
Abstract
Intravenous (IV) ketamine has been shown to have rapid and robust antidepressant effects in adults with treatment-resistant depression (TRD). Urological toxicity has been observed in chronic ketamine abusers as evidenced by dysuria, urgency, and hematuria. The foregoing observation provides the basis for evaluating whether ketamine-induced urological toxicity (KIUT) is associated with sub-anesthetic doses of ketamine (0.5–1.0 mg/kg) in adults with mood disorders. The overarching objective of this article is to identify potential mechanisms of KIUT which appears to be dose and frequency dependent. Available research indicates that high-frequency ketamine is associated with disruption of the urothelial barrier as well as direct ketamine toxicity (i.e., decreased expression of junction proteins) in KIUT of the bladder. Chronic and high-frequency ketamine use is also associated with bladder inflammation mediated via neurogenic and IgE inflammation. Other non-mutually exclusive causes are nerve hyperplasia, hypersensitivity, cell apoptosis, microvascular damage, and overexpression of carcinogenic genes. Notwithstanding the evidence of KIUT in ketamine abusers, there is no evidence that ketamine and/or esketamine treatment in adults with mood disorders is associated with KIUT. However, all patients receiving ketamine/esketamine for mood disorder treatment should be queried about genitourinary symptoms during acute and, where applicable, maintenance dosing.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ketamine is a dissociative anesthetic that has been used globally for over 50 years (Morgan et al. 2012). Over the past two decades, intravenous (IV) ketamine has been shown to have rapid and robust antidepressant effects in patients with treatment-resistant depression (TRD) at sub-anesthetic doses (0.5–1.0 mg/kg infused over 40 min) (McIntyre et al. 2020). TRD is defined by the lack of response to two or more trials of antidepressant drugs, and it impairs productivity, social functioning, and overall health of depressed individuals (Rush et al. 2006; Walker et al. 2015). More recently, intranasal (IN) esketamine, the S-enantiomer of ketamine, was approved for unipolar TRD and has also demonstrated short-term antidepressant effects (28–84 mg administered 2 times/week for < 6 months) (Daly et al. 2018; Kim et al. 2019). Antidepressant effects may be observed after a single ketamine infusion (0.5–1.0 mg/kg); however, effects are lost after 1–2 weeks, requiring repeated infusions (0.5–1.0 mg/kg every 1–4 weeks) to maintain antidepressant effects (Kryst et al. 2020). For IN esketamine, the response appears to sustain for more than 2 months even with a lower dosing frequency (1 time/week or every 2 weeks) (Daly et al. 2018). While the safety and tolerability of short-term doses have been well characterized, the safety profile of long-term IV ketamine and IN esketamine treatment (≥ 6 months) at antidepressant doses (ketamine: 0.5–1.0 mg/kg every 1–4 weeks; esketamine: 28–84 mg administered 2 times/week) has yet to be established (Na and Kim 2021; Rodrigues et al. 2020; Short et al. 2018).
Recommendations for prescribing ketamine/esketamine in adults with mood disorders in both acute and maintenance phases provide the impetus for a better understanding of specific safety concerns. Ketamine-induced urological toxicity (KIUT) has been associated with chronic and frequent high-dose exposure in ketamine abusers (dose: ≥ 0.125 g/session; frequency: ≥ 1 day/month) (Winstock et al. 2012). Ketamine-induced urological toxicity secondary to ketamine abuse was first described in 2007. The evidence appeared that chronic ketamine abusers experienced KIUT as evidenced by urinary frequency, urgency, hematuria, and dysuria (Shahani et al. 2007). Chronic ketamine abuse is also reported to affect the upper urinary tract, where hydronephrosis is observed, and is associated with vesicoureteral reflux and ureteral stenosis (Chu et al. 2008; Lai et al. 2012). Winstock et al. (2012) have reported 26.6% of ketamine abusers (n = 1285) experienced urological symptoms. The risk of urological complications is directly related to ketamine dose and frequency, where higher doses (≥ 0.25 g/session) and frequency of use (≥ 5 days/month) are associated with higher rates of lower abdominal pain, hematuria, and burning sensation during urination (Winstock et al. 2012). Complicating the interpretation of KIUT in individuals who are abusing ketamine is a possibility that other substances of abuse are often co-abused which may further contribute to KIUT in this population. Disparate mechanisms likely are contributing to KIUT in polysubstance abusers; below, we reviewed mechanisms implicated for KIUT specifically related to ketamine.
While urological side effects have been observed in chronic ketamine abusers, the relative risk of urological complications of IV ketamine (0.5–1.0 mg/kg every 1–4 weeks) and IN esketamine (28–84 mg administered 2 times/week) at long-term (≥ 6 months) antidepressant doses appears to be minimal. Survey data of IV ketamine providers suggests that urological complications are rare with short-term depression maintenance treatment (prevalence = 0.1%, duration: < 6 months). There is also no evidence suggesting that short-term ketamine treatment in mood disorders is associated with urological toxicity. Similarly, two clinical studies have reported a low occurrence (< 10%) of urological complications in patients with TRD when examining the safety and efficacy of short-term IN esketamine treatment (Daly et al. 2019; Wajs et al. 2020). Albeit the low prevalence of urological toxicity in short-term clinical trials, it is still unknown whether such safety profiles could be maintained in long-term studies or clinical interventions. Currently, there are limited studies that investigate the long-term safety (≥ 6 months) of IV ketamine and IN esketamine treatments (Feifel et al. 2020; Findeis et al. 2020; Short et al. 2018).
