
Abstract
Microglial cells coordinate immune responses in the central nervous system. Carvedilol (CVL) is a non-selective β-blocker with anti-inflammatory and anti-oxidant effects. This study aims to investigate the anti-inflammatory effects and the underlying mechanisms of CVL on lipopolysaccharide (LPS)-induced inflammation in microglial BV2 cells. BV2 cells were stimulated with LPS, and the protective effects of CVL were investigated via measurement of cell viability, reactive oxygen species (ROS), and interleukin (IL)-1β liberation. The protein levels of some inflammatory cascade, Notch, and peroxisome proliferator-activated receptor (PPAR)-γ pathways and relative markers of M1/M2 microglial phenotypes were assessed. Neuroblastoma SH-SY5Y cells were cultured with a BV2-conditioned medium (CM), and the capacity of CVL to protect cell viability was evaluated. CVL displayed a protective effect against LPS stress through reducing ROS and down-regulating of nuclear factor kappa B (NF-κB) p65, NLR family pyrin domain containing-3 (NLRP3), and IL-1β proteins. LPS treatment significantly increased the levels of the M1 microglial marker inducible nitric oxide synthase (iNOS) and M1-associated cleaved-NOTCH1 and hairy and enhancer of split-1 (HES1) proteins. Conversely, LPS treatment reduced the levels of the M2 marker arginase-1 (Arg-1) and PPAR-γ proteins. CVL pre-treatment reduced the protein levels of iNOS, cleaved-NOTCH1, and HES1, while increased Arg-1 and PPAR-γ. CM of CVL-primed BV2 cells significantly improved SH-SY5Y cell viability as compared with the LPS-induced cells. CVL suppressed ROS production and alleviated the expression of inflammatory markers in LPS-stimulated BV2 cells. Our results demonstrated that targeting Notch and PPAR-γ pathways as well as directing BV2 cell polarization toward the M2 phenotype may provide a therapeutic strategy to suppress neuroinflammation by CVL.
Graphical Abstract

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neuroinflammation is a complex innate immune reaction in the central nervous system (CNS) that occurs in response to ischemic stroke, neurodegenerative diseases, and mechanical trauma such as spinal cord injury (Roe 2021). The pathological hallmark of neuroinflammation is mediated by a variety of cytokines, chemokines, and reactive oxygen species (ROS) produced by resident CNS glia (microglia and astrocytes) or infiltrated immune cells (DiSabato et al. 2016).
Previous studies revealed that the production of pro-inflammatory cytokines by activated immune cells plays a major role in upregulating the expression of inflammatory mediators. In particular, tumor necrosis factor-alpha (TNF-α) is tightly connected with the initial induction of inflammatory responses and oxidative stress (Becher et al. 2017; Ludwig et al. 2017). Downstream of the TNF-α signaling is the nuclear factor kappa B (NF-κB), which is a well-known transcription factor of the inflammatory process (Becher et al. 2017). NF-κB promotes the transcription of cyclooxygenase-2 (COX-2), inducible nitric oxide synthase (iNOS), interleukin (IL)-1β, IL-6, and IL-18. There is a strong association between the expression of inflammatory mediators and oxidative stress-mediated neurotoxicity.
