of APMM approaches, which relates to surfaces and nanostructures. Accessing the values of their distributed multipoles and polarizabilities, along with the handling of Periodic Boundary Conditions (PBC) in SIBFA, and following validations against QC in model systems, could pave the way for numerous studies on adsorption events.
-
Modeling of nucleic acid s. The application of APMM to nucleic acids constitutes another virtually uncharted ground [see, e.g., Refs. 207–211]. Yet NA’s are an ideal domain of application for such approaches, considering the polyanionic nature of the sugar-phosphate backbone and the strongly polar and polarizable nature of the bases. Several issues can be mentioned: the structure, organization, and dynamics of the water networks in the groove, the dynamics of binding of metal cations to the backbone, the groove, and/or the water networks; the amplitude of stacking energies of successive base-pairs, from which the sequence-dependent conformation of NA’s depend; and the conformational properties of the phosphodiester backbone needing to handle properly polarization and anomeric effects.
-
Abbreviations
- AMOEBA:
-
Atomic multipoles optimized energetics for biological applications
- APMM:
-
Anisotropic polarizable molecular mechanics
- ATP:
-
Adenosine triphosphate
- aug-cc-pVTZ:
-
Augmented correlation-consistent polarized valence triple dzeta
- BO:
-
Born-Oppenheimer
- CDP:
-
Claverie, Dreyfus, Pullman
- CEP:
-
Coreless effective potential
- CP:
-
Car-Parrinello
- CSOV:
-
Constrained space orbital variation
- CTU:
-
Carboxythiourea
- dd-Cosmo:
-
Domain-decomposition conductor screening model
- DIIS:
-
Direct inversion of iterative space
- EFP:
-
Effective fragment potential
- ELF:
-
Electron localization function
- EM:
-
Energy-minimization
- EVG:
-
Elvitegravir
- FAK:
-
Focal adhesion kinase
- FEP:
-
Free energy perturbation
- GEM:
-
Gaussian electrostatic model
- GPU:
-
Graphics processor unit
- GS:
-
Garmer and Stevens
- HF:
-
Hartree-Fock
- IEHT:
-
Iterative extended Huckel theory
- I-NoLLS:
-
Interactive non linear least squares
- INT:
-
Integrase
- KM:
-
Kitaura-Morokuma
- LC:
-
Langlet-Claverie
- LMO:
-
Localized molecular orbitals
- MC:
-
Monte-Carlo
- MD:
-
Molecular dynamics
- MEP:
-
Molecular electrostatic potential
- MO:
-
Molecular orbitals
- NA:
-
Nucleic acids
- NRP1:
-
Neuropilin-1
- OPEP:
-
Optimized partitioning of electrostatic properties
- PBC:
-
Periodic boundary conditions
- PME:
-
Particle Mesh Ewald
- PMI:
-
Phosphomannose isomerase
- PMM:
-
Polarizable molecular mechanics
- QC:
-
Quantum chemistry
- QM/MM:
-
Quantum mechanics/molecular mechanics
- RVS:
-
Reduced variational space
- SAPT:
-
Symmetry-adapted perturbation theory
- SIBFA:
-
Sum of interactions between fragments Ab initio computed
- SOD:
-
Superoxide dismutase
- vdW:
-
van der Waals
References
Car R, Parrinello M (1985) Phys Rev 55:2471
Kuhne TD, Krack M, Mohamed FR, Parrinello M (2007) Phys Rev Letts 98:066401
