
Abstract
Rationale
Anticipatory nausea (AN) is a poorly controlled side effect experienced by chemotherapy patients. Currently, pharmacotherapy is restricted to benzodiazepine anxiolytics, which have limited efficacy, have significant sedative effects and induce dependency. The non-psychoactive phytocannabinoid, cannabidiolic acid (CBDA), has shown considerable efficacy in pre-clinical AN models, however determination of its neuromotor tolerability profile is crucial to justify clinical investigation. Provisional evidence for appetite-stimulating properties also requires detailed investigation.
Objectives
This study aims to assess the tolerability of CBDA in locomotor activity, motor coordination and muscular strength tests, and additionally for ability to modulate feeding behaviours.
Methods
Male Lister Hooded rats administered CBDA (0.05–5 mg/kg; p.o.) were assessed in habituated open field (for locomotor activity), static beam and grip strength tests. A further study investigated whether these CBDA doses modulated normal feeding behaviour. Finally, evidence of anxiolytic-like effects in the habituated open field prompted testing of 5 mg/kg CBDA for anxiolytic-like activity in unhabituated open field, light/dark box and novelty-suppressed feeding (NSF) tests.
Results
CBDA had no adverse effects upon performance in any neuromotor tolerability test, however anxiolytic-like behaviour was observed in the habituated open field. Normal feeding behaviours were unaffected by any dose. CBDA (5 mg/kg) abolished the increased feeding latency in the NSF test induced by the 5-HT1AR antagonist, WAY-100,635, indicative of anxiolytic-like effects, but had no effect on anxiety-like behaviour in the novel open field or light/dark box.
Conclusions
CBDA is very well tolerated and devoid of the sedative side effect profile of benzodiazepines, justifying its clinical investigation as a novel AN treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chemotherapy treatment commonly causes distressing and debilitating side effects in cancer patients, including acute and delayed vomiting (Martin 1996); acute, delayed and anticipatory nausea (Rock et al. 2014b); reduced food intake and bodyweight (Hainsworth and Hesketh 1992); and fatigue (Ahlberg et al. 2003). These chemotherapy-induced nausea and vomiting (CINV) symptoms are highly distressing for patients, adversely affecting quality of life to the point where some will delay and even consider refusing future cycles of chemotherapy treatment (Janelsins et al. 2013). It is estimated that, without prophylaxis, CINV would be experienced by up to 80 % of patients, with prevalence and severity varying according to the individual chemotherapy regimen employed (dos Santos et al. 2012). Many commonly prescribed chemotherapy drugs are classified as highly emetogenic within typical dose ranges, including cisplatin, cyclophosphamide (>1500 mg/m2) and carmustine, all of which lead to CINV in >90 % of patients without effective prophylaxis (Hesketh 2008; Roila et al. 2010). Cisplatin, the most extensively studied highly emetogenic chemotherapy drug, elicits a biphasic CINV response, comprising an acute phase (within 24 h) and delayed phase (24–120 h), each with distinct pathogeneses and sensitivities to anti-emetic treatments (Martin 1996).
The effective control of the acute phase of CINV is achieved in approximately half of patients undergoing highly emetic chemotherapy using 5-HT3 receptor antagonists (e.g. ondansetron) in combination with a glucocorticoid (e.g. dexamethasone). However, the delayed phase of CINV remains poorly controlled by this combination of drugs (Hickok et al. 2003). More recently, it has been shown that adjunctive use of neurokinin 1 (NK1) receptor antagonists (e.g. apripitant) with conventional anti-emetic treatment regimens can significantly reduce the incidence of delayed vomiting (Navari et al. 1999; Campos et al. 2001; Hesketh et al. 2003). Indeed, the combination therapy of NK1 receptor antagonist, 5HT3R antagonist and dexamethasone is now strongly recommended for CINV prophylaxis in highly emetogenic regimes as it provides complete control of vomiting in both the acute and delayed phases of CINV in 60–70 % of patients (Kris et al. 2006; Roila et al. 2010). Despite these advances, the control of delayed nausea, and the consequences of incomplete control of acute and/or delayed vomiting, remains problematic and requires new antiemetic strategies (Hesketh 2008; Janelsins et al. 2013).
Incomplete or ineffective control of nausea can cause increased anxiety, depression and the development of anticipatory nausea in patients (Rock et al. 2014b). Anticipatory nausea (AN) manifests as nausea (sometimes accompanied by vomiting) prior to administration of chemotherapy, and occurs in up to 20 % of patients before any one chemotherapy cycle and in up to 30 % of patients by the fourth cycle (Roscoe et al. 2011). AN is widely considered to be a form of Pavlovian classical conditioning, in which the cues of the clinical environment become associated with the nausea and vomiting induced by chemotherapy (Nesse et al. 1980; Matteson et al. 2002) and, as such, is not controlled by standard antiemetic treatments (Janelsins et al. 2013; Kamen et al. 2014). Furthermore, once AN has developed, patients also report more severe acute nausea following subsequent cycles of chemotherapy (Bovbjerg 2006).
At present, treatment options for AN remain limited, with clinical recommendations focussed on prophylaxis against the initial manifestation of AN through adequate control of acute nausea and vomiting (Basch et al. 2012). In patients who develop AN due to a failure of adequate control, recommendations are limited to behavioural interventions such as systemic desensitisation and progressive muscle relaxation (Figueroa-Moseley et al. 2007) or the use of non-specific benzodiazepine anxiolytic drugs (Kamen et al. 2014). While behavioural interventions, in particular systemic desensitisation, are considered the most promising option currently available, a systematic review has highlighted the limited evidence for their efficacy (Lotfi-Jam et al. 2008). Furthermore, a lack of suitably trained personnel in treatment settings has been identified as an ongoing difficulty for the implementation of such interventions (Roscoe et al. 2011). The use of benzodiazepine anxiolytics is supported by two small clinical trials. Razavi et al. (1993) investigated the use of alprazolam as an adjunct to psychological support to prevent AN in 57 women undergoing adjuvant chemotherapy for breast cancer. They found a significant reduction in AN rate at second assessment (0 vs 18 %), concluding alprazolam treatment delays the occurrence of AN. In a larger trial where lorazepam was administered with anti-emetic therapy (metoclopramide, clemastine and dexamethasone) in 180 patients receiving high-dose cisplatin, Malik et al. (1995) reported a significantly higher complete response to AN in patients receiving lorazepam (52 vs 35 %); however, these patients also experienced significantly higher occurrences of sedation (92 vs 37 %) and amnesia (32 vs 1 %). In addition to the debilitating side effects and dependency induced by benzodiazepine treatment, their efficacy against AN is also reduced during multiple chemotherapy cycles (Roila et al. 2010). Thus, there remains an unmet clinical need for convenient, effective and well-tolerated pharmacotherapies for AN.