The release of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) from damaged tissues leads to the induction of inflammation. These signals may activate downstream signaling receptors called pattern recognition receptors (PRRs), which increase the secretion of inflammatory cytokines. ROS plays a critical role in the pathogenesis of neuroinflammation by stimulating the nucleotide oligomerization domain (NOD)-like receptors (NLRs) and activation of inflammasome complexes such as NLR family pyrin domain containing-3 (NLRP3) (Mortezaee et al. 2018). The inflammasome is a multi-protein complex that finely regulates the activation of caspase-1 and mediates the secretion of pro-inflammatory cytokines (Luo et al. 2014; He et al. 2016). NLRP3 inflammasome is highly expressed in the resident innate immune cells of CNS, i.e., microglia. After damage to the CNS, microglial cells are activated and undergo morphological and phenotypic alterations. Various studies have reported that during neural damage, activated microglia polarize toward the pro-inflammatory (M1) or anti-inflammatory (M2) phenotypes in response to different micro-environmental disturbances. Both M1 and M2 microglia are involved in the pathogenesis of neurodegeneration. M1 promotes inflammation and subsequent neurotoxicity, and M2 leads to neuroprotection by reducing or inhibiting inflammation (Kwon and Koh 2020). NF-κB and Notch signaling pathways are involved in regulating M1 microglia polarization (Fazio and Ricciardiello 2016). Peroxisome proliferator-activated receptor (PPAR)-γ is a nuclear receptor that is highly expressed in microglia. Several studies showed that the PPAR-γ acts as a crucial regulator in M2 polarization by inactivating NF-κB during inflammatory reactions and up-regulating of M2 marker Arg-1 (Korbecki et al. 2019).
Various drugs influence microglia polarization through modulation of signaling pathways (Guo et al. 2022). Carvedilol (CVL) is a non-selective β-blocker that has been widely used as a therapeutic drug in hypertension and ischemic cardiac disease. It has been demonstrated that microglia express high levels of β2-receptors (Lechtenberg et al. 2019; Damo et al. 2023). Tozaki-Saitoh and colleagues have shown that pharmacological inhibition of α1, β1, and especially β2 receptors decreases lipopolysaccharide (LPS)-induced inflammation in murine microglial BV2 cells. They found that exchange protein directly activated by cyclic adenosine monophosphate (EPAC) and tumor progression locus-2 (TPL2) are involved in β2-receptor-mediated IL-1β production in microglial cells (Tozaki-Saitoh et al. 2020). Furthermore, CVL exhibits potent antioxidant activity that is not related to its ability to block adrenergic receptors. It has been demonstrated that CVL is a strong inhibitor of toll-like receptor-4 (TLR4) signaling (Xu et al. 2018). The TLR4/NF-kB/NLPR3 axis plays a crucial role in innate immunity induction and IL-1β release (Yang et al. 2019). The anti-inflammatory and antioxidant effects of CVL have been reported in many studies (Xu et al. 2022). CVL can reduce the release of pro-inflammatory cytokines and oxidative stress, and alleviate the neuroinflammation in Alzheimer’s disease and brain ischemia (Lysko et al. 1992; Wang et al. 2011). CVL could play a protective role in spinal cord injury by decreasing the levels of ROS and inflammatory cytokines (Karatas et al. 2015; Liu et al. 2015). CVL treatment significantly decreases TNF-α, IL-1β, IL-6, and NF-κB p65 levels in hypoxia-induced inflammation in BV2 microglia cells (Gao et al. 2017). Furthermore, it has been shown that CVL alleviates NF-κB, TNF-α, IL-6, nitric oxide (NO), and NLRP3 expression in LPS-induced J774A.1 murine macrophage cells (Wong et al. 2018). The present study aimed to investigate the protective effect of CVL against the LPS-induced activation and inflammatory responses of BV2 microglial cells in vitro.
Materials and methods
Cell culture
Murine BV2 and human neuroblastoma SH-SY5Y cell lines were obtained from the National Cell Bank of Iran (Pasteur Institute of Tehran, Iran) and were used as in vitro models of microglial and neuronal cells, respectively. Cells were cultured in high-glucose Dulbecco’s Modified Eagle Medium (DMEM; Capricorn: DMEM-HPA-P10) containing 10% fetal bovine serum (FBS; Panbioteh: P30-3033), 2 mM L-glutamine (Gibco: 25,030), 100 mg/mL streptomycin, 10,000 U/mL penicillin (Panbiotech: P06-07100), and non-essential amino acid (NEAA; Gibco: 11,140,050) at 37 °C in a humidified atmosphere of 5% CO2. Cells were passaged every 2–3 days to maintain growth.