Schlegel HB, Iyengar S, Li X, Millam JM, Voth GA, Scuseria GE, Frisch M (2002) J Chem Phys 117:8694
Tongraar A, Liedl KR, Rode BM (1998) J Phys Chem A 102:10340
Titov AV, Ufimtsev IS, Luehr N, Martinez TJ (2013) J Chem Theory Comput 9:213
Ufimtsev IS, Luehr N, Martinez TJ (2011) J Phys Chem Lett 2:1789
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA (1993) J Comput Chem 14:1347
York DM, Lee T-S, Yang W (1998) Phys Rev 80:5011
Scuseria G (1999) J Phys Chem A 103:4782
Nam K, Gao J, York DM (2005) J Chem Theory Comput 1:2
Yang W, Lee T-S (1996) J Chem Phys 103:5674
Monard G, Bernal-Uruchurtu MI, van der Waart A, Merz KM, Ruiz-Lopez M (1998) J Phys Chem A 109:3425
VandeVondele J, Borstnik U, Hutter J (2012) J Chem Theory Comput 8:3565
Warshel A, Levitt M (1976) J Mol Biol 103:227
Maseras F, Morokuma K (1995) J Comput Chem 16:1170
Senn HM, Thiel W (2009) Angewandte Chem 48:1198
Rod TH, Ryde U (2005) Phys Rev Lett 94:138
Monari A, Rivail J-L, Assfeld X (2013) Acc Chem Res 46:596
Ferre N, Assfeld X, Rivail J-L (2002) J Comput Chem 23:610
Very T, Despax S, Hebraud P, Monari A, Assfeld X (2012) Phys Chem Chem Phys 14:12496
Antonczak S, Monard G, Ruiz-Lopez M, Rivail J-L (1998) J Am Chem Soc 120:8834
Ryde U, Olsen L, Nilsson K (2002) J Comput Chem 23:1058
Gresh N, de Courcy B, Piquemal JP, Foret J, Courtiol-Legourd S, Salmon L (2011) J Phys Chem B 115:8304
Gresh N (2006) Curr Pharm Des 12:2121
Dehez F, Angyan JG, Soteras Gutierrez IS, Luque FJ, Schulten K, Chipot C (2007) J Chem Theory Comput 3:1914
Gresh N, Cisneros AG, Darden TA, Piquemal J-P (2007) J Chem Theory Comput 3:1960
Warshel A, Kato M, Pisliakov AV (2007) J Chem Theory Comput 3:2034
Cieplak P, Dupradeau F-Y, Duan Y, Wang J (2009) J Phys: Condens Matter 21:33102
Ponder JW, Wu C, Ren PY, Pande VS, Chodera JD, Mobley DL, Schnieders MJ, Haque I, Lambrecht DS, DiStasio RA Jr, Head-Gordon M, Clark GNI, Johnson ME, Head-Gordon T (2010) J Phys Chem B 114:2549
Rick SW, Stuart SJ, Berne BJ (1994) J Chem Phys 101:6141
Stern HA, Rittner F, Berne BJ, Friesner RA (2001) J Chem Phys 115:2237
Chelli R, Procacci P (2002) J Chem Phys 117:9175
Lamoureux G, MacKerell AD Jr, Roux B (2003) J Chem Phys 119:5185
Anisimov V, Vorobyov IV, Roux B, MacKerell AD Jr (2007) J Chem Theory Comput 3:1927
Pullman B, Claverie P, Caillet J (1967) Proc Natl Acad Sci USA 57:1663
Rein R, Claverie P, Pollack M (1968) Int J Quantum Chem 2:1129
Claverie P, Rein R (1969) Int J Quantum Chem 3:537
Gresh N, Claverie P, Pullman A (1984) Theor Chim Acta 66:1
Gresh N, Pullman A, Claverie P (1985) Theor Chim Acta 67:11
Price SL, Stone AJ (1984) Mol Phys 51:569
Price SL, Stone AJ (1992) J Chem Soc Faraday Soc 88:1755
Day P, Jensen JH, Gordon MS, Webb SP, Stevens WJ, Krauss M, Garmer DR, Basch H, Cohen D (1996) J Chem Phys 105:1968
Slipchenko LV, Gordon MS (2007) J Comput Chem 28:276
Gordon MS, Fedorov DG, Pruitt SR, Slipchenko LV (2012) Chem Rev 112:632