Therefore, it would be useful to point out the potential urological complications in long-term depression maintenance treatment and identify possible mechanisms of KIUT in chronic ketamine abusers. This would inform health care practitioners of the possibility of urological complications in long-term studies and interventions, and allow them to take appropriate measures when such symptoms arise. Of note, the current review focused on the mechanisms of ketamine, rather than esketamine. However, due to the similarity between the two substances, mechanisms and risk are likely to have significant overlap with ongoing research in this area (Findeis et al. 2020).
Pharmacology of ketamine
Ketamine is a lipophilic molecule that is rapidly absorbed in the human body through oral, nasal, and parenteral routes (Castellani et al. 2020; Peltoniemi et al. 2016; Zanos et al. 2018). Upon absorption, ketamine crosses the blood-brain barrier (BBB) and binds to the phencyclidine site of NMDARs in neurons, thus blocking the transmembrane ion flux and acting as a non-competitive antagonist (Malinovsky et al. 1996; Zanos et al. 2018). The anesthetic effect of ketamine is widespread in the central nervous system (CNS) as NMDARs are present in many tissues, such as the cerebral cortex, cerebellum, brainstem, and spinal cord, that are important in neuronal signal transduction (Myers et al. 2016).
The primary site of ketamine biotransformation is in the liver, where it is metabolized by cytochrome P450 enzymes to norketamine through the N-demethylation pathway (Chen and Chen 2010). Norketamine is then excreted in bile and expelled in the urine (Peltoniemi et al. 2016). As a result of the first-pass liver metabolism, oral administration of ketamine has a poor bioavailability (17%), whereas bioavailability via the parenteral route is close to 100% (Grant et al. 1981; Malinovsky et al. 1996). For intranasal administration, the bioavailability is approximately 50% (Peltoniemi et al. 2016).
Clinical findings of ketamine-induced urological toxicity
The clinical symptoms of KIUT vary greatly with respect to disease progression (Jhang et al. 2015). Common symptoms of KIUT are dysuria, hematuria, urinary urgency, urinary frequency, and post-micturition dribble (Shahani et al. 2007). Ultrasound studies have reported reduced bladder capacity with thickened bladder wall, bilateral hydronephrosis, and hydroureter following chronic abuse of ketamine (Pappachan et al. 2014; Lee et al. 2009). Cystoscopic findings from chronic ketamine abusers with KIUT (n = 16) have detailed fragile bladder mucosa, mucosal laceration after bladder distention, and glomerular hemorrhage (CL Lee et al. 2013). Hematoxylin-eosin stains of KIUT bladder have also revealed denuded urothelium, alongside mild inflammation in the mucosa with eosinophils, lymphocytes, and plasma cells infiltration (Wei et al. 2013). Ketamine-induced urological toxicity has been associated with chronic and frequent high-dose exposure in ketamine abusers (dose: ≥ 0.125 g/session; frequency: ≥ 1 days/month) (Winstock et al. 2012). The severity of symptoms was observed to be dose and frequency dependent, where higher doses (≥ 0.25 g/session) and frequency of use (≥ 5 days/month) were associated with increased rates of urinary frequency, hematuria, and burning sensation in urination (Winstock et al. 2012). Although there is no observed sex difference in KIUT symptom prevalence, female ketamine abusers tend to have more serious urinary symptoms than males (9.7% vs 3.8%, n = 1614) (Chen et al. 2014).
Potential mechanisms of KIUT
The extant literature suggests multiple non-mutually exclusive possible mechanisms of KIUT in chronic ketamine abusers, including urothelial barrier disruption, bladder inflammation, ketamine direct toxicity, nerve hyperplasia and hypersensitivity, cell apoptosis, microvascular damage, and overexpression of carcinogenic genes (Table 1).
Urothelial barrier disruption
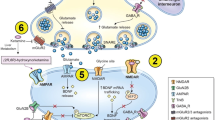
Damage of the urothelial barrier has been postulated as a possible mechanism of KIUT. A decreased expression of intercellular junction proteins (E-cadherins and zonula occludens-1) has been reported in bladder tissues of chronic ketamine abusers and rats with KIUT, which suggests a disruption in cell-cell junction stability, thereby disrupting the urothelial barrier (Duan et al. 2017; Wang et al. 2017). Intercellular junction proteins are crucial in cell adhesions and the maintenance of epithelial barrier function by preventing substances from entering the bladder wall (Castellani et al. 2020). Thus, chronic exposure to ketamine in the bladder may lead to urothelial barrier damage by reducing the expression of junction proteins, which allows urinary irritants to enter the compromised bladder wall and provoke inflammatory responses (Fig. 1) (Jhang et al. 2015). Exploring the relationship between chronic ketamine abuse and reduced expression of junction proteins in the urothelial barrier will be useful in elucidating pathophysiological mechanisms in KIUT.

Disruption of the urothelial barrier by ketamine and its metabolites. Ketamine could damage the urothelial barrier by reducing the expression of junction proteins (E-cadherins and zonula occludens-1). Downregulation of these proteins would allow the entry of urinary irritants to the bladder wall and induce inflammation
The downregulation of junction proteins could also lead to epithelial-to-mesenchymal transition (EMT), a process where the urothelium loses apical-basal polarity and cell-cell adhesion, experiences phenotypical changes, and develops invasive properties (Lamouille et al. 2014; Thiery et al. 2009). It has been shown that the loss of E-cadherin is paralleled by the acquisition of mesenchymal markers such as vimentin and fibronectin, indicating the development of EMT in KIUT of the bladder (Wang et al. 2017). The exposure of mesenchymal cells to the surroundings may promote their activation and further result in fibrosis, a condition defined by the excessive accumulation of extracellular matrix (ECM) components like collagen and fibronectin (Wynn and Ramalingam 2012). Additionally, studies have revealed that transforming growth factor-β1 (TGF-β1) signaling activates several transcription factors (Snail, Slug, and Twist) and induces EMT, which may contribute to fibrosis in KIUT of the bladder (Lamouille et al. 2014; Thiery et al. 2009). Hence, more studies are needed to elucidate the implication of downregulated junction proteins and how these factors can initiate EMT progression in KIUT via the TGF-β1 pathway.