Cytotoxicity assay
The toxicity of LPS and CVL on BV2 cells was estimated using 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay. Briefly, BV2 cells were seeded into each well of 96-well plates at a density of 5 × 103 cells per well in 200 µL. The cells were then treated with increasing concentrations of LPS (1 to 500 µg/mL) and CVL (0.01 to 25 µM) for 48 h. After the completion of treatment, 20 µL of MTT (MTT; Sigma: M2128) solution (5 mg/mL in PBS) was added to 180 µL medium in each culture well and incubated for 4 h at 37 °C. The formazan crystals were then dissolved in dimethyl sulfoxide (DMSO; Sigma: D4540). The absorbance was measured at 570 nm using an ELISA reader (Synergy HT, BioTek) (Niapour et al. 2015, 2016). The viability of cells in each group was verified as the percentage of cell viability relative to the control. The median inhibitory concentrations (IC50) of each group were estimated by performing the four parametric nonlinear regressions in SigmaPlot software V12 (Systat Software, Inc., San Jose, CA, USA) (Niapour et al. 2019). The entire experiment was repeated three times from three independent samples. Based on our findings and in agreement with previous research (Dai et al. 2015; Bao et al. 2019; Peng et al. 2021; Guo et al. 2022), 0.5 μM of CVL and 10 µg/mL of LPS were chosen as the optimal concentrations for the rest of this study.
Measurement of intracellular ROS
Intracellular ROS levels were directly measured using the DCFDA/DCF Cellular ROS Assay Kit (Abcam: ab113851). Following the manufacturer’s instructions, BV2 cells were seeded into a 96-well plate at a density of 25 × 103 cells per well in 200 µL and incubated overnight. Cells were pre-treated with 0.5 µM of CVL in 150 µL/well of 1X supplemented buffer for 6 h. BV2 cells were then post-treated with LPS at 10 µg/mL in 150 µL/well of 1X supplemented buffer for 18 h. 2X dilution (40 µM) 2′, 7′-dichlorofluorescein diacetate (DCFDA) in 150 µL of 1X supplemented buffer was overlaid on top of the treated cells 45 min before the end of the experiment. The fluorescence intensity of the 2′,7′-dichloro-fluorescein (DCF) was measured by a fluorescence microplate reader (Synergy HT, Bio-Tek) at Ex/Em = 485/535 nm. Tertbutyl hydrogen peroxide (TBHP) at 150 µM was used as a positive control (Mahmoudinia and Niapour 2019). The entire experiment was run in duplicate from three independent samples.
Measurement of IL-1β release
The protein level of IL-1β was measured with a commercially available enzyme-linked immunosorbent assay (ELISA) kit (R&D: DY401). The optical density was determined at 450 nm using a microplate reader (Synergy HT, Bio-Tek). The absolute value of each protein was calculated based on the standard curve, and data was reported as a percentage of the control (Saadati et al. 2021). The entire experiment was run in duplicate from three independent samples.
Western blotting
BV2 cells were collected and washed twice with PBS. Subsequently, the cells were homogenized in a lysis buffer containing the following components: 0.5% Nonidet P-40, 1% Triton X-100, 50 mM Tris–HCl (pH = 7.4), 150 mM NaCl, 1 mM dithiothreitol (DTT), 1 mM ethylenediaminetetraacetic acid (EDTA), 0.1% sodium dodecyl-sulfate (SDS), 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 2 mM phenylmethylsulfonyl fluoride (PMSF), and 1% protease inhibitor cocktail. After quantification of proteins, the samples (20 μg proteins) were separated by 12% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel and transferred to polyvinylidene difluoride (PVDF) membranes. Nonspecific binding was blocked with Tris-buffered saline (TBS), and membranes were then incubated with the primary antibodies (Table 1) at 4 °C overnight. After further washing with tris-buffered saline with Tween 20 (TTBS), the membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Table 1) at room temperature for 1.5 h. The bands were visualized using the enhanced chemiluminescence (ECL) detection kit (Niapour et al. 2023). To serve as an internal control, β-actin profile was also detected alongside the target proteins. Band intensities were determined using the ImageJ software version 1.53, developed by the (National Institutes of Health (NIH) in Bethesda, USA). The entire experiment was repeated three times using independent samples.