Recently, a number of pre-clinical studies have identified the non-psychoactive phytocannabinoid, cannabidiolic acid (CBDA), as a potential novel pharmacotherapy for the treatment of AN (Bolognini et al. 2013; Rock and Parker 2013a; Rock and Parker 2013b; Rock et al. 2014a). Parker and colleagues assessed the ability of CBDA and a number of other phytocannabinoids to prevent cisplatin- or lithium chloride (LiCl)-induced vomiting (a model of acute vomiting) in house musk shrews and in rats to prevent LiCl-induced gaping (a model of acute nausea) or context-induced gaping (a conditioned model of AN). The potential for these drugs to enhance saccharin palatability was also assessed in the latter model (see Rock et al. 2014b for review of animal models). In studies using CBDA, low doses (0.01–0.5 mg/kg; i.p.) attenuated acute vomiting in shrews, and both acute and anticipatory nausea in rats, with the latter effect blocked by the 5-HT1AR antagonist WAY-100,635 (Bolognini et al. 2013). The same study reported an enhancement of saccharin palatability, as measured by unconditioned hedonic reactions. Further studies demonstrated that subthreshold doses of CBDA (0.1–0.5 μg/kg) potentiated the suppression of acute nausea by the anti-emetics ondansetron or metoclopramide (Rock and Parker 2013a; Rock and Parker 2013b). When CBDA was compared to the anti-emetic ondansetron or the benzodiazepine anxiolytic chlordiazepoxide (CDP) in the rat model of AN, both CBDA and CDP showed considerable efficacy, while ondansetron was ineffective (Rock et al. 2014a). Interestingly, in this study, rats were tested in an activity chamber for 15 min immediately following the AN trial, which demonstrated the expected benzodiazepine-induced suppression of locomotor activity in rats treated with CDP, but not in those with CBDA. These studies demonstrate that, in rodent models, CBDA is a highly potent treatment for both AN and acute nausea and vomiting. They also provide limited data to suggest CBDA may stimulate aspects of feeding under non-pathological conditions and lack the sedative effects of benzodiazepine anxiolytics.
To justify clinical investigation of CBDA as a novel AN treatment, it is crucial that the neuromotor tolerability profile is investigated in detail, to determine whether or not it elicits the side effects which have compromised the utility of benzodiazepines for this indication. Rock et al. (2014a) have shown that CBDA does not suppress spontaneous locomotor activity at doses ≤1 mg/kg; however, this represents the sum total of published tolerability data for CBDA. To provide a more complete assessment of CBDA tolerability, the first part of our study administered CBDA to rats across a greater dose range (0.05–5 mg/kg), after which they completed a battery of tests for effects on locomotor activity, balance, fine motor control and muscular strength. The previously reported observation that CBDA enhanced unconditioned saccharin palatability raises the intriguing possibility that CBDA may directly stimulate feeding behaviour, and thus may have additional therapeutic potential for the attenuation of chemotherapy-induced anorexia and/or cancer cachexia. In the second part of our study, we directly assessed the potential hyperphagic actions of CBDA using a well-established pre-feed paradigm for investigation of hyperphagic activity, which we have previously demonstrated for a number of other phytocannabinoids (Williams et al. 1998; Williams and Kirkham 2002; Farrimond et al. 2012a; Farrimond et al. 2012b).
Although the primary aims of this study were to determine the neuromotor tolerability and feeding behaviour profiles of CBDA, an additional follow-up experiment was also conducted to assess the anxiolytic effects of CBDA. During our battery of locomotor tasks, there was the suggestion of putative anxiolytic-like effects seen in the habituated open field test. As a final experiment therefore, using three tests of anxiety-like behaviour, CBDA was assessed alone and in combination with WAY-100,635, a 5-HT1AR silent antagonist, as this receptor has previously been shown to block the effects of CBDA in models of acute and anticipatory nausea (Bolognini et al. 2013).
Methods
Drugs
CBDA (GW Pharmaceuticals, UK) was dissolved directly into sesame oil (by sonication at room temperature) to a maximal working concentration of 5 mg/ml. Working solutions of 0.5 and 0.05 mg/ml were prepared by serial dilution in sesame seed oil. WAY-100,635 (Sigma-Aldrich, UK) was dissolved directly into sterile 0.9 % saline vehicle (by vortex at room temperature), with a working concentration of 0.1 mg/ml prepared from frozen aliquots of 1 mg/ml stock solution. All drugs were prepared freshly each test day and protected from light until administration.
CBDA or sesame seed oil vehicle were administered per orem (p.o.) via a syringe placed into the cheek pouch at 1 ml/kg dosing volume, while WAY-100,635 or saline vehicle were administered intraperitoneally (i.p.) at an injection volume of 1 ml/kg.
Animals
Young adult male Lister Hooded rats (Harlan, UK), weighing 200–225 g on delivery, were housed in pairs in temperature- and humidity-controlled rooms with reversed light cycles (dim red light 12:00–24:00), with standard laboratory chow and water available ad libitum. A total of 60 rats were used in these experiments. All experiments were performed at the University of Reading in accordance with the principles of laboratory animal care, UK Home Office regulations [Animals (Scientific Procedures) Act 1986] and the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al. 2010; McGrath et al. 2010).
Experimental designs
Experiments 1 and 2 (neuromotor tolerability and acute feeding tests) were conducted using a within-subjects design, with all experimental units (individual animals) receiving 0.05, 0.5 and 5.0 mg/kg CBDA and vehicle according to a pseudo-random, counterbalanced, Latin square protocol. All animals received doses separated by a minimum 48 h washout period. On test days, animals were administered CBDA or vehicle 60 min prior to commencement of testing, consistent with our previously published studies of oral cannabinoid administration (Williams et al. 1998).
Experiment 3 (anxiety-like behavioural tests) was conducted using a between-subjects 2 × 2 design. Animals received either WAY-100,635 (0.1 mg/kg) or saline and either CBDA (5 mg/kg) or sesame seed oil vehicle to yield four groups: saline/vehicle, saline/CBDA, WAY/vehicle and WAY/CBDA. WAY-100,635 or saline were administered 15 min prior to CBDA or vehicle (as per Parker and colleagues’ protocol), with a further 60 min allowed for drug assimilation prior to commencement of testing. Animals were randomly allocated to the four treatment groups, and then further divided into five equally distributed blocks for daily testing, such that two animals from each group were tested on each day of the week, then again 1 week later. During the first week, animals completed the open field and light/dark box tests consecutively in a single session, followed by the novelty-suppressed feeding test 7 days later. The test order of groups was counterbalanced across the five test days each week.
Experiment 1 procedure (neuromotor tolerability)
Prior to testing, animals (n = 12) were subjected to a 5-day habituation process, consisting of daily handling, vehicle drug administration and habituation to open field and static beam test procedures. On test days, all procedures were conducted during the first half of the dark period (12:00–18:00) in the same room as the animals were housed. All test equipment was cleaned with 70 % ethanol and allowed to dry completely between animals. All tasks were presented in the following order with animals having a 5 min rest period in their home cage between tasks.
Open field
This consists of a 1.1 × 1.1 × 0.4 m black acrylic-lined box, delineated into 25 equal squares to form a 3 × 3 central sector and a single square wide peripheral sector. The open field was illuminated by dim red light (~10 lx). Animals were placed in the corner of the open field and left for 5 min with behaviour video recorded for offline coding using Observer XT software (Noldus, Netherlands). Locomotor activity was quantified based on the number of times animals crossed the lines on the open field floor, with time spent in the central area of the field and latency to first entry used to quantify anxiety-like behaviour (i.e. degree of thigmotaxis). It should be noted that the habituation animals received for this test is necessary for within-subjects assessment of drug-induced changes of locomotor activity; however as a consequence, the aversive/novel nature of the environment is attenuated. As such, a novel (i.e. unhabituated) open field test, as conducted in experiment 3, is more typically used when investigation of anxiety-like behaviour is the primary purpose of the test.