Bladder inflammation
Ketamine-induced urological toxicity of the bladder commonly displays a denuded epithelium, infiltration of macrophages, neutrophils, eosinophils, mast cells in the mucosa, increased collagen deposition, and elevated serum interleukin (IL) 1, 5, 6, and 17, which demonstrate the activation of inflammatory processes in bladder tissue (Castellani et al. 2020). Inflammation is further confirmed by the presence of inflammatory markers including, cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), and matrix metallopeptidase-9 (MMP-9), identified through immunohistochemical staining (Gu et al. 2014; Lin et al. 2015). Among the 3 markers, iNOS is particularly important since it is associated with many inflammatory mediators, such as cytokines, prostaglandin E2, and the COX pathway, leading to robust downstream inflammatory responses and possibly interstitial fibrosis in the bladder (Jhang et al. 2015).
Furthermore, the transcription factor NF-κB is found to be responsible in this process by regulating downstream COX-2 gene expression (Juan et al. 2015). It has been postulated that the stimulation of ketamine on bladder cells leads to the disinhibition of NF-κB by BAY 11-7082 (Fig. 2). Subsequently, nuclear translocation of NF-κB occurs at the promoter region of the COX-2 gene and upregulates its gene expression. The upregulation of COX-2 then results in the secretion of prostaglandin, leading to inflammation of the bladder (Juan et al. 2015). It should be noted that available evidence regarding this pathway is mixed with some studies suggesting that COX-2 acts upstream of NF-κB, where it activates the transcription factor (Chen et al. 2013). Therefore, future research will be required to elucidate the role of enzymes involved in the inflammatory response to ketamine.

Upregulation of COX-2 gene expression through NF-κB signaling pathway. Ketamine stimulation of bladder cells leads to disinhibition of NF-κB by BAY 11-7082. Nuclear translocation of NF-κB occurs subsequently where it binds to the promoter region of the COX-2 gene and upregulates its gene expression. The upregulation of the COX-2 gene then results in the secretion of prostaglandin, leading to inflammation of the bladder
Ketamine-mediated toxicity
Direct toxic effects of ketamine in urine have been considered as a plausible mechanism for the bladder damage present in KIUT (Shahani et al. 2007). After the biotransformation process in the liver, ketamine and its metabolites can last in the urine for 14 days and be exposed to bladder tissue continuously (Parkin et al. 2008). Studies have proposed ketamine in urine might trigger direct damage and inflammation in the bladder (Chuang et al. 2013). The presence of denuded urothelium, ulceration, and fibrosis could explain the direct bladder injury by ketamine, which is similar to cystitis caused by cytotoxic agents like cyclophosphamide (CP) and mitomycin-C (Payne et al. 2013).
Baker et al. (2016) has demonstrated ketamine can also induce toxicity in the bladder by provoking a large release of calcium (Ca2+) ions from the endoplasmic reticulum (ER) into the cytosol. This damage is dose and time dependent. The sustained increase in intracellular Ca2+ level can cause stress in the mitochondria and the ER, which results in cell apoptosis (Fig. 3). Furthermore, it has been found that hydroquinone, a ketamine metabolite, directly fragments DNA and chromosomes in mouse bladder cells (Enguita and Leitão 2013; Tan et al. 2011; Wai et al. 2013). As a result, hydroquinone may play an important role in the pathogenesis of KIUT. Further investigations on the concentration of hydroquinone in urine are required to indicate its role in KIUT in humans.

Endoplasmic reticulum and mitochondrial stress induced by elevated intracellular Ca2+ levels. Ketamine may exert toxicity to the bladder by provoking the release of Ca2+ from the ER into the cytosol. The increase in intracellular Ca2+ would cause stress to the mitochondria and ER, which results in cell apoptosis
Nerve hyperplasia and hypersensitivity
The immune system is believed to be involved in the pathogenesis of KIUT. Apart from eosinophil infiltration, the IgE level is also found to be higher in blood and bladder tissue of chronic ketamine abusers with KIUT (Jhang et al. 2016). It has been speculated that an antigen-IgE antibody complex is formed upon entry of ketamine metabolites into the bladder, which recruits eosinophils and further enhances the inflammatory response (Teegavarapu et al. 2005). Moreover, a separate study has suggested elevated IgE levels are strongly associated with hypersensitivity, where chronic ketamine abusers with KIUT experience more intense bladder pain (Jhang et al. 2014).
In addition, nerve hyperplasia has been noticed in the bladder tissue of chronic ketamine abusers with KIUT and could be the cause of hypersensitivity (Jhang et al. 2019). Significant nerve bundles and stromal neurogenesis are observed in the deep lamina propria of the urothelium (Baker et al. 2013; Jhang et al. 2018). The expression levels of neurotrophins, such as nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), growth-associated protein 43 (GAP-43), tropomyosin receptor kinase A (Trk A), and their receptors are upregulated in the bladder mucosa of chronic ketamine abusers with KIUT (Jhang et al. 2019). Neurotrophins in the bladder are responsible for regulating sensory afferents and modulating the growth of neurons; hence, their upregulation could result in nerve hyperplasia and ultimately hypersensitivity (Cruz 2014; Jhang et al. 2019). The increased levels of neurotrophins are also shown to be correlated with bladder pain in chronic ketamine abusers with KIUT (Jhang et al. 2019). Nonetheless, some studies have shown opposite results where the expression levels of neurotrophins are lower in chronic ketamine abusers with KIUT (Ke et al. 2014). The difference in results may be due to the small sample size acquired in each study. Thus, future studies involving a larger sample size are warranted.