Treatment of SH-SY5Y cells with the conditioned media of BV2 cells
BV2 cells were primed with 0.5 μM of CVL in 200 µL for 6 h and then treated with 10 µg/mL of LPS in 200 µL for the next 18 h. The culture media were collected either from co-treated cells or individual treatments of CVL and LPS. After centrifugation at 1800 revolutions per minute (RPM) for 10 min and removal of cell debris, the supernatant was used as BV2 microglia-conditioned medium (CM) (Cacci et al. 2008).
Each well of 96-well plates was seeded with 5 × 103 of SH-SY5Y cells in 200 µL and incubated overnight. SH-SY5Y cells were then incubated with 200 µL of CM of BV2 cells for 48 h in 5% CO2 at 37 °C, and neuronal viability was determined using MTT assay. The entire experiment was repeated three times from three independent samples.
Statistical analysis
Values of experiments in this study were presented as means ± standard deviation (SD) and were analyzed using SPSS software version 20. Comparisons among groups were conducted by one-way analysis of variance (ANOVA) followed by Tukey’s test for post hoc analysis. A p value of < 0.05 represented a statistically significant difference.
Results
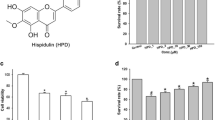
CVL and LPS affect the viability of BV2 cells at higher concentrations
MTT assay was carried out to assess the viability of cells following treatment with various concentrations of CVL and LPS. BV2 cellular viability was not affected by concentrations up to 5 μM of CVL. Exposure to 1–10 µg/mL of LPS resulted in a minimal reduction in cellular viability. However, increasing the concentrations of both CVL and LPS led to a significant reduction in BV2 cellular viability in a dose-dependent manner (Fig. 1A) (Supplementary file 1). The estimated IC50 for LPS- and CVL-received cells was 30.2 ± 1.83 μg/mL and 6.89 ± 0.05 μM, respectively. Furthermore, our results indicated that pre-treatment with 0.5 µM of CVL for 6 h followed by post-treatment with 10 µg/mL of LPS for 18 h does not exert a significant effect on cellular viability as compared with CVL alone and LPS alone groups (Fig. 1B) (Supplementary file 1).

Cytotoxicity of CVL and LPS on BV2 microglial cells. BV2 cells were treated with various concentrations of CVL and LPS, and their dose–response curves were shown (A). Viability of BV2 cells following pre-and post-treatment with a selected dose of CVL and LPS was illustrated (B). *p < 0.05, **p < 0.01, and ***p < 0.001 vs. control group
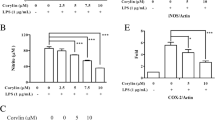
CVL suppressed the LPS‐induced ROS generation and IL-1β release in BV2 cells
In our study, CVL treatment reduced ROS and IL-1β compared to the control group. However, stimulation of BV2 cells resulted in a significant increase in the levels of ROS and IL-1β. In the presence of CVL, there was a significant (p < 0.05) reduction in the production of ROS and IL-1β in LPS-stimulated BV2 cells (Fig. 2A, B) (Supplementary file 2).

Effect of CVL on ROS and IL-1β levels. The levels of intracellular ROS and IL-1β in different groups were plotted (A, B), respectively. TBHP was used as a positive control in ROS assay. *p < 0.05 and ***p < 0.001 vs. control group. ##p < 0.01 and ###p < 0.001 vs. LPS group
CVL modulates Notch and PPAR-γ pathways and promotes M2 polarization of microglial cells
Western blot results showed that LPS led to a significant increase in the NF-κB p65 and NLRP3 protein levels as compared with the control group. However, CVL alone and pre-treatment with CVL significantly ameliorated NF-κB p65 and NLRP3 proteins as compared with the control group and LPS-stimulated BV2 cells, respectively (Fig. 3A, B) (Supplementary file 3).