Static beam
The apparatus consisted of a 3.2 cm diameter cylindrical beam, 1 m long and 0.5 m above floor level, with a bright light positioned at the start and an enclosed goal box at the end. Animals were placed at the start of the beam and allowed a maximum of 5 min to successfully traverse its length to reach the goal box. Animals were then given a 2 min rest period in home cages prior to repeating the test. Tests were video recorded for offline coding using Observer XT software (Noldus, Netherlands). In the static beam test, performance generated four outcome measures, based on successful completion or length of beam traversed prior to falling (pass rate and distance travelled), number of times paws were fully extended past the beam (foot slips) and time taken to traverse the middle 50 cm of beam (speed).
Forelimb grip strength
Animals completed two repeats of the forelimb grip strength test, separated by a 30 s rest period. Animals were placed with forelimbs gripping a trapeze bar connected to a digital force gauge (FH50, Sauter GmbH, Germany), then uniformly pulled by the tail base away from bar along the horizontal plane until grip was released and peak force recorded.
Analysis
All behavioural coding was conducted by an experimenter blinded to treatment allocation. For static beam and forelimb grip strength outcome measures, where animals were subjected to two tests during the battery, data represent the mean of the two technical repeats, with the exception of pass rate on static beam in which a score of 0–2 was allocated based on number of successfully completed tests. All continuous data were analysed using SPSS 18 (IBM, UK) by one-way repeated measures ANOVA (ordinal pass rate data were analysed by Friedman’s ANOVA), with degrees of freedom and p values corrected where assumptions of sphericity were violated (using Greenhouse-Geisser correction). When significant overall dose effects were observed, planned comparisons of all dose groups vs vehicle group were conducted to reveal any significant pairwise comparisons. Results were considered significant if p < 0.05.
Experiment 2 procedure (acute feeding)
Acute feeding experiments were conducted in pre-satiated animals according to a well-established paradigm for the detection of hyperphagia following administration of cannabinoids (Williams et al. 1998). Animals were habituated to handling (10 days), vehicle dosing and the pre-feed procedure (7 days), and the testing apparatus (5 days) prior to commencement of testing. The pre-feed procedure was conducted at the onset of the dark period, when animals (n = 8) were transferred to individual cages containing 30.5 ± 0.5 g of highly palatable wet-mash food. The wet-mash comprised 1 part Rat and Mouse Expanded Ground Diet (SDS, Witham, UK) and 1.25 parts tap water. Animals were allowed 2 h to consume the wet-mash, following which they were returned to their home cages and quantity of wet-mash consumed was measured. Animals were habituated to this pre-feed procedure until a stable consumption level was reached, as indicated by a non-significant main effect of test day by one-way ANOVA across four consecutive habituation days (F 3, 28 = 0.653, p = 0.588).
On test days, the pre-feed procedure was again conducted, immediately after which animals were administered CBDA or vehicle and replaced in home cages for 1 h for drug assimilation, during which time food was unavailable. Animals were then placed into feeder cages for 2 h, during which time food consumption and locomotor activity were recorded on automated food intake and infrared photobeam activity systems (TSE Systems, Germany and Ugo Basile, Italy respectively) and behaviour was video recorded. Animals were then returned to home cages at the end of the experiment, with food available ad libitum until the following test procedure ≥48 h later. Quantity of food consumed was confirmed manually by weighing the remaining chow pellets in food hoppers and any crumbs in spillage trays below the cages, and subtracting these from the initial weight of chow in the hopper. The automated food intake system provided data output on the time, duration and size of each feeding bout, which were confirmed from video recordings as genuine feeding episodes as opposed to exploratory interactions with food hoppers. Feeding bouts were combined into ‘meals’, defined as feeding bouts consuming ≥0.5 g and separated by ≥900 s, criteria previously shown to more accurately reflect the natural process of food consumption (Williams and Kirkham 2002; Farrimond et al. 2012b).
Analysis
Data were analysed to provide measures of appetitive and consummatory behaviours, using the parameters of latency to first meal (appetitive) and meal sizes and durations (consummatory) in addition to total intake amounts. Ambulatory locomotor activity was quantified over the test duration using the number of infrared beam breaks. All continuous data were analysed using SPSS 18 (IBM, UK) by one-way repeated measures ANOVA, with degrees of freedom and p values corrected where assumptions of sphericity were violated (using Greenhouse-Geisser correction). When significant overall dose effects were observed, planned comparisons of all dose groups vs vehicle group were conducted to reveal any significant pairwise comparisons. Results were considered significant if p < 0.05.
Experiment 3 procedure (unconditioned anxiety test battery)
Animals (n = 40) were habituated to home environment and handling for 10 days prior to testing, and additionally to p.o. vehicle dosing and transfer to individual holding cages on the last 2 days of habituation. One day prior to the start of testing, all animals were tested for baseline levels of spontaneous locomotor activity, in which ambulatory activity was measured in an infrared photobeam activity cage (Ugo Basile, Italy) for 5 min. These data confirmed that randomisation to treatment group (as detailed above) had been successful, due to non-significant effects of treatment group on baseline activity (F 3, 36 = 1.342, p = 0.276) or bodyweight (F 3, 36 = 0.4829, p = 0.695).
All testing was completed during the first half of the dark period (12:00–18:00) in the same room as the animals were normally housed. On test days, animals were administered drugs at 30 min intervals from the onset of the dark period, such that all animals commenced testing 60 min after receiving CBDA or vehicle. Following drug administration, animals were placed in individual holding cages for the drug assimilation and inter-test rest periods. During testing in week 1, animals completed the open field test followed by the light/dark box test, separated by a 5-min rest period. During testing in week 2 (novelty-suppressed feeding), animals were food deprived in their home cages for 16–18 h prior to testing (dependent on test order).
Open field
The open field test was conducted exactly as described for experiment 1; however, animals had not previously been habituated to the procedure/apparatus so the field represented a novel environment. Test data were analysed as described for experiment 1.
Light/dark box
The apparatus consisted of an enclosed, black acrylic chamber (40 × 40 × 20 cm) connected via a small entrance hole to an open, white acrylic chamber of the same dimensions. The light sector was illuminated by a 60 W white lamp such that light levels were ~500 lx, in contrast to ~5 lx in the dark sector. Animals were placed into the light chamber facing the entrance hole and behaviour was video recorded for 5 min. Animals were then returned to home cages and equipment was cleaned with 70 % ethanol and allowed to dry completely. Movement between the sectors was recorded via an overhead digital video camera for subsequent offline coding using The Observer XT software (Noldus, The Netherlands), blinded to treatment group, with the number of entries and duration spent within the light sector quantified.
Novelty-suppressed feeding
This task was conducted in a 1.1 × 1.1 × 0.4 m white-walled arena with a sawdust-covered floor. The field was illuminated by bright white light (~450 lx) and 10 standard chow pellets were placed on a large circular piece of filter paper in the centre. Animals were placed in the corner facing the centre and allowed a maximum of 10 min to begin feeding. Latency to onset of feeding (defined as pellet held in both paws and animals sat on haunches while eating) was timed manually and subsequently confirmed from the digital video recording of the test. As soon as an animal began feeding, it was removed from the open field and placed in an individual holding cage containing a weighed quantity of standard laboratory chow. It was allowed to feed ad libitum for 30 min, after which the quantity of food consumed was recorded and the animal was returned to its home cage. The test thus generated outcome measures of latency to feeding onset and post-test food intake.