Gordon MS, Smith QA, Xu P, Slipchenko LV (2013) Annu Rev Phys Chem 64:553
Millot CJ, Soetens JC, Martins Costa NTC, Hodges MP, Stone AJ (1998) J Phys Chem A 102:754
Karlstrom G, Linse P, Wallqvist A, Jonsson B (1983) J Am Chem Soc 105:3777
Hermida-Ramon JM, Brdarski S, Karlstrom G, Berg U (2003) J Comput Chem 24:161
Ren PY, Ponder JW (2003) J Phys Chem B 107:5933
Shi Y, Xia Z, Zhang J, Best R, Wu C, Ponder JW, Ren PY (2012) J Chem Theory Comput 9:4046
Dreyfus M (1970) PhD Thesis, University of Paris
Claverie P (1973) PhD Thesis, Paris CNRS library number AO 8214
Claverie P Localization and delocalization in quantum chemistry. In: Chalvet O, Daudel R, Diner S, Malrieu JP (eds) Reidel, Dordrecht Vol II, p 127
Pullman A, Zarkzewska C, Perahia D (1979) Int J Quantum Chem 16:395
Pullman A, Perahia D (1979) Theoret Chim Acta 50:351
Perahia D, Pullman A, Pullman B (1979) Theoret Chim Acta 51:349
Pullman B, Perahia D, Cauchy D (1979) Nucleic Acids Res 10:3821
Lavery R, Pullman A, Pullman B, de Oliveria M (1980) Nucleic Acids Res 8:5095
Lavery R, Pullman B, Corbin S (1981) Nucleic Acids Res 9:6539
Lavery R, Pullman B, Zakrzewska K (1982) Biophys Chem 15:343
Lavery R, Pullman A, Wen YK (1982) Int J Quantum Chem 24:353
Zarkzewska C, Pullman A (1982) Int J Quantum Chem Quantum Biol Symp 9:411
Etchebest C, Lavery R, Pullman A (1982) Studia Biophys 90:7
Gresh N, Pullman A (1983) Int J Quantum Chem Quantum Biol Symp 10:215
Gresh N, Claverie P, Pullman A (1979) Int J Quantum Chem Symp 13:243
Gresh N (1980) Biochim Biophys Acta 597:345
Gresh N, Pullman B (1980) Biochim Biophys Acta 625:356
Gresh N, Etchebest C, de la Luz Rojas O, Pullman A (1981) Int J Quantum Chem Quantum Biol Symp 8:109
Gresh N, Pullman A (1982) Int J Quantum Chem 22:709
Gresh N, Pullman B (1984) Mol Pharmacol 25:452
Gresh N (1985) Biopolymers 24:1527
Vigné-Maeder F, Claverie P (1988) J Chem Phys 88:4934
Rein R, Rabinowitz JR, Swissler TJ (1972) J Theor Biol 34:215
Rein R (1973) Adv Quantum Chem 7:335
Stone AJ (1981) Chem Phys Lett 83:233
Stone AJ, Alderton M (1985) Mol Phys 56:1047
Sokalski WA, Poirier RA (1983) Chem Phys Lett 98:86
Sokalski WA, Sawaryn AJ (1987) Chem Phys 87:526
Andersson M, Karlstrom G (1985) J Phys Chem 89:4957
Angyan JG, Chipot C, Dehez F, Hattig C, Jansen G, Millot C (2003) J Comput Chem 24:997
Angyan JG, Chipot C (2005) New J Chem 29:411
Freitag M, Gordon MS, Jensen JH, Stevens WJ (2000) J Chem Phys 112:7300
Gordon MS, Freitag MA, Bandyopadhyay A, Jensen JH, Kairys V, Stevens WJ (2001) J Phys Chem A 105:293
Piquemal J-P, Gresh N, Giessner-Prettre C (2003) J Phys Chem A 107:10353
Slipchenko LV, Gordon MS (2006) J Comput Chem 28:276
Wang B, Truhlar DG (2010) J Chem Theory Comput 6:3330
Stone AJ (2011) J Phys Chem A 115:7017
Garmer DR, Stevens WJ (1989) J Phys Chem 93:8263
Gresh N, Leboeuf M, Salahub DR (1994) Modeling the hydrogen bond American Chemical Society Symposia No 569 82 D A Smith Ed