To further evaluate the effect of CVL on the phenotypical switch of BV2 cells, the expression levels of iNOS as an M1-associated microglial marker and Arg-1 as a marker of microglial M2 polarization were assessed. CVL alone significantly altered the levels of iNOS and Arg-1 compared to the control. The results showed that treatment with LPS increases the expression of iNOS and decreases the expression of Arg-1. Pre-treatment with CVL showed the opposite effect and led to a decrease in the expression of iNOS and an increase in the expression of Arg-1 in LPS-stimulated BV2 cells (Fig. 3A, C) (Supplementary file 3).
Our results revealed that LPS induced key elements of the Notch signaling pathway while CVL alone and pre-treatment with CVL decreased cleaved-NOTCH1 and hairy and enhancer of split-1 (HES1) protein levels as compared to the control and LPS-stimulated groups. Furthermore, PPAR-γ as important anti-inflammatory signaling was reduced in LPS-induced cells. CVL alone and CVL pre-treatment significantly increased PPAR-γ levels as compared with the control and LPS-treated BV2 cells, respectively (Fig. 3A, D) (Supplementary file 3).

Effect of CVL on inflammatory cascade and microglial polarization. The expression profile of various proteins in individual and combinational treatments was illustrated (A). The expression levels of NF-κB p65 and NLRP3 were quantified (B). The quantification of iNOS and Arg-1 as microglial M1/M2 dynamics was shown (C). The cleaved-NOTCH1, HES1, and PPAR-γ levels were illustrated as a columnar graph (D). **p < 0.01, and ***p < 0.001 vs. control group. ##p < 0.01 and ###p < 0.001 vs. LPS group
CVL protects SH-SY5Y cells against the deleterious effects of microglial activation by LPS
Incubation of SH-SY5Y cells with the CM of LPS-induced BV2 cells resulted in a significant decrease in cell viability (p < 0.001). However, the viability of SH-SY5Y cells was significantly improved by the cultivation with the CM of CVL-primed/LPS-induced BV2 cells (Fig. 4) (Supplementary file 4).

Viability of SH-SY5Y following incubation with BV2-conditioned medium. CM of BV2 cells that were acutely stimulated with LPS and those that were pretreated with CVL and then treated with LPS were collected and used on SH-SY5Y cells. The viability of SH-SY5Y cells was measured using MTT assay and summarized. *p < 0.05, ***p < 0.001 vs. SH-SY5Y group. ##p < 0.01 vs. LPS-induced CM group
Discussion
Activation of microglia is a common pathological feature of several neurodegenerative diseases and traumatic injuries of CNS. Several studies showed that the reduction of neuroinflammation may require specific treatment with simultaneous reduction of the M1 phenotype and promotion of the M2 phenotype (Hu et al. 2015). Therefore, regulation of activated microglia is considered as a therapeutic approach. Numerous studies have emphasized the beneficial effects of CVL on Alzheimer’s disease and traumatic brain and spinal cord injury through reducing oxidative stress and proinflammatory cytokines (Wang et al. 2011; Liu et al. 2015; Amirshahrokhi and Niapour 2022). The immortalized BV2 cell line is generally used as an in vitro microglial inflammatory model system. Various experiments have been carried out to determine the anti-inflammatory effects of drugs on hypoxia and LPS-activated microglial cell lines (Streit et al. 2004; Peng et al. 2020; Wu et al. 2021). Gao et al. reported that treating with 10 µM of CVL for 24 h can reduce ROS, IL-1β, NF-κB, and TNF-α in hypoxia-induced BV2 cells (Gao et al. 2017). It has been shown that CVL at a dose of 5 µM exerts a protective effect against 6-hydroxydopamine (6-OHDA) toxicity in rat pheochromocytoma PC12 cells (Wang et al. 2014). Another research has indicated that 10 µg/mL of LPS activates BV2 cells, triggers inflammation, and reduces PC12 cell viability (Dai et al. 2015). In the present work, we speculated 0.5 μM of CVL and 10 µg/mL of LPS as the appropriate concentrations. The implemented concentration of CVL was selected based on the physiologic concentrations of CVL (0.5–1 µM) (Peng et al. 2021; Guo et al. 2022) as well as the absence of cytotoxicity up to 5 µM. Our data indicated that CVL at minimum serum concentration could elicit its anti-inflammatory functions. Lower concentrations of LPS generally act as a cytostatic rather than cytotoxic agent (Zhuang and Wogan 1997). Concentrations of LPS in the range of 1 to 10 µg/mL have been widely used in published research (Dai et al. 2015, 2019; Bao et al. 2019). The concentration of LPS which can induce an inflammatory response was important in our study. The results of our preliminary study indicated that a concentration of 10 µg/mL of LPS was sufficient to induce IL-1β with minimal effects on cell viability.