Cell apoptosis
Apoptosis of bladder tissue is another possible mechanism of KIUT. Increased apoptosis is observed in the bladder tissue of patients with interstitial cystitis/painful bladder syndrome (IC/PBS), overactive bladder of rats, and CP-induced cystitis (Jezernik et al. 2003; Lee et al. 2011; Shie et al. 2012). Similarly, there is a higher number of apoptotic cells in the bladder tissue of chronic ketamine abusers with KIUT (Lee et al. 2013). In addition, Song et al. (2016) reported that an overexpression of apoptotic factor B cell leukemia/lymphoma-2-associated protein (BAX) in the bladders of rats with KIUT. Apoptotic cells in the bladder may undergo secondary necrosis, a process where the cell membrane is permeable to macromolecules that eventually leads to inflammatory responses (Jhang et al. 2015). However, the causal link between cell apoptosis and KIUT-associated bladder inflammation remains undetermined. Further investigations on this relationship are required.
Microvascular damage
In a human KIUT bladder biopsy, the tortuous shape of microvessels and a thicker basement membrane are seen in the endothelium (Lin et al. 2016). The NMDARs are also found on endothelial cells in the bladder; thus, it has been speculated that the antagonism of these receptors by ketamine results in vessel impairment and chronic inflammation (Lin et al. 2016). Microvascular injury in the bladder may cause local ischemia as well, leading to dysuria and pelvic pain (Lin et al. 2016). To date, there is limited data on this possible mechanism of KIUT. More studies are needed to illustrate the importance of microvascular damage in the pathogenesis of KIUT.
Overexpression of carcinogenic genes
Currently, no evidence suggests the development of bladder cancer in chronic ketamine abusers with KIUT, although there are particular features in KIUT that are associated with urothelial carcinoma (Jhang et al. 2015). Urothelial atypia has been found in some chronic ketamine abusers with KIUT, where the enlargement and loss of polarity in urothelial cells are observed and resemble carcinoma in situ (Oxley et al. 2009). Furthermore, moderate to high immunoreactivity of tumor suppressor p53 and cell proliferation marker Ki-67 is found in bladders affected by KIUT, which indicates the presence of carcinoma in situ (Lopez-Beltran et al. 2013; Yin et al. 2006).
Gu et al. (2013) has reported a higher expression of phosphorylated transgelin bladder tissue in animals with KIUT. Transgelin, an actin-binding protein that regulates the actin cytoskeleton, is usually present in smooth muscle and some epithelial tissue (Assinder et al. 2009; Yin et al. 2019). Transgelin might also act as a tumor suppressor, where its loss of expression is seen in prostate, breast, and colon cancers (Assinder et al. 2009). It has been speculated that phosphorylation of transgelin would inactivate the actin-binding process and possibly the tumor-suppressing function of the protein, which could lead to the development of cancer in the organ (Leung et al. 2011).
Lastly, the expression of intercellular junction proteins (E-cadherin) is reduced in bladders affected by KIUT, which may lead to urothelial barrier damage and subsequent inflammatory responses (Wang et al. 2017). Previous studies have observed that the low expression of E-cadherin is correlated with cancer progression (Popov et al. 2000). Carcinogenic factors could thus be a downstream response to persistent inflammation in KIUT of the bladder. Long-term follow-up in chronic ketamine abusers with KIUT will be necessary to explore the causal relationship between KIUT and urothelial carcinoma.
Discussion
Herein, we identified multiple mechanisms that may be involved in the pathophysiology of KIUT, such as urothelial barrier disruption, inflammation in the bladder, ketamine direct toxicity, nerve hyperplasia and hypersensitivity, cell apoptosis, microvascular damage, and overexpression of carcinogenic genes. The foregoing constellation of possible pathophysiological mechanisms that may contribute to KIUT indicates that the actual pathophysiology of KIUT is complex and may result from a combination of several pathways.
The critical pathological change in KIUT is persistent bladder inflammation. It is believed that ketamine in the urine leads to elevated neurotrophin levels and subsequently neurogenic inflammation, induces IgE-mediated inflammation, and stimulates the NOS-COX-prostaglandin pathway (Castellani et al. 2020; Jhang et al. 2015). Chronic inflammation in the bladder could result in collagen deposition and fibrosis, which may cause severe bladder pain and increased urinary frequency. Chronic ketamine abuse could also exert direct toxicity to the bladder, damage the urothelium barrier function, and induce cell apoptosis. Furthermore, the overexpression of carcinogenic genes may be a downstream response to KIUT bladder inflammation.
Nevertheless, there is a limitation to the generalizability of KIUT to the urological symptoms observed in IV ketamine and IN esketamine trials. Chronic ketamine abusers often use multiple substances besides ketamine such as amphetamine, cocaine, and alcohol (Winstock et al. 2012). Thus, it is unclear how much of the urological symptoms can be attributed to ketamine purely.