Jensen JH, Day PN, Gordon MS, Basch H, Cohen D, Garmer DR, Krauss M, Stevens WJ Ibid p 139
Murrell JN, Teixeira-Dias JJC (1970) Mol Phys 19:521
Murrell JN, Randic M, Williams DR (1966) Proc R Soc Lond Ser A 284:566
Gresh N, Claverie P, Pullman A (1982) Int J Quantum Chem 22:199
Gresh N, Claverie P, Pullman A (1986) Int J Quantum Chem 29:101
Creuzet S, Langlet J, Gresh N (1991) J Chim Phys Phys Chim Biol 88:2399
Stevens WJ, Fink W (1987) Chem PhysLett 139:15
Bagus PS, Hermann K, Bauschlicher C (1984) J Chem Phys 80:4378
Jeziorski B, Moszynski R, Szalewicz K (1984) Chem Rev 94:1887
Piquemal J-P, Hubbard BW, Fey N, Deeth R, Gresh N, Giessner-Prettre C (2003) J Comput Chem 24:1963
Dudev T, Devereux M, Meuwly M, Lim C, Piquemal J-P, Gresh N (2015) J Comput Chem 36:285
Buckingham AD, Fowler PW (1983) J Chem Phys 79:6426
Garmer DR, Gresh N (1994) J Am Chem Soc 116:3556
Gresh N, Garmer DR (1996) J Comput Chem 17:1481
Gresh N (1995) J Comput Chem 16:856
Piquemal J-P, Chevreau H, Gresh N (2007) J Chem Theory Comput 3:824
Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X, Jiang H, Zhu W (2009) J Med Chem 52:2854
Hernandes MZ, Cavalcanti SM, Moreira DR, de Azevedo WF, Jr Leite AC (2010) Curr Drug Targets 11:303
Matter H, Nazaré M, Güssregen S, Will DW, Schreuder H, Bauer A, Urmann M, Ritter K, Wagner M, Wehner V (2009) Angew Chem Int Ed 48:2911
Clark T, Hennemann M, Murray JS, Politzer P (2007) J Mol Mod 13:291
Murray JS, Lane P, Politzer P (2009) J Mol Mod 15:723
Politzer P, Murray JS, Clark T (2010) Phys Chem Chem Phys 12:7748
Ibrahim MA (2011) J Comput Chem 32:2564
Jorgensen WL, Schyman P (2012) J Chem Theory Comput doi:101021/ct300180w
Kolar M, Hobza P (2012) J Chem Theory Comput 8:1325
El Hage K, Piquemal J-P, Hobaika Z, Maroun RG, Gresh N (2013) J Comput Chem 34:1125
Gourlaouen C, Piquemal J-P, Parisel O (2006) J Chem Phys 124:174311
Gourlaouen C, Parisel O (2007) Angew Chem Int Ed 46:553
van Severen M-C, Piquemal J-P, Parisel O (2009) Chem Phys Lett 478:17
Devereux M, van Severen M-C, Parisel O, Piquemal J-P, Gresh N (2011) J Chem Theory Comput 7:138
Tiraboschi G, Roques BP, Gresh N (1999) J Comput Chem 20:1379
Tiraboschi G, Gresh N, Giessner-Prettre C, Pedersen LG, Deerfield DW (2000) J Comput Chem 21:1011
Garmer DR, Gresh N, Roques BP (1998) Proteins Struct Funct Genet 31:42
Gresh N (1997) J Phys Chem A 101:8690
Masella M, Gresh N, Flament JP (1998) J Chem Soc, Faraday Trans 94:2745
Piquemal J-P, Chelli R, Procacci P, Gresh N (2007) J Phys Chem A 111:8170
Guo H, Gresh N, Roques BP, Salahub DR (2000) J Phys Chem B 104:9746
Gresh N, Piquemal J-P, Krauss M (2005) J Comput Chem 26:1113
Gresh N, Policar C, Giessner-Prettre C (2002) J Phys Chem A 106:5660
Ohrn A, Karlstrom G (2007) J Chem Theory Comput 3:1993
Beachy MD, Chasman D, Murphy RB, Halgren TA, Friesner RA (1997) J Am Chem Soc 119:5908
Gresh N, Kafafi SA, Truchon J-F, Salahub DR (2004) J Comput Chem 25:823
Rogalewicz F, Gresh N, Ohanessian G (2000) J Comput Chem 21:963