Oxidative stress, as a result of an imbalance between free radical production and effective scavenging, can influence cell viability and metabolic activity. Inflammation caused by injury and LPS leads to mitochondrial dysfunction and the production of ROS and NO (Wong et al. 2018). A previous study reported that treatment with CVL decreases the levels of ROS and inflammatory cytokines in LPS-treated human bronchial epithelial cells (BEAS2B) (Xu et al. 2022). Although a higher concentration of LPS was applied in our study, similar results were detected in BV2 cells. These data support the antioxidant properties of CVL.
We demonstrated that pre-treatment of BV2 cells with CVL leads to the reduction of NF-κB p65, IL-1β, and NLRP3 protein levels. LPS functions as a ligand for the TLR4 receptor and leads to the up-regulation of IL-1β gene expression (Swanson et al. 2019). The inhibitory effect of CVL on TLR4 signaling has been demonstrated (Xu et al. 2018). Our findings confirm the reduction of NF-κB p65 and IL-1β in CVL pre-treated cells as compared with LPS-primed BV2 cells. It has been demonstrated that LPS and NF-κB p65 activate NLRP3 inflammasome and mediate the secretion of mature IL-1β (Swanson et al. 2019). Therefore, we propose the inhibitory effect of CVL on NLRP3 activation through its impact on TLR4/NF-κB priming signals and its ability to reduce ROS production, which serves as the second signal for NLRP3 assembly and activation. The present work is in agreement with prior research demonstrating the anti-inflammatory effects of CVL by reducing NLRP3 inflammasome expression (Wong et al. 2018).
Microglia polarization and the ratio between M1/M2 phenotypes play essential roles in their cooperation during tissue repair. It was shown that the M1 microglia release a large number of harmful substances, such as inflammatory agents and oxygen-free radicals during the early stages of inflammation which damage the structure of neurons and impair their normal functions. However, the M2 phenotype of microglia secretes neurotrophic substances in the inflammatory process, removes necrotic or apoptotic neuron fragments, and subsequently leads to tissue repair (Zendedel et al. 2016; Dai et al. 2019). We verified that treatment with 0.5 μM of CVL can lead to a decrease in the expression of iNOS protein as a marker of M1 phenotype and an increase in the expression of Arg-1 protein as a marker of M2 polarization. Treatment with CVL increased this ratio toward Arg-1 and promoted the polarization of microglial cells to the M2 phenotype. To our knowledge, it is the first time we have demonstrated the impact of CVL on the polarization of microglia toward the M2 phenotype. Therefore, CVL reduces tissue damage due to the reduction of inflammatory factors and oxygen-free radicals.