In recent years, IV ketamine has shown acute antidepressant effects in TRD when administered in sub-anesthetic, repeat doses (0.5–1.0 mg/kg every 1–4 weeks) (aan het Rot et al. 2010; Rodrigues et al. 2020; Singh et al. 2016). It appears to be well-tolerated and effective in the short term (< 6 months). However, the safety profile of long-term IV ketamine treatment (≥ 6 months) in persons with TRD are currently unknown. To date, there is no evidence suggesting ketamine treatment in mood disorders is associated with urological toxicity. For IN esketamine, two clinical studies have reported a low occurrence (< 10%) of urological complications at antidepressant doses (28–84 mg) (Daly et al. 2019; Wajs et al. 2020). Nonetheless, extant data on long-term safety profile (≥ 6 months) of IN esketamine are also limited. To ensure they are feasible, long-term treatment options, more research is needed to examine the safety of long-term IV ketamine and IN esketamine administration (≥ 6 months). Additionally, a more comprehensive understanding of KIUT mechanisms would further inform safety measures in clinical trials and clinical interventions with IV ketamine/IN esketamine and TRD. Therefore, further investigations are required to address these issues. In the interim, the risk of KIUT should be discussed as part of informed consent for long-term IV ketamine treatment (≥ 6 months), along with routine monitoring of urological signs and symptoms by ketamine providers (Sanacora et al. 2017).
Conclusion
Prior research suggests that the pathogenesis of KIUT is comprised of complex interactions between several physiological pathways. One key pathological change in KIUT is bladder inflammation, which could be the result of neurogenic inflammation, IgE-mediated inflammation, and stimulation of the NOS-COX-prostaglandin pathway. Ketamine could also inflict damage to the urothelium directly and enhance cell apoptosis. Notwithstanding the observations in persons using high-frequency ketamine recreationally, there is no evidence that ketamine or esketamine treatment in mood disorders is associated with KIUT. Despite the absence of KIUT in persons with mood disorders receiving ketamine/esketamine treatment, clinicians are encouraged to inform patients of the potential risks and should specifically probe patients for symptoms and signs that are suggestive of KIUT.
References
aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, Mathew SJ (2010) Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry 67:139–145. https://doi.org/10.1016/j.biopsych.2009.08.038
Assinder SJ, Stanton JAL, Prasad PD (2009) Transgelin: an actin-binding protein and tumour suppressor. Int J Biochem Cell Biol 41:482–486. https://doi.org/10.1016/j.biocel.2008.02.011
Baker SC, Stahlschmidt J, Oxley J, Hinley J, Eardley I, Marsh F, Gillatt D, Fulford S, Southgate J (2013) Nerve hyperplasia: A unique feature of ketamine cystitis. Acta Neuropathol Commun 1:64. https://doi.org/10.1186/2051-5960-1-64
Baker SC, Shabir S, Georgopoulos NT, Southgate J (2016) Ketamine-induced apoptosis in normal human urothelial cells: a direct, N-methyl-d-aspartate receptor-independent pathway characterized by mitochondrial stress. Am J Pathol 186:1267–1277. https://doi.org/10.1016/j.ajpath.2015.12.014
Castellani D, Pirola GM, Gubbiotti M, Rubilotta E, Gudaru K, Gregori A, Dellabella M (2020) What urologists need to know about ketamine-induced uropathy: a systematic review. Neurourol Urodyn 39:1049–1062. https://doi.org/10.1002/nau.24341
Chen JT, Chen RM (2010) Mechanisms of ketamine-involved regulation of cytochrome P450 gene expression. Expert Opin Drug Metab Toxicol 6:273–281. https://doi.org/10.1517/17425250903505108
Chen Z, Liu M, Liu X, Huang S, Li L, Song B, Li H, Ren Q, Hu Z, Zhou Y, Qiao L (2013) COX-2 regulates E-cadherin expression through the NF-κB/Snail signaling pathway in gastric cancer. Int J Mol Med 32:93–100. https://doi.org/10.3892/ijmm.2013.1376
Chen WY, Huang MC, Lin SK (2014) Gender differences in subjective discontinuation symptoms associated with ketamine use. Subst Abuse Treat Prev Policy 9:39. https://doi.org/10.1186/1747-597X-9-39
Chu PSK, Ma WK, Wong SCW, Chu RWH, Cheng CH, Wong S, Tse JML, Lau FL, Yiu MK, Man CW (2008) The destruction of the lower urinary tract by ketamine abuse: a new syndrome? BJU Int 102:1616–1622. https://doi.org/10.1111/j.1464-410X.2008.07920.x
Chuang SM, Liu KM, Li YL, Jang MY, Lee HH, Wu WJ, Chang WC, Levin RM, Juan YS (2013) Dual involvements of cyclooxygenase and nitric oxide synthase expressions in ketamine-induced ulcerative cystitis in rat bladder. Neurourol Urodyn 32:1137–1143. https://doi.org/10.1002/nau.22367
Cruz CD (2014) Neurotrophins in bladder function: what do we know and where do we go from here? Neurourol Urodyn 33:39–45. https://doi.org/10.1002/nau.22438
Daly EJ, Singh JB, Fedgchin M, Cooper K, Lim P, Shelton RC, Thase ME, Winokur A, Van Nueten L, Manji H, Drevets WC (2018) Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression. JAMA Psychiatry 75:139–148. https://doi.org/10.1001/jamapsychiatry.2017.3739