Analysis
For all outcome measures, data were analysed by two-way independent ANOVA (CBDA × WAY). Where significant interactions were observed, follow-up analysis by one-way independent ANOVA and Tukey’s post-hoc comparisons were conducted. To avoid attrition bias resulting from missing data points due to technical errors in data capture (two animals in light/dark box and two in novelty-suppressed feeding), data were analysed on an intention-to-treat basis, with missing data replaced by simple imputation methods (group means). Analysis on a per protocol basis with all animals with missing data excluded did not alter the experiment’s conclusions. Results were considered significant at p < 0.05.
Results
Experiment 1: neuromotor tolerability tests
To determine the viability of CBDA as a potential clinical candidate for the treatment of AN without the sedative effects typical of benzodiazapines, we first assessed its neuromotor tolerability profile using a battery of tests designed to reveal any effects on locomotor activity, balance and fine motor control and muscular strength. In addition to assessing locomotor activity, the habituated open field can provide an indication of any putative anxiolytic or anxiogenic activity.
Open field test
CBDA had no effect on locomotor activity at any dose when assessed in the open field test (Fig. 1a), with no significant overall effect of dose observed for the number of lines crossed (F 3, 33 = 0.405, p = 0.750). However, a significant attenuation of anxiety-like behaviour was apparent, with total time spent in the central sector (Fig. 1b) increased with increasing CBDA dose (F 3, 33 = 8.40, p < 0.0005). Planned comparisons revealed a significantly increased time spent in the central sector by both 0.5 mg/kg (p = 0.005) and 5.0 mg/kg (p < 0.0005) groups compared to vehicle-treated animals. In contrast, CBDA treatment had no effect upon latency to first entry into the central sector, a further measure of anxiety-like behaviour (F 3, 33 = 0.769, p = 0.52).

Effects of CBDA treatment on behavioural parameters in habituated open field test, conducted as part of the neuromotor tolerability test battery (experiment 1). Ambulatory locomotor activity (a) as measured by number of line crosses was unaffected by any dose, however anxiolytic-like effects, as measured by increased time spent in central sector (b), were observed following 0.5 and 5.0 mg/kg CBDA treatment. Data presented as means ± SEM and analysed by one-way repeated measures ANOVA and planned comparisons (all groups vs vehicle), all groups n = 12, **p < 0.01, ***p < 0.001
Static beam test
CBDA had no effect at any dose on any measure of balance or motor coordination as assessed in the static beam test (Table 1). Neither balance, as assessed by pass rate (F r 3 = 3.522, p = 0.318), nor distance travelled (F 3, 33 = 0.673, p = 0.574) was affected by CBDA treatment. Fine motor coordination was similarly unaffected, with CBDA treatment having no effect at any dose upon the number of foot slips made (F 3, 33 = 0.605, p = 0.617) or time to cross the beam (F 3, 33 = 1.105, p = 0.361).
Grip strength test
The forelimb grip strength test (Table 1) for muscular strength and functional neurotoxicity revealed no significant overall dose effect of CBDA (F 1.5, 16.2 = 1.109, p = 0.335).
The results from experiment 1 demonstrate that CBDA, at doses up to 5 mg/kg, is well tolerated and exerts no deleterious effects on locomotor activity, balance, fine motor control or muscular strength. Furthermore, the dose-dependent increase in central sector duration suggests that CBDA may possess anxiolytic-like properties. These findings support its viability as a novel treatment of anticipatory nausea, without the neuromotor side effects typical of the benzodiazepine anxiolytics currently in clinical use. In light of this favourable tolerability profile, the ability of CBDA to stimulate feeding behaviours was investigated using the same dose range employed in experiment 1.
Experiment 2: test of hyperphagia in pre-satiated rats
To determine whether previously reported increases in saccharin palatability following CBDA administration were indicative of hyperphagic properties, we investigated the effects of CBDA on feeding behaviour in pre-satiated rats. As shown in Fig. 2a, CBDA dose exerted no significant overall effect on total food intake during the 2 h test period (F 1.4, 9.5 = 0.336, p = 0.641). There was also no significant overall effect of CBDA dose on ambulatory locomotor activity (Fig. 2b) within the feeding chambers (F 3, 21 = 0.309, p = 0.819), further validating our findings in the habituated open field test.

Food intake and ambulatory locomotor activity during 2 h feeding test in pre-satiated rats (experiment 2). CBDA had no effect on total chow consumed (a) or total locomotor activity (b) at any dose. Data presented as mean ± SEM and analysed by one-way repeated measures ANOVA, all groups n = 8
A more granular analysis of meal pattern microstructure parameters (Fig. 3) revealed no significant overall effect of CBDA dose on latency to meals 1 or 2 (F 3, 21 = 0.348, p = 0.791 and F 3, 21 = 0.546, p = 0.656 respectively), size of meals (meal 1: F 3, 21 = 0.709, p = 0.557; meal 2: F 3, 21 = 0.541, p = 0.659) or duration of meals (meal 1: F 1.5, 10.4 = 0.832, p = 0.429; meal 2: F 3, 21 = 0.821, p = 0.399). That the latency to first feeding episode was approximately 90 min into the test session for all groups demonstrates that the pre-satiation procedure was effective, and further corroborates the lack of CBDA effect on total food intake.

Graphic summary of meal pattern microstructure parameters from experiment 2. Left edge of boxes positioned along x-axis according to meal latencies, box widths scaled to meal durations and meal sizes (in g) given above. CBDA had no effect on any of these measures at any dose. Note that no animals consumed a second meal in the 0.05 mg/kg group, hence this box is omitted from the figure, and group mean meal sizes below the 0.5-g meal criteria reflect that a number of animals consumed only ≤1 meal. Data presented as means and analysed by one-way repeated measures ANOVA, all groups n = 8
The results of experiment 2 demonstrate that CBDA, in the dose range tested, did not modulate any aspect of feeding behaviour in pre-satiated rats. Based on the results from experiments 1 and 2, a further study was conducted to assess whether the putative anxiolytic-like effect of CBDA could be validated in the novel open field and light/dark box tests. Additionally, CBDA was assessed in the novelty-suppressed feeding test, to investigate whether motivation to eat could be increased under anxiogenic-like conditions which typically suppress feeding behaviour. The 5-HT1AR activation-dependent mechanism previously reported for CBDA in the AN model (Bolognini et al. 2013) led us to further investigate whether any anxiolytic-like effects were sensitive to 5-HT1AR antagonist challenge in these tests.