The Notch signaling pathway is initiated by several factors including oxidative stress. NOTCH1 regulates IL-6 expression in activated macrophages through the NF-κB pathway. It was shown that NOTCH1 promotes the polarization of the M1 phenotype of microglial cells (Wu et al. 2018; Kong et al. 2022). NOTCH1 has also been identified as a stimulator of the NLRP3 inflammasome signaling pathway (Lee et al. 2020; Guo et al. 2022). It has been demonstrated that NLRP3 activation can switch microglia states toward the M1 phenotype (Ibrahim et al. 2023). Hence, the Notch/NF-κB/NLRP3 axis appears to be crucial in driving microglia toward the M1 phenotype. Our findings indicate that the administration of CVL may promote the polarization of microglia from M1 to M2 by suppressing the Notch/NF-κB/NLRP3 axis. A previous study reported that the administration of 30 mg/kg CVL for 1 week can reduce the expression of NOTCH1, HES1, NF-κB, and TNF-α in an animal model of ulcerative colitis (Nasser et al. 2022). PPAR-γ activation decreases inflammatory mechanisms by inhibiting the expression of TNF-α and influencing the activity of NF-κB (Nasser et al. 2022). The data presented in this report demonstrate, for the first time, that LPS increased the expression of NOTCH1 and HES1 proteins in BV2 cells while decreased the expression of PPAR-γ. Interestingly, treatment with 0.5 μM CVL for 6 h mitigated the protein expressions of NOTCH1 and HES1 and increased the expression of PPAR-γ. It has been shown that PPAR-γ agonists attenuate NLRP3 inflammasome activation in macrophages (Yang et al. 2021).
We showed that the CM of LPS-activated BV2 cells reduced SH-SY5Y cell viability. This finding suggests that microglial cells spread inflammation to neurons through cytokines after initial activation. This process occurs naturally after inflammatory and ischemic conditions. Additionally, our findings demonstrated that incubation with the CM of CVL-primed BV2 cells significantly improved the viability of SH-SY5Y cells as compared with the LPS-induced cells. Interestingly, CVL pre-treatment did not compensate for the diminished viability of BV2 cells treated with LPS (Supplementary file 5). In line with our findings, it was shown that the exposure of PC12 cells to the CM of LPS-induced BV2 cells elevates apoptosis mediators and decreases cell viability. IL-1β released from activated BV2 microglia is responsible for the damage of neurons (Dai et al. 2015). Another study documented that incubation of SH-SY5Y cells with the CM of LPS-stimulated BV2 cells reduces SH-SY5Y cell survival and increases apoptosis. Liu and colleagues have also shown that butein can attenuate the toxic effects of LPS-induced BV2 cells on SH-SY5Y cells through the reductions of the activation of NF-κB p65 and extracellular signal-regulated kinase (ERK) signaling pathways (Liu et al. 2020). Cyanidin-3-O-glucoside was able to reduce the toxic effect of LPS-induced BV2 cells on PC12 cells (Kaewmool et al. 2020). Based on these findings, CVL may exert its neuroprotective effects by reducing the production of inflammatory cytokines in microglial cells. Although several studies have investigated the impact of microglial activation on SH-SY5Y cell viability, some limitations should be considered. The SH-SY5Y cell line is derived from human neuroblastoma. The cancerous properties of SH-SY5Y cells may influence their differentiation, viability, proliferation, and metabolic activity (Peng et al. 2021). The mice and human microglia express a set of conserved sensing genes that regulate microglial responses to environmental changes. However, some soluble factors secreted by each cell type may not have appropriate receptors on cells of different animal origins (Abels et al. 2021). This finding highlights the need for further research to better understand the differences in microglia response between species.