Tiraboschi G, Fournie-Zaluski MC, Roques BP, Gresh N (2001) J Comput Chem 22:1038
Gresh N, Shi G-B (2004) J Comput Chem 25:160
Faerman CH, Price SL (1990) J Am Chem Soc 112:4915
Chaudret R, Gresh N, Parisel O, Piquemal J-P (2011) J Comput Chem 31:2949
Barr MP, Byrne AM, Duffy AM, Condron CM, Devocelle M, Harriott P, Bouchier-Hayes DJ, Harmey JH (2005) Br J Cancer 92:328
Borriello L, Montès M, Lepelletier Y, Leforban B, Liu W-Q, Demange L, Delhomme B, Pavoni S, Jarray R, Boucher JL, Dufour S, Hermine O, Garbay C, Hadj-Slimane R, Raynaud F (2014) Cancer Lett 349:120
van der Kooi C, Jusino MA, Perman B, Neau DB, Bellamy HD, Leahy D (2007) Proc Natl Acad Sci USA 104:6152
Jarvis A, Allerston CK, Jia H, Herzog B, Garza-Garcia A, Winfield N, Ellard K, Aqil R, Lynch R, Chapman C, Hartzoulakis B, Nally J, Stewart M, Cheng L, Menon M, Tickner M, Djordjevic S, Driscoll PC, Zachary I, Selwood DL (2010) J Med Chem 53:2215
Goldwaser E, de Courcy B, Demange L, Garbay C, Raynaud F, Hadj-Slimane R, Piquemal J-P, Gresh N (2014) J Mod Mol 20:2472
Silvi B, Savin A (1994) Nature 371:683
Piquemal J-P, Pilme J, Parisel O, Gerard H, Fourre I, Berges J, Gourlaouen C, De La Lande A, Van Severen M-C, Silvi B (2008) Int J Quantum Chem 108:1951
Evangelakis GA, Rizos JP, Lagaris IE, Demetropoulos IN (1987) Comput Phys Commun 46:401
Zheng J, Yu T, Papajak E, Alecu IM, Mielke S, Truhlar DG (2011) Phys Chem Chem Phys 13:10885
Tafipolsky M, Schmid R (2005) J Comput Chem 26:1579
Marchand C, Maddali K, Metifiot M, Pommier Y (2009) Curr Top Med Chem 9:1016
Klibanov OM (2009) Curr Opin Invest Drugs 10:190
Satom M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, Kawakami H, Matsuzaki Y, Watanabe W, Yamataka K et al (2006) J Med Chem 49:1506
Schafer JJ, Squires KE (2010) Ann Pharmacother 44:145
Hare S, Gupta S, Valkov E, Engelman A, Cherepanov P (2010) Nature 464:232
El Hage K, Piquemal J-P, Hobaika Z, Maroun RG, Gresh N (2015) J Comput Chem 36:210
El Hage K, Piquemal J-P, Hobaika Z, Maroun RG, Gresh N (2014) J Phys Chem A 118:9772
Kitaura K, Morokuma K (1976) Int J Quantum Chem 10:325
McCord JM, Fridovich I (1969) J Biol Chem 244:6049
Somwar R, Erdjumen-Bromage H, Larsson E, Shum D, Lockwood WW, Yang G, Sander C, Ouerfelli O, Tempst PJ, Djaballah H, Varmus HE (2011) Proc Natl Acad Sci USA 108:16375
Glasauer A, Sena LA, Diebold LP, Mazar AP, Chandell NS (2014) J Clin Invest 124:117
Bijeire L, Elias B, Souchard J-P, Gicquel E, Moucheon C, Kirsch-de Mesmaeker A, Vicendo P (2006) Biochem 45:6160
Hough MA, Hasnain SS (2003) Structure 11:937
Tainer JA, Getzoff ED, Richardson JS, Richardson DC (1983) Nature 306:284
Gresh N, El Hage KE, Perahia D, Piquemal J-P, Berthomieu C, Berthomieu D (2014) J Comput Chem 35:2096
Lipparini F, Lagardere L, Stamm B, Cances E, Schnieders M, Ren P, Maday Y, Piquemal J-P (2014) J Chem Theory Comput 10:1638
Krause DS, Van Etten RA (2005) N Engl J Med 353:172
Nikolopoulos S, Giancotti FG (2005) Cell Cycle 4:e131