Experiment 3: anxiety-like behaviour tests
Open field test
The total time spent in the central sector of the open field did not show significant main effects of either CBDA (F 1, 36 = 0.177, p = 0.676) or WAY-100,635 administration (F 1, 36 = 0.156, p = 0.695), nor was any interaction observed (F 1, 36 = 0.042, p = 0.838). Locomotor activity within the open field, as measured by the number of line crosses, again did not show significant main effects of either CBDA (F1, 36 = 2.908, p = 0.097) or WAY-100,635 administration (F 1, 36 = 0.613, p = 0.439), nor was any interaction observed (F 1, 36 = 0.391, p = 0.536). However, a significant interaction was observed between the effects of WAY-100,635 and CBDA (F 1, 36 = 5.270, p = 0.028) on latency to first entry into the central sector. Further analysis of this interaction using one-way ANOVA did not show a significant overall effect of treatment group (F 3, 36 = 1.879, p = 0.151) or any significant pairwise comparisons, indicating a lack of meaningful drug effect.
Light/dark box test
The number of entries into the light sector of the box did not show significant main effects of CBDA (F 1, 36 = 1.677, p = 0.204) or WAY-100,635 (F 1, 36 = 0.995, p = 0.325), nor was any interaction observed (F 1, 36 = 0.379, p = 0.542). The total time spent in the light sector of the box did not show significant main effects of CBDA (F 1, 36 = 1.096, p = 0.302) or WAY-100,635 (F 1, 36 = 0.237, p = 0.629), nor was any interaction observed (F 1, 36 = 0.501, p = 0.484).
Novelty-suppressed feeding test
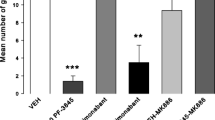
A significant interaction was observed between the effects of CBDA and WAY-100,635 (F 1, 36 = 7.551, p = 0.009) on latency to onset of feeding (Fig. 4a). Follow-up analysis revealed a significant overall effect of treatment group (F 3, 36 = 10.619, p < 0.0005), due to an increased latency to feed in animals treated with WAY-100,635 alone vs vehicle control animals (p = 0.017) or those treated with CBDA alone (p < 0.0005). This increased latency was completely abolished in animals treated with both CBDA and WAY-100,635 (p < 0.0005). Post-test food intake in home cages (Fig. 4b) did not show significant main effects of either CBDA (F1, 36 = 1.266, p = 0.268) or WAY-100,635 administration (F 1, 36 = 2.056, p = 0.160), nor was any interaction observed (F 1, 36 = 1.811, p = 0.187). The lack of effect of either drug on post-test food intake indicates that their effect on latency to feed was due to modulation of anxiety-like behaviour alone, and not confounded by effects on appetite.

Effects of CBDA (5 mg/kg) and the 5-HT1AR antagonist WAY-100,635 (0.1 mg/kg) in the novelty-suppressed feeding test, conducted in a modified open field as part of the anxiety-like behaviour test battery (experiment 3). Treatment with WAY-100,635 alone elicited an anxiogenic-like effect by increasing latency to feeding onset, which was abolished by co-treatment with CBDA (a). Home cage food intake in the 30 min following the test was unaffected by either drug (b). Data presented as means ± SEM and analysed by two-way ANOVA, followed by one-way ANOVA and Tukey’s post-hoc comparisons, all groups n = 10, *p < 0.05, ***p < 0.001
Discussion
Our results suggest CBDA is well-tolerated, since it failed to produce any neuromotor side effects at any dose tested. In the same dose range, CBDA also had no modulatory effect on feeding behaviour in healthy, pre-satiated rats. However, CBDA did abolish the potentiated suppression of feeding behaviour in the NSF test induced by the 5-HT1AR antagonist WAY-100,635. Thus, CBDA does not appear to increase appetite per se, but may selectively stimulate feeding under putatively anxiogenic conditions which suppress feeding behaviour, possibly via 5-HT1AR-mediated mechanisms.
The battery of neuromotor tolerability tests used in this study has previously been utilised to assess other phytocannabinoids against drugs with known clinical neuromotor side effects (Hill et al. 2012; Hill et al. 2013). The benzodiazepine class of drugs, which are used clinically to attenuate AN, cause significant sedative side effects, decrease activity in the OFT (reviewed in Prut and Belzung 2003) and impair performance in the static beam (Stanley et al. 2005) and forelimb grip strength assays (Meyer et al. 1979; Ferguson and Paule 1996). Thus, these tests have predictive validity for assessment of the neuromotor tolerability profile of novel compounds for AN treatment. In our experiments, CBDA did not affect activity in the open field or cause any detrimental effects on any performance measure in either static beam or grip strength tests at any dose tested, the range of which was comparable to that used in previous studies in models of acute and anticipatory nausea. The lack of effect on locomotor activity in the OFT is consistent with a previously published report that CBDA (0.0001-1 mg/kg; i.p.) did not affect distance travelled in a 15-min activity chamber test (Rock et al. 2014a), although doses of the benzodiazepine CDP which suppressed AN (5–10 mg/kg) exerted a sedative effect in this test. The observation that ambulatory locomotor activity during the duration of the feeding test (experiment 2) was also unaffected by any dose of CBDA further confirms the lack of sedative effect, even over an extended test period (2 h) -considerably longer than that typically used for activity tests (Curzon et al. 2009). The present study extended the investigation of potential sedative effects to include measures of motor coordination, using the static (walking) beam assay, which can more sensitively predict clinical sedative effects than the more commonly used rotarod test (Stanley et al. 2005; Hill et al. 2012). CBDA had no effect at any dose on performance measures of balance or fine motor control in this test. The final component of the tolerability test battery, the forelimb grip strength test, demonstrated that CBDA administration did not result in drug-induced muscle relaxation at any dose. These results validate and considerably extend the preliminary evidence for the lack of sedative effects of CBDA, supporting its potential as a novel treatment for AN unlikely to have the compromised clinical utility of benzodiazepines (Malik et al. 1995; Rock et al. 2014b).
A previously published study of the effects of CBDA in the AN model reported increased unconditioned hedonic reactions to saccharin (i.e. increased palatability), which the authors speculated could indicate an appetite-enhancing effect (Bolognini et al. 2013). Such an effect could have an additional clinical utility by attenuating the comorbid anorectic effects of chemotherapy treatment (Hainsworth and Hesketh 1992) and/or cancer anorexia-cachexia syndrome (Stephens and Fearon 2008).
To investigate whether this suggestion of an appetite-enhancing effect could first be validated in healthy rats under more naturalistic feeding conditions (than intraoral cannula-delivered saccharin responses), experiment 2 was conducted using a well-established test of hyperphagia. The acute feeding test in pre-satiated rats has been utilised in many previous studies in our lab to sensitively determine hyperphagic actions of pharmacological compounds, providing detailed information on both food intake and the microstructure of meal patterns (Williams et al. 1998; Williams and Kirkham 2002; Farrimond et al. 2010a; Farrimond et al. 2010b; Farrimond et al. 2012a). In vehicle-dosed rats, feeding behaviour during the test period is minimal, typically comprising one to two small meals with a total consumption of ≤1 g, occurring after ~90 min (Farrimond et al. 2012b). Consistent with this typical baseline level of consumption, no significant effect was seen on total food intake following administration of any dose of CBDA, with rats consuming 0.4–1 g over 2 h. The latency to consumption of the first meal, a measure of appetitive feeding behaviour (motivation to eat), was similarly unaffected by CBDA treatment, and neither were consummatory behaviour measures of meal size or duration. These data indicate that, at oral doses of 0.05–5 mg/kg, CBDA does not modulate total food intake or any aspects of meal microstructure. This is in contrast to the reported effect on saccharin palatability, however it should be noted that the previously reported effect was only seen at 0.01 mg/kg, but not at 0.1–5 mg/kg, and furthermore the behavioural model and route of administration were also different (Bolognini et al. 2013). It therefore remains possible that CBDA may have appetite-stimulating effects only at very low doses, or selectively for hedonic foods over regular chow, however the present data does not support any effects on feeding behaviour at doses ≥0.05 mg/kg. However, in light of the effects seen in the NSF test presented here, it may be the case that (at least at higher doses) CBDA selectively stimulates feeding under putatively anxiogenic conditions, which is more consistent with the positive effects seen in the AN model, and may be more clinically useful. As such, further investigation of CBDA actions on feeding in models of chemotherapy- or anxiety-induced anorexia is warranted.