Daly EJ, Trivedi MH, Janik A, Li H, Zhang Y, Li X, Lane R, Lim P, Duca AR, Hough D, Thase ME, Zajecka J, Winokur A, Divacka I, Fagiolini A, Cubala WJ, Bitter I, Blier P, Shelton RC, Molero P, Manji H, Drevets WC, Singh JB (2019) Efficacy of esketamine nasal spray plus oral antidepressant treatment for relapse prevention in patients with treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry 76:893–903. https://doi.org/10.1001/jamapsychiatry.2019.1189
Duan Q, Wu T, Yi X, Liu L, Yan J, Lu Z (2017) Changes to the bladder epithelial barrier are associated with ketamine-induced cystitis. Exp Ther Med 14:2757–2762. https://doi.org/10.3892/etm.2017.4913
Enguita FJ, Leitão AL (2013) Hydroquinone: environmental pollution, toxicity, and microbial answers. Biomed Res Int 2013:542168–542114. https://doi.org/10.1155/2013/542168
Feifel D, Dadiomov D, Lee C K (2020) Safety of repeated administration of parenteral ketamine for depression. Pharmaceuticals (Basel) 13. https://doi.org/10.3390/ph13070151
Findeis H, Sauer C, Cleare A, Bauer M, Ritter P (2020) Urothelial toxicity of esketamine in the treatment of depression. Psychopharmacology (Berl). 237:3295–3302. https://doi.org/10.1007/s00213-020-05611-y
Grant IS, Nimmo WS, Clements JA (1981) Pharmacokinetics and analgesic effects of i.m. and oral ketamine. Br J Anaesth 53:805–810. https://doi.org/10.1093/bja/53.8.805
Gu D, Huang J, Shan Z, Yin Y, Zheng S, Wu P (2013) Effects of long-term ketamine administration on rat bladder protein levels: a proteomic investigation using two-dimensional difference gel electrophoresis system. Int J Urol 20:1024–1031. https://doi.org/10.1111/iju.12100
Gu D, Huang J, Yin Y, Shan Z, Zheng S, Wu P (2014) Long-term ketamine abuse induces cystitis in rats by impairing the bladder epithelial barrier. Mol Biol Rep 41:7313–7322. https://doi.org/10.1007/s11033-014-3616-5
Jezernik K, Romih R, Mannherz HG, Koprivec D (2003) Immunohistochemical detection of apoptosis, proliferation and inducible nitric oxide synthase in rat urothelium damaged by cyclophosphamide treatment. Cell Biol Int 27:863–869. https://doi.org/10.1016/s1065-6995(03)00175-6
Jhang JF, Hsu YH, Jiang YH, Kuo HC (2014) Elevated serum IgE may be associated with development of ketamine cystitis. J Urol 192:1249–1256. https://doi.org/10.1016/j.juro.2014.05.084
Jhang JF, Hsu YH, Kuo HC (2015) Possible pathophysiology of ketamine-related cystitis and associated treatment strategies. Int J Urol 22:816–825. https://doi.org/10.1111/iju.12841
Jhang JF, Hsu YH, Jiang YH, Kuo HC (2016) The role of immunoglobulin E in the pathogenesis of ketamine related cystitis and ulcerative interstitial cystitis: an immunohistochemical study. Pain Physician 19:E581–E587
Jhang JF, Hsu YH, Jiang YH, Lee CL, Kuo HC (2018) Histopathological characteristics of ketamine-associated uropathy and their clinical association. Neurourol Urodyn 37:1764–1772. https://doi.org/10.1002/nau.23514
Jhang JF, Wang HJ, Hsu YH, Birder LA, Kuo HC (2019) Upregulation of neurotrophins and transforming growth factor-β expression in the bladder may lead to nerve hyperplasia and fibrosis in patients with severe ketamine-associated cystitis. Neurourol Urodyn 38:2303–2310. https://doi.org/10.1002/nau.24139
Juan YS, Lee YL, Long CY, Wong JH, Jang MY, Lu JH, Wu WJ, Huang YS, Chang WC, Chuang SM (2015) Translocation of NF-κB and expression of cyclooxygenase-2 are enhanced by ketamine-induced ulcerative cystitis in rat bladder. Am J Pathol 185:2269–2285. https://doi.org/10.1016/j.ajpath.2015.04.020
Ke X, Ding Y, Xu K, He H, Zhang M, Wang D, Deng X, Zhang X, Zhou C, Liu Y, Ning Y, Fan N (2014) Serum brain-derived neurotrophic factor and nerve growth factor decreased in chronic ketamine abusers. Drug Alcohol Depend 142:290–294. https://doi.org/10.1016/j.drugalcdep.2014.06.043
Kim J, Farchione T, Potter A, Chen Q, Temple R (2019) Esketamine for treatment-resistant depression—first FDA-approved antidepressant in a new class. N Engl J Med 381:1–4. https://doi.org/10.1056/NEJMp1903305
Kryst J, Kawalec P, Mitoraj AM, Pilc A, Lasoń W, Brzostek T (2020) Efficacy of single and repeated administration of ketamine in unipolar and bipolar depression: a meta-analysis of randomized clinical trials. Pharmacol Rep 72:543–562. https://doi.org/10.1007/s43440-020-00097-z
Lai Y, Wu S, Ni L, Chen Z, Li X, Yang S, Gui Y, Guan Z, Cai Z, Ye J (2012) Ketamine-associated urinary tract dysfunction: an underrecognized clinical entity. Urol Int 89:93–96. https://doi.org/10.1159/000338098
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15:178–196. https://doi.org/10.1038/nrm3758
Lee P, Ong T, Chua C, Lei C, Teh G (2009) Street ketamine-associated bladder dysfunction: an emerging health problem. Malays Fam Physician 4:15–18
Lee WC, Chuang YC, Chiang PH, Chien CT, Yu HJ, Wu CC (2011) Pathophysiological studies of overactive bladder and bladder motor dysfunction in a rat model of metabolic syndrome. J Urol 186:318–325. https://doi.org/10.1016/j.juro.2011.03.037