McLean GW, Carragher NO, Avizienyte VG, Frame MC (2005) Nat Cancer 5:505
Cohen LA, Guan JL (2005) Curr Cancer Drug Targets 5:629
Lietha D, Cai X, Ceccarelli DFJ, Li Y, Schaller MD, Eck M (2007) J Cell 129:1177
Choi HS et al (2006) Bioorgan Med Chem Lett 16:2689
de Courcy B, Piquemal J-P, Garbay C, Gresh N (2010) J Am Chem Soc 132:3312
Langlet J, Claverie P, Caillet J, Pullman A (1998) J Phys Chem 92:1617
Claverie P, Daudey J-P, Langlet J, Pullman B, Piazzola D (1978) J Phys Chem 8:405
Mundim KC, Tsallis C (1996) Int J Quantum Chem 58:373
Moret MA, Pascutti PG, Bisch PM, Mundim KC (1998) J Comput Chem 19:647
Jiao D, Golubkov PA, Darden TA, Ren P (2008) Proc Natl Acad Sci USA 105:6290
Lietha D, Eck M (2008) J PLOS one. doi:10.1371/journalpone0003800
Gracy RW, Noltmann EA (1968) J Biol Chem 243:3161
Patterson JH, Waller RF, Jeevarajah D, Billman-Jacobe H, McConville M (2003) J Biochem J 372:77
Garami A, Ilg T (2001) J Biol Chem 276:6566
Shinabarger D, Berry A, May TB, Rothmel R, Fialho A, Chakrabarty AM (1991) J Biol Chem 266:2080
Roux C, Lee JH, Jeffery CJ, Salmon L (2004) Biochemistry 43:2926
Cleasby A, Wonacott A, Skarzynski T, Hubbard RE, Davies GJ, Proudfoot AEI, Bernard AR, Payton MA, Wells TNC (1996) Nat Struct Biol 3:470
Roux C, Gresh N, Perera LE, Piquemal J-P, Salmon L (2007) J Comput Chem 28:938
Stevens WJ, Basch H, Krauss M (1984) J Chem Phys 81:6026
Stevens WJ, Krauss M, Basch H, Jasien PG (1992) Can J Chem 70:612
Dunning TH (1989) J Chem Phys 90:1007
Feller D (1996) J Comput Chem 17:1571
Law MM, Hutson JM (1997) Comp Phys Commun 102:252
Devereux M, Meuwly M (2010) J Chem Inf Model 50:349
Devereux M, Gresh N, Piquemal J-P, Meuwly M (2014) J Comput Chem 35:1577
Foster JM, Boys SF (1960) Rev Mod Phys 32:300
Chaudret R, Gresh N, Cisneros GA, Scemama A, Piquemal J-P (2013) Can J Chem 91:804
Piquemal J-P, Cisneros GA, Reinhardt P, Gresh N, Darden TA (2006) J Chem Phys 124:104101
Chaudret R, Gresh N, Narth C, Lagardere L, Darden TA, Cisneros GA, Piquemal J-P (2014) J Phys Chem A 118:7598
Wang Q, Rackers JA, He C, Qi R, Narth C, Lagardere L, Gresh N, Ponder JW, Piquemal JP, Ren PY (2015) J Chem Theory Comput 11:2609
Lagardère L, Lipparini F, Polack E, Stamm B, Cances E, Schnieders M, Ren PY, Maday Y, Piquemal JP (2015) J Chem Theory Comput 11:2589
Lipparini F, Lagardère L, Raynaud C, Stamm B, Cances E, Schnieders M, Ren PY, Mennucci B, Maday Y, Piquemal JP (2015) J Chem Theory Comput 11:623
Antony J, Gresh N, Olsen L, Hemmingsen L, Schofield CJ, Bauer R (2002) J Comput Chem 23:1281
Antony J, Piquemal J-P, Gresh N (2005) J Comput Chem 26:1131
Gresh N, Derreumaux P (2003) J Phys Chem B 107:4862
Miller Jenkins LM, Hara T, Durell SR, Hayashi R, Inman JK, Piquemal J-P, Gresh N, Appella E (2007) J Am Chem Soc 129:11067
Gresh N, Audiffren N, Piquemal J-P, de Ruyck J, Ledecq M, Wouters J (2010) J Phys Chem B 114:4884
Zhang J, Yang W, Piquemal J-P, Ren P (2012) J Chem Theory Comput 8:1314