An interesting observation made during the neuromotor tolerability study was the dose-dependent increase in the time rats spent in the central sector of the habituated open field. This test was primarily designed as a test of sedative/stimulant effects, and hence rats were habituated to the open field to achieve stable baseline activity prior to CBDA administration. However, the lack of locomotor activity modulation in this test (as measured by line crosses) suggests this observation may still be indicative of an anxiolytic-like effect. Cannabidiol (CBD), produced by spontaneous decarboxylation of CBDA (Cluny et al. 2011), has well-documented anxiolytic-like effects in both animals and humans (reviewed by de Schier et al. 2012) which appears to be primarily facilitated by 5-HT1AR-mediated neurotransmission (Campos et al. 2012). However, to the best of our knowledge, only a single study of the anxiolytic-like effects of CBDA has been published to date. In this study, CBDA (0.001–1 mg/kg, i.p.) was assessed for the ability to attenuate conditioned freezing to a shock-paired tone; however, expression of conditioned freezing was not modified by any dose (Rock et al. 2014a). The suggestion of an anxiolytic-like effect of CBDA in the habituated open field test, and the paucity of published data in anxiety-like behavioural models for this cannabinoid, prompted us to further investigate the effects of CBDA in three typical models of unconditioned anxiety-like behaviour. As the greatest effect in the habituated open field was seen following administration of 5 mg/kg CBDA, and previous reports implicated indirect 5-HT1AR activation in AN models (Bolognini et al. 2013; Rock et al. 2014a), we investigated this dose with and without pre-treatment with the selective 5-HT1AR antagonist WAY-100,635, at the same dose used by Parker and colleagues as a behaviourally silent antagonist in their AN studies. In the novel (unhabituated) open field test, the more aversive nature of the environment was apparent from both the reduced central sector duration in control rats (16 vs 24 s in the habituated OFT) and number of line crosses (96 vs 157 in the habituated OFT). However, in this test, CBDA had no effect on central sector duration or number of line crosses, suggesting that in this more aversive environment CBDA did not have significant anxiolytic-like effects, and thus CBDA has limited, if any, efficacy within this test. Consistent with the results from the novel open field, in the light/dark box test, which is another test based on the conflict between rats’ exploratory drive and fear of bright or exposed areas (Bourin and Hascoët 2003), CBDA also had no effect on either number of entries or duration spent in the light sector, which would be indicative of an anxiolytic-like effect. In both tests, administration of 0.1 mg/kg WAY-100,635, alone or in combination with CBDA, also had no effect on any measure of anxiety-like behaviour or general locomotor activity. This indicates that this dose, which was behaviourally silent in previous AN studies, was also appropriate as a silent antagonist challenge in the open field and light/dark box tests, and that no interaction occurred with CBDA relevant to behavioural outcomes in these tests.
A third test of anxiety-like behaviour was conducted using the novelty-suppressed feeding (or hyponeophagia) test, which differs from the open field and light/dark box tests in that the conflict arises between the innate aversion to bright unfamiliar spaces and the desire to feed (following a period of food deprivation) rather than to explore a novel environment (Britton and Britton 1981; Dulawa and Hen 2005). The NSF test is sensitive to numerous drugs with known anxiolytic activity, including the 5-HT1AR agonist 8-OH-DPAT (Rex et al. 1998; Zhang et al. 2010), and also demonstrates the anxiogenic-like activity of the 5-HT1AR antagonist NAN-190 (Zhang et al. 2010) and increased anxiety-like behaviour in 5-HT1AR knockout mice (Gross et al. 2000). In the present study, treatment with CBDA alone did not affect the latency to feed, however WAY-100,635 treatment alone significantly increased latency, indicative of an anxiogenic-like effect. That administration of WAY-100,635 alone had an anxiogenic-like effect in this test was unexpected, given that the dose of this compound was chosen as a behaviourally silent antagonist challenge, which had no effect in either previous AN studies (Bolognini et al. 2013) or in the novel open field or light/dark box tests in the present study. While this increased latency is consistent with the work of Zhang et al. (2010) using the 5-HT1AR antagonist NAN-190, it should also be noted that a 0.3-mg/kg dose of WAY-100,635 given to mice in the NSF test was behaviourally silent (Duvvuri et al. 2009), and that in other tests of anxiety-like behaviour this compound can be anxiogenic or even anxiolytic dependent on dose and test type (Sánchez 1996; Griebel et al. 1999; Griebel et al. 2000). Interestingly, in rats which were administered both CBDA and WAY-100,635 in the present NSF test, this anxiogenic-like effect of WAY-100,635 was completely abolished. The results from the post-test home cage intake test did not show significant effects of either drug or their combination, ruling out confounding effects on appetite, consistent with results from the acute feeding study. Similarly, the lack of effect of either drug on the number of line crosses in the novel open field test rules out possible confounding effects of locomotor activity modulation. It thus appears that this dose of WAY-100,635, while behaviourally silent in the open field and light/dark box tests, elicits an anxiogenic-like response in the NSF test, and that this response is antagonised by CBDA, despite this cannabinoid having no anxiolytic-like effect when administered alone. Such pharmacological effects, while seemingly robust in terms of the data obtained, are less than straightforward to interpret based on the present experiments alone.
Previous studies demonstrated that while the ability of CBDA to attenuate nausea is abolished by pretreatment with WAY-100,635, in vitro binding experiments suggested this is via an indirect enhancement of 5-HT1AR activation rather than direct activation (Bolognini et al. 2013). The results from experiment 3 thus provides some further support for the notion that CBDA has limited efficacy as a typical 5-HT1AR agonist anxiolytic, but under certain anxiogenic conditions does possess anxiolytic-like activity, presumably via indirect modulation of 5-HT1AR-mediated neurotransmission. While beyond the scope of the present study, it may be valuable to further characterise the locus and mechanism of this activity.
The present report provides vital further data in support of CBDA as a novel treatment for anticipatory nausea, which is unlikely to elicit the compromising sedative effects of the benzodiazepine anxiolytics currently in clinical use. CBDA appears to have some anxiolytic-like activity, specific to models of feeding suppression or nausea involving alterations in 5-HT1AR-dependent neurotransmission. CBDA did not modulate feeding behaviour in healthy rats, however these and previous data suggest beneficial effects on feeding may occur under pathological anxiogenic conditions, further investigation of which is warranted. While such investigations may provide evidence of further therapeutic potential for such conditions, the tolerability data presented here strongly supports clinical investigation of CBDA as a non-sedative alternative to benzodiazepine anxiolytics for the treatment of AN.