Lee CL, Jiang YH, Kuo HC (2013) Increased apoptosis and suburothelial inflammation in patients with ketamine-related cystitis: a comparison with non-ulcerative interstitial cystitis and controls. BJU Int 112:1156–1162. https://doi.org/10.1111/bju.12256
Leung WKC, Ching AKK, Chan AWH, Poon TCW, Mian H, Wong AST, To KF, Wong N (2011) A novel interplay between oncogenic PFTK1 protein kinase and tumor suppressor TAGLN2 in the control of liver cancer cell motility. Oncogene 30:4464–4475. https://doi.org/10.1038/onc.2011.161
Lin HC, Lee HS, Chiueh TS, Lin YC, Lin HA, Lin YC, Cha TL, Meng E (2015) Histopathological assessment of inflammation and expression of inflammatory markers in patients with ketamine-induced cystitis. Mol Med Rep 11:2421–2428. https://doi.org/10.3892/mmr.2014.3110
Lin CC, Lin ATL, Yang AH, Chen KK (2016) Microvascular injury in ketamine-induced bladder dysfunction. PloS One 11:e0160578. https://doi.org/10.1371/journal.pone.0160578
Lopez-Beltran A, Montironi R, Vidal A, Scarpelli M, Cheng L (2013) Urothelial dysplasia of the bladder: diagnostic features and clinical significance. Anal Quant Cytopathol Histpathol 35:121–129
Malinovsky JM, Servin F, Cozian A, Lepage JY, Pinaud M (1996) Ketamine and norketamine plasma concentrations after i.v., nasal and rectal administration in children. Br J Anaesth 77:203–207. https://doi.org/10.1093/bja/77.2.203
McIntyre RS, Rodrigues NB, Lee Y, Lipsitz O, Subramaniapillai M, Gill H, Nasri F, Majeed A, Lui LMW, Senyk O, Phan L, Carvalho IP, Siegel A, Mansur RB, Brietzke E, Kratiuk K, Arekapudi AK, Abrishami A, Chau EH et al (2020) The effectiveness of repeated intravenous ketamine on depressive symptoms, suicidal ideation and functional disability in adults with major depressive disorder and bipolar disorder: Results from the Canadian Rapid Treatment Center of Excellence. J Affect Disord 274:903–910. https://doi.org/10.1016/j.jad.2020.05.088
Morgan CJA, Curran HV, Independent Scientific Committee on Drugs (2012) Ketamine use: a review. Addiction 107:27–38. https://doi.org/10.1111/j.1360-0443.2011.03576.x
Myers FA, Bluth MH, Cheung WW (2016) Ketamine: a cause of urinary tract dysfunction. Clin Lab Med 36:721–744. https://doi.org/10.1016/j.cll.2016.07.008
Na KS, Kim YK (2021) Increased use of ketamine for the treatment of depression: benefits and concerns. Prog Neuropsychopharmacol Biol Psychiatry 104:110060. https://doi.org/10.1016/j.pnpbp.2020.110060
Oxley JD, Cottrell AM, Adams S, Gillatt D (2009) Ketamine cystitis as a mimic of carcinoma in situ. Histopathology 55:705–708. https://doi.org/10.1111/j.1365-2559.2009.03437.x
Pappachan JM, Raj B, Thomas S, Hanna FW (2014) Multiorgan dysfunction related to chronic ketamine abuse. Proc (Bayl Univ Med Cent) 27:223–225. https://doi.org/10.1080/08998280.2014.11929117
Parkin MC, Turfus SC, Smith NW, Halket JM, Braithwaite RA, Elliott SP, Osselton MD, Cowan DA, Kicman AT (2008) Detection of ketamine and its metabolites in urine by ultra high pressure liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 876:137–142. https://doi.org/10.1016/j.jchromb.2008.09.036
Payne H, Adamson A, Bahl A, Borwell J, Dodds D, Heath C, Huddart R, McMenemin R, Patel P, Peters JL, Thompson A (2013) Chemical- and radiation-induced haemorrhagic cystitis: Current treatments and challenges. BJU Int 112:885–897. https://doi.org/10.1111/bju.12291
Peltoniemi MA, Hagelberg NM, Olkkola KT, Saari TI (2016) Ketamine: a review of clinical pharmacokinetics and pharmacodynamics in anesthesia and pain therapy. Clin Pharmacokinet 55:1059–1077. https://doi.org/10.1007/s40262-016-0383-6
Popov Z, Gil-Diez de Medina S, Lefrere-Belda MA, Hoznek A, Bastuji-Garin S, Abbou CC, Thiery JP, Radvanyi F, Chopin DK (2000) Low E-cadherin expression in bladder cancer at the transcriptional and protein level provides prognostic information. Br J Cancer 83:209–214. https://doi.org/10.1054/bjoc.2000.1233
Rodrigues NB, McIntyre RS, Lipsitz O, Lee Y, Cha DS, Nasri F, Gill H, Lui LMW, Subramaniapillai M, Kratiuk K, Lin K, Ho R, Mansur RB, Rosenblat JD (2020) Safety and tolerability of IV ketamine in adults with major depressive or bipolar disorder: results from the Canadian rapid treatment center of excellence. Expert Opin Drug Saf 19:1031–1040. https://doi.org/10.1080/14740338.2020.1776699
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, McGrath PJ, Rosenbaum JF, Sackeim HA, Kupfer DJ, Luther J, Fava M (2006) Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry 163:1905–1917. https://doi.org/10.1176/ajp.2006.163.11.1905
Sanacora G, Frye MA, McDonald W, Mathew SJ, Turner MS, Schatzberg AF, Summergrad P, Nemeroff CB, American Psychiatric Association (APA) Council of Research Task Force on Novel Biomarkers and Treatments (2017) A consensus statement on the use of ketamine in the treatment of mood disorders. JAMA Psychiatry 74:399–405. https://doi.org/10.1001/jamapsychiatry.2017.0080