Nicolini P, Frezzato D, Gellini C, Bizzarri M, Chelli R (2013) J Comput Chem 34:1561
Marjolin A, Gourlaouen C, Clavaguéra C, Ren P, Wu J, Gresh N, Dognon J-P, Piquemal J-P (2012) Theor Chem Acc 131:1199
Muddana H, Gilson MK (2012) J Chem Theory Comput 8:2023
Grimme S (2012) Chem Eur J 18:9955
Gresh N, Sponer J (1999) J Phys Chem B 103:1415
Gresh N, Sponer JE, Spackova N, Leszczynski J, Sponer J (2003) J Phys Chem B 107:8669
Ghosh D, Kosenkov D, Vanovschi V, Williams CR, Herbert JM, Gordon MS, Schmidt MW, Slipchenko LV, Krylov A (2010) J Phys Chem A 114:12739
Smith QA, Gordon MS, Slipchenko LV (2011) J Phys Chem A 115:11269
Savelyev A, MacKerell AD (2014) J Comput Chem 16:1219
Acknowledgments
We wish to thank Dr. Michael Devereux (University of Basel, Switzerland) for his recent contributions on Pb(II) modeling, and for enabling to derive INollS-optimized SIBFA parameters in conjunction with the aug-cc-pVTZ(-f) basis set. Several colleagues are thanked for rewarding collaborations in the last few years: Tom Darden (Open Eyes, Santa Fe, USA), G. Andres Cisneros (University of Detroit, USA), Aude Marjolin (Quantum Theory Project, Pittsburgh, USA), Marie Ledecq (Union Chimiques Belges, Brussels, Belgium), Johan Wouters (Universites Notre-Dame de la Paix, Namur, Belgium), Dr. Dorothée Berthomieu (Dorothée Berthomieu, Institut Charles-Gerhardt, Université de Montpellier, France) and Markus Meuwly (University of Basel, Switzerland). We would also like to mention, regarding the experimental work, Prof. Christiane Garbay (Université ParisDescartes, Paris, France) and Prof. Laurent Salmon (Institut de Chimie Moléculaire d’Orsay, France).
We wish to thank the Grand Equipement National de Calcul Intensif (GENCI): Institut du Développement et des Ressources en Informatique Scientifique (IDRIS), Centre Informatique de l’Enseignement Supérieur (CINES), France, project No. x2009-075009), and the Centre de Ressources Informatiques de Haute Normandie (CRIHAN, Rouen, France), project 1998053. This work was supported in part by the French state funds managed by CALSIMLAB and the ANR within the Investissements d’Avenir program under reference ANR-11-IDEX-0004-02. F.L. We also wish to thank the Association Philippe Jabre (Beirut, Lebanon) for financing the PhD Thesis of Krystel El Hage.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Gresh, N. et al. (2015). Addressing the Issues of Non-isotropy and Non-additivity in the Development of Quantum Chemistry-Grounded Polarizable Molecular Mechanics. In: Rivail, JL., Ruiz-Lopez, M., Assfeld, X. (eds) Quantum Modeling of Complex Molecular Systems. Challenges and Advances in Computational Chemistry and Physics, vol 21. Springer, Cham. https://doi.org/10.1007/978-3-319-21626-3_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-21626-3_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-21625-6
Online ISBN: 978-3-319-21626-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)