Abbreviations
- CBDA:
-
Cannabidiolic acid
- CBD:
-
Cannabidiol
- pCB:
-
Phytocannabinoid
- CINV:
-
Chemotherapy-induced nausea and vomiting
- AN:
-
Anticipatory nausea
- CDP:
-
Chlordiazepoxide
- NK1:
-
Neurokinin 1
- 5-HT:
-
5-Hydroxytryptamine
- 5-HT1AR:
-
5-Hydroxytryptamine receptor type 1A
References
Ahlberg K, Ekman T, Gaston-Johansson F, Mock V (2003) Assessment and management of cancer-related fatigue in adults. Lancet 362:640–650. doi:10.1016/S0140-6736(03)14186-4
Basch E, Prestrud AA, Hesketh PJ et al (2012) Antiemetic use in oncology: updated guideline recommendations from ASCO. Am Soc Clin Oncol Educ Book 532:532–540. doi:10.14694/EdBook_AM.2012.32.532
Bolognini D, Rock EM, Cluny NL et al (2013) Cannabidiolic acid prevents vomiting in Suncus murinus and nausea-induced behaviour in rats by enhancing 5-HT1A receptor activation. Br J Pharmacol 168:1456–1470. doi:10.1111/bph.12043
Bourin M, Hascoët M (2003) The mouse light/dark box test. Eur J Pharmacol 463:55–65. doi:10.1016/S0014-2999(03)01274-3
Bovbjerg DH (2006) The continuing problem of post chemotherapy nausea and vomiting: contributions of classical conditioning. Auton Neurosci 129:92–98. doi:10.1016/j.autneu.2006.07.016
Britton DR, Britton KT (1981) A sensitive open field measure of anxiolytic drug activity. Pharmacol Biochem Behav 15:577–582. doi:10.1016/0091-3057(81)90212-4
Campos AC, Moreira FA, Gomes FV et al (2012) Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Philos Trans R Soc B Biol Sci 367:3364–3378. doi:10.1098/rstb.2011.0389
Campos D, Pereira JR, Reinhardt RR et al (2001) Prevention of cisplatin-induced emesis by the oral neurokinin-1 antagonist, MK-869, in combination with granisetron and dexamethasone or with dexamethasone alone. J Clin Oncol 19:1759–1767
Cluny NL, Naylor RJ, Whittle BA, Javid FA (2011) The effects of cannabidiolic acid and cannabidiol on contractility of the gastrointestinal tract of Suncus murinus. Arch Pharm Res 34:1509–1517. doi:10.1007/s12272-011-0913-6
Curzon P, Zhang M, Radek R, Fox G (2009) The behavioral assessment of sensorimotor processes in the mouse: acoustic startle, sensory gating, locomotor activity, rotarod, and beam walking. Methods Behav. Anal, Neurosci
Dos Santos LV, Souza FH, Brunetto AT et al (2012) Neurokinin-1 receptor antagonists for chemotherapy-induced nausea and vomiting: a systematic review. J Natl Cancer Inst 104:1280–1292. doi:10.1093/jnci/djs335
Dulawa SC, Hen R (2005) Recent advances in animal models of chronic antidepressant effects: the novelty-induced hypophagia test. Neurosci Biobehav Rev 29:771–783. doi:10.1016/j.neubiorev.2005.03.017
Duvvuri V, Risbrough VB, Kaye WH, Geyer MA (2009) 5-HT1A receptor activation is necessary for 5-MeODMT-dependent potentiation of feeding inhibition. Pharmacol Biochem Behav 93:349–353. doi:10.1016/j.pbb.2009.05.014
Farrimond JA, Hill AJ, Whalley BJ, Williams CM (2010a) Cannabis constituents modulate δ9-tetrahydrocannabinol-induced hyperphagia in rats. Psychopharmacology (Berl) 210:97–106. doi:10.1007/s00213-010-1821-z
Farrimond JA, Whalley BJ, Williams CM (2012a) Non-Δ9tetrahydrocannabinol phytocannabinoids stimulate feeding in rats. Behav Pharmacol 23:113–117. doi:10.1097/FBP.0b013e32834ed832
Farrimond JA, Whalley BJ, Williams CM (2012b) Cannabinol and cannabidiol exert opposing effects on rat feeding patterns. Psychopharmacology (Berl) 223:117–129. doi:10.1007/s00213-012-2697-x
Farrimond JA, Whalley BJ, Williams CM (2010b) A low-Δ9tetrahydrocannabinol cannabis extract induces hyperphagia in rats. Behav Pharmacol. doi:10.1097/FBP.0b013e328340a062
Ferguson SA, Paule MG (1996) Effects of chlorpromazine and diazepam on time estimation behavior and motivation in rats. Pharmacol Biochem Behav 53:115–122. doi:10.1016/0091-3057(95)02002-0
Figueroa-Moseley C, Jean-Pierre P, Roscoe JA et al (2007) Behavioral interventions in treating anticipatory nausea and vomiting. J Natl Compr Canc Netw 5:44–50
Griebel G, Rodgers RJ, Perrault G, Sanger DJ (1999) Behavioural profiles in the mouse defence test battery suggest anxiolytic potential of 5-HT 1A receptor antagonists. Psychopharmacology (Berl) 144:121–130. doi:10.1007/s002130050984
Griebel G, Rodgers RJ, Perrault G, Sanger DJ (2000) The effects of compounds varying in selectivity as 5-HT1A receptor antagonists in three rat models of anxiety. Neuropharmacology 39:1848–1857. doi:10.1016/S0028-3908(00)00074-5
Gross C, Santarelli L, Brunner D et al (2000) Altered fear circuits in 5-HT(1A) receptor KO mice. Biol Psychiatry 48:1157–1163. doi:10.1016/S0006-3223(00)01041-6
Hainsworth JD, Hesketh PJ (1992) Single-dose ondansetron for the prevention of cisplatin-induced emesis: efficacy results. Semin Oncol 19:14–19
Hesketh PJ (2008) Chemotherapy-induced nausea and vomiting. N Engl J Med 358:2482–2494. doi:10.1056/NEJMra0706547
Hesketh PJ, Grunberg SM, Gralla RJ et al (2003) The oral neurokinin-1 antagonist aprepitant for the prevention of chemotherapy-induced nausea and vomiting: a multinational, randomized, double-blind, placebo-controlled trial in patients receiving high-dose cisplatin—the Aprepitant Protocol 052 Study Gr. J Clin Oncol 21:4112–4119. doi:10.1200/JCO.2003.01.095
Hickok JT, Roscoe JA, Morrow GR et al (2003) Nausea and emesis remain significant problems of chemotherapy despite prophylaxis with 5-hydroxytryptamine-3 antiemetics: a University of Rochester James P. Wilmot Cancer Center Community Clinical Oncology Program Study of 360 cancer patients treated in treated in the community. Cancer 97:2880–2886. doi:10.1002/cncr.11408
Hill AJ, Mercier MS, Hill TDM et al (2012) Cannabidivarin is anticonvulsant in mouse and rat. Br J Pharmacol 167:1629–1642. doi:10.1111/j.1476-5381.2012.02207.x
Hill TDM, Cascio M-G, Romano B et al (2013) Cannabidivarin-rich cannabis extracts are anticonvulsant in mouse and rat via a CB1 receptor-independent mechanism. Br J Pharmacol 170:679–692. doi:10.1111/bph.12321
Janelsins MC, Tejani MA, Kamen C et al (2013) Current pharmacotherapy for chemotherapy-induced nausea and vomiting in cancer patients. Expert Opin Pharmacother 14:757–766. doi:10.1517/14656566.2013.776541