Shahani R, Streutker C, Dickson B, Stewart RJ (2007) Ketamine-associated ulcerative cystitis: a new clinical entity. Urology 69:810–812. https://doi.org/10.1016/j.urology.2007.01.038
Shie JH, Liu HT, Kuo HC (2012) Increased cell apoptosis of urothelium mediated by inflammation in interstitial cystitis/painful bladder syndrome. Urology 79:484.e7-13. https://doi.org/10.1016/j.urology.2011.09.049
Short B, Fong J, Galvez V, Shelker W, Loo CK (2018) Side-effects associated with ketamine use in depression: a systematic review. Lancet Psychiatry 5:65–78. https://doi.org/10.1016/S2215-0366(17)30272-9
Singh JB, Fedgchin M, Daly EJ, De Boer P, Cooper K, Lim P, Pinter C, Murrough JW, Sanacora G, Shelton RC, Kurian B, Winokur A, Fava M, Manji H, Drevets WC, Van Nueten L (2016) A double-blind, randomized, placebo-controlled, dose-frequency study of intravenous ketamine in patients with treatment-resistant depression. Am J Psychiatry 173:816–826. https://doi.org/10.1176/appi.ajp.2016.16010037
Song M, Yu HY, Chun JY, Shin DM, Song SH, Choo MS, Song YS (2016) The fibrosis of ketamine, a noncompetitive N-methyl-d-aspartic acid receptor antagonist dose-dependent change in a ketamine-induced cystitis rat model. Drug Chem Toxicol 39:206–212. https://doi.org/10.3109/01480545.2015.1079916
Tan S, Chan WM, Wai MSM, Hui LKK, Hui VWK, James AE, Yeung LY, Yew DT (2011) Ketamine effects on the urogenital system—changes in the urinary bladder and sperm motility. Microsc Res Tech 74:1192–1198. https://doi.org/10.1002/jemt.21014
Teegavarapu PS, Sahai A, Chandra A, Dasgupta P, Khan MS (2005) Eosinophilic cystitis and its management. Int J Clin Pract 59:356–360. https://doi.org/10.1111/j.1742-1241.2004.00421.x
Thiery JP, Acloque H, Huang RYJ, Nieto MA (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139:871–890. https://doi.org/10.1016/j.cell.2009.11.007
Wai MSM, Luan P, Jiang Y, Chan WM, Tsui TYM, Tang HC, Lam WP, Fan M, Yew DT (2013) Long term ketamine and ketamine plus alcohol toxicity—what can we learn from animal models? Mini Rev Med Chem 13:273–279. https://doi.org/10.2174/1389557511313020009
Wajs E, Aluisio L, Holder R, Daly EJ, Lane R, Lim P, George JE, Morrison RL, Sanacora G, Young AH, Kasper S, Sulaiman AH, Li CT, Paik JW, Manji H, Hough D, Grunfeld J, Jeon HJ, Wilkinson ST, Drevets WC, Singh JB (2020) Esketamine nasal spray plus oral antidepressant in patients with treatment-resistant depression: assessment of long-term safety in a phase 3, open-label study (SUSTAIN-2). J Clin Psychiatry 81. https://doi.org/10.4088/JCP.19m12891
Walker ER, McGee RE, Druss BG (2015) Mortality in mental disorders and global disease burden implications: a systematic review and meta-analysis. JAMA Psychiatry 72:334–341. https://doi.org/10.1001/jamapsychiatry.2014.2502
Wang J, Chen Y, Gu D, Zhang G, Chen J, Zhao J, Wu P (2017) Ketamine-induced bladder fibrosis involves epithelial-to-mesenchymal transition mediated by transforming growth factor-β1. Am J Physiol Renal Physiol 313:F961–F972. https://doi.org/10.1152/ajprenal.00686.2016
Wei YB, Yang JR, Yin Z, Guo Q, Liang BL, Zhou KQ (2013) Genitourinary toxicity of ketamine. Hong Kong Med J 19:341–348. https://doi.org/10.12809/hkmj134013
Winstock AR, Mitcheson L, Gillatt DA, Cottrell AM (2012) The prevalence and natural history of urinary symptoms among recreational ketamine users. BJU Int 110:1762–1766. https://doi.org/10.1111/j.1464-410X.2012.11028.x
Wynn TA, Ramalingam TR (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med 18:1028–1040. https://doi.org/10.1038/nm.2807
Yin H, He Q, Li T, Leong ASY (2006) Cytokeratin 20 and Ki-67 to distinguish carcinoma in situ from flat non-neoplastic urothelium. Appl Immunohistochem Mol Morphol 14:260–265. https://doi.org/10.1097/00129039-200609000-00002
Yin LM, Ulloa L, Yang YQ (2019) Transgelin-2: Biochemical and clinical implications in cancer and asthma. Trends Biochem Sci 44:885–896. https://doi.org/10.1016/j.tibs.2019.05.004
Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, Pereira EFR, Albuquerque EX, Thomas CJ, Zarate CA, Gould TD (2018) Ketamine and ketamine metabolite pharmacology: insights into therapeutic mechanisms. Pharmacol Rev 70:621–660. https://doi.org/10.1124/pr.117.015198
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Roger McIntyre has received research grant support from CIHR/GACD/Chinese National Natural Research Foundation; speaker/consultation fees from Lundbeck, Janssen, Purdue, Pfizer, Otsuka, Allergan, Takeda, Neurocrine, Sunovion, Minerva, Intra-Cellular, Abbvie, and Eisai. Dr. Roger McIntyre is a shareholder and CEO of Champignon.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ng, J., Lui, L.M.W., Rosenblat, J.D. et al. Ketamine-induced urological toxicity: potential mechanisms and translation for adults with mood disorders receiving ketamine treatment. Psychopharmacology 238, 917–926 (2021). https://doi.org/10.1007/s00213-021-05767-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-021-05767-1