Kamen C, Tejani MA, Chandwani K et al (2014) Anticipatory nausea and vomiting due to chemotherapy. Eur J Pharmacol 722:172–179. doi:10.1016/j.ejphar.2013.09.071
Kilkenny C, Browne W, Cuthill IC et al (2010) Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160:1577–1579. doi:10.1111/j.1476-5381.2010.00872.x
Kris MG, Hesketh PJ, Somerfield MR et al (2006) American Society of Clinical Oncology guideline for antiemetics in oncology: update 2006. J Clin Oncol 24:2932–2947. doi:10.1200/JCO.2006.06.9591
Lotfi-Jam K, Carey M, Jefford M et al (2008) Nonpharmacologic strategies for managing common chemotherapy adverse effects: a systematic review. J Clin Oncol 26:5618–5629. doi:10.1200/JCO.2007.15.9053
Malik IA, Khan WA, Qazilbash M et al (1995) Clinical efficacy of lorazepam in prophylaxis of anticipatory, acute, and delayed nausea and vomiting induced by high doses of cisplatin. A prospective randomized trial. Am J Clin Oncol 18:170–175
Martin M (1996) The severity and pattern of emesis following different cytotoxic agents. Oncology 53(Suppl 1):26–31. doi:10.1159/000227637
Matteson S, Roscoe J, Hickok J, Morrow GR (2002) The role of behavioral conditioning in the development of nausea. Am J Obstet Gynecol 186:S239–S243. doi:10.1067/mob.2002.122597
McGrath JC, Drummond GB, McLachlan EM et al (2010) Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol 160:1573–1576. doi:10.1111/j.1476-5381.2010.00873.x
Meyer OA, Tilson HA, Byrd WC, Riley MT (1979) A method for the routine assessment of fore- and hindlimb grip strength of rats and mice. Neurobehav Toxicol 1:233–236
Navari RM, Reinhardt RR, Gralla RJ et al (1999) Reduction of cisplatin-induced emesis by a selective neurokinin-1-receptor antagonist. L-754,030 Antiemetic Trials Group. N Engl J Med 340:190–195. doi:10.1056/NEJM199901213400304
Nesse RM, Carli T, Curtis GC, Kleinman PD (1980) Pretreatment nausea in cancer chemotherapy: a conditioned response? Psychosom Med 42:33–36
Prut L, Belzung C (2003) The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: a review. Eur J Pharmacol 463:3–33. doi:10.1016/S0014-2999(03)01272-X
Razavi D, Delvaux N, Farvacques C et al (1993) Prevention of adjustment disorders and anticipatory nausea secondary to adjuvant chemotherapy: a double-blind, placebo-controlled study assessing the usefulness of alprazolam. J Clin Oncol 11:1384–1390
Rex A, Voigt JP, Voits M, Fink H (1998) Pharmacological evaluation of a modified open-field test sensitive to anxiolytic drugs. Pharmacol Biochem Behav 59:677–683. doi:10.1016/S0091-3057(97)00461-9
Rock E, Parker L (2013a) Effect of low doses of cannabidiolic acid and ondansetron on LiCl-induced conditioned gaping (a model of nausea-induced behaviour) in rats. Br J Pharmacol. doi:10.1111/bph.12162
Rock EM, Limebeer CL, Navaratnam R et al (2014a) A comparison of cannabidiolic acid with other treatments for anticipatory nausea using a rat model of contextually elicited conditioned gaping. Psychopharmacology (Berl). doi:10.1007/s00213-014-3498-1
Rock EM, Limebeer CL, Parker LA (2014b) Anticipatory nausea in animal models: a review of potential novel therapeutic treatments. Exp Brain Res. doi:10.1007/s00221-014-3942-9
Rock EM, Parker LA (2013b) Suppression of lithium chloride-induced conditioned gaping (a model of nausea-induced behaviour) in rats (using the taste reactivity test) with metoclopramide is enhanced by cannabidiolic acid. Pharmacol Biochem Behav 111:84–89. doi:10.1016/j.pbb.2013.08.012
Roila F, Herrstedt J, Aapro M et al (2010) Guideline update for MASCC and ESMO in the prevention of chemotherapy- and radiotherapy-induced nausea and vomiting: results of the Perugia consensus conference. Ann Oncol 21(Suppl 5):v232–v243. doi:10.1093/annonc/mdq194
Roscoe JA, Morrow GR, Aapro MS et al (2011) Anticipatory nausea and vomiting. Support Care Cancer 19:1533–1538. doi:10.1007/s00520-010-0980-0
Sánchez C (1996) 5-HT(1A) receptors play an important role in modulation of behavior of rats in a two-compartment black and white box. Behav Pharmacol 7:788–797
de Schier ARM, Ribeiro NP, Silva AC et al (2012) Cannabidiol, a Cannabis sativa constituent, as an anxiolytic drug. Rev Bras Psiquiatr 34(Suppl 1):S104–S110. doi:10.1590/S1516-44462012000500008
Stanley JL, Lincoln RJ, Brown TA et al (2005) The mouse beam walking assay offers improved sensitivity over the mouse rotarod in determining motor coordination deficits induced by benzodiazepines. J Psychopharmacol 19:221–227. doi:10.1177/0269881105051524
Stephens N, Fearon K (2008) Anorexia, cachexia and nutrition. Medicine (Baltimore) 36:78–81. doi:10.1016/j.mpmed.2007.11.004
Williams CM, Kirkham TC (2002) Observational analysis of feeding induced by Delta9-THC and anandamide. Physiol Behav 76:241–250. doi:10.1016/S0031-9384(02)00725-4
Williams CM, Rogers PJ, Kirkham TC (1998) Hyperphagia in pre-fed rats following oral delta9-THC. Physiol Behav 65:343–346. doi:10.1016/S0031-9384(98)00170-X
Zhang J, Huang X-Y, Ye M-L et al (2010) Neuronal nitric oxide synthase alteration accounts for the role of 5-HT1A receptor in modulating anxiety-related behaviors. J Neurosci 30:2433–2441. doi:10.1523/JNEUROSCI.5880-09.2010
Acknowledgments
This research was supported by grants to CMW and BJW by GW Pharmaceuticals and Otsuka Pharmaceuticals, and in part by the University of Reading Research Endowment Trust Fund to DIB.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The work reported was funded in part by grants to BJW and CMW from GW Pharmaceuticals and Otsuka Pharmaceuticals. The original study concept was discussed with the sponsor (GW Pharmaceuticals) although all subsequent study design, data collection, analysis and interpretation were conducted independently by the authors. The report was approved by the sponsor company prior to submission, and the authors retain full control of all primary data.
Rights and permissions
About this article

Cite this article
Brierley, D.I., Samuels, J., Duncan, M. et al. Neuromotor tolerability and behavioural characterisation of cannabidiolic acid, a phytocannabinoid with therapeutic potential for anticipatory nausea. Psychopharmacology 233, 243–254 (2016). https://doi.org/10.1007/s00213-015-4100-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-015-4100-1