Conclusion
The present study indicated that CVL reduces ROS levels and key inflammatory machinery such as NF-κB p65, modulates Notch and PPAR-γ signaling pathways, and promotes the polarization of microglial cells toward the M2 phenotype. CVL also exhibits neuroprotective effects on SH-SYY cells when treated with CM of LPS-induced BV2 cells. Further evaluations are needed to understand cellular and molecular mechanisms of CVL against neuro-inflammation.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Abels ER, Nieland L, Hickman S, Broekman MLD, El Khoury J, Maas SLN (2021) Comparative analysis identifies similarities between the human and Murine microglial sensomes. Int J Mol Sci 22(3):1495. https://doi.org/10.3390/ijms22031495
Amirshahrokhi K, Niapour A (2022) Carvedilol attenuates brain damage in mice with hepatic encephalopathy. Int Immunopharmacol 111:109119
Bao Y, Zhu Y, He G, Ni H, Liu C, Ma L, Zhang L, Shi D (2019) Dexmedetomidine attenuates neuroinflammation in LPS-stimulated BV2 microglia cells through upregulation of miR-340. Drug Des Dev Ther 13:3465–3475
Becher B, Spath S, Goverman J (2017) Cytokine networks in neuroinflammation. Nat Rev Immunol 17:49–59
Cacci E, Ajmone-Cat MA, Anelli T, Biagioni S, Minghetti L (2008) In vitro neuronal and glial differentiation from embryonic or adult neural precursor cells are differently affected by chronic or acute activation of microglia. Glia 56:412–425
Dai XJ, Li N, Yu L, Chen ZY, Hua R, Qin X, Zhang YM (2015) Activation of BV2 microglia by lipopolysaccharide triggers an inflammatory reaction in PC12 cell apoptosis through a toll-like receptor 4-dependent pathway. Cell Stress Chaperones 20:321–331
Dai W, Wang X, Teng H, Li C, Wang B, Wang J (2019) Celastrol inhibits microglial pyroptosis and attenuates inflammatory reaction in acute spinal cord injury rats. Int Immunopharmacol 66:215–223
Damo E, Agarwal A, Simonetti M (2023) Activation of beta2-adrenergic receptors in microglia alleviates neuropathic hypersensitivity in mice. Cells 12(2):284. https://doi.org/10.3390/cells12020284
DiSabato DJ, Quan N, Godbout JP (2016) Neuroinflammation: the devil is in the details. J Neurochem 139(Suppl 2):136–153
Fazio C, Ricciardiello L (2016) Inflammation and Notch signaling: a crosstalk with opposite effects on tumorigenesis. Cell Death Dis 7:e2515
Gao X, Wu B, Fu Z, Zhang Z, Xu G (2017) Carvedilol abrogates hypoxia-induced oxidative stress and neuroinflammation in microglial BV2 cells. Eur J Pharmacol 814:144–150
Guo S, Wang H, Yin Y (2022) Microglia polarization from M1 to M2 in neurodegenerative diseases. Frontiers in Aging Neuroscience 14:815347
He Y, Hara H, Núñez G (2016) Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci 41:1012–1021
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Chen J (2015) Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol 11:56–64
Ibrahim WW, Skalicka-Wozniak K, Budzynska B, El Sayed NS (2023) NLRP3 inflammasome inhibition and M1-to-M2 microglial polarization shifting via scoparone-inhibited TLR4 axis in ovariectomy/D-galactose Alzheimer’s disease rat model. Int Immunopharmacol 119:110239
Kaewmool C, Udomruk S, Phitak T, Pothacharoen P, Kongtawelert P (2020) Cyanidin-3-O-glucoside protects PC12 cells against neuronal apoptosis mediated by LPS-stimulated BV2 microglial activation. Neurotox Res 37:111–125
Karatas Y, Cengiz SL, Esen H, Toker A, Savas C (2015) Effect of carvedilol on secondary damage in experimental spinal cord injury in rats. Turk Neurosurg 25:930–935
Kong P, Cui ZY, Huang XF, Zhang DD, Guo RJ, Han M (2022) Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct Target Ther 7:131
Korbecki J, Bobiński R, Dutka M (2019) Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflammation Research: Official Journal of the European Histamine Research Society [et Al] 68:443–458
Kwon HS, Koh SH (2020) Neuroinflammation in Neurodegenerative Disorders: the Roles of Microglia and Astrocytes 9:42
Lechtenberg KJ, Meyer ST, Doyle JB, Peterson TC, Buckwalter MS (2019) Augmented beta2-adrenergic signaling dampens the neuroinflammatory response following ischemic stroke and increases stroke size. J Neuroinflammation 16:112
Lee S, Kim SK, Park H, Lee YJ, Park SH, Lee KJ, Lee DG, Kang H, Kim JE (2020) Contribution of autophagy-notch1-mediated NLRP3 inflammasome activation to chronic inflammation and fibrosis in keloid fibroblasts. Int J Mol Sci 21(21):8050. https://doi.org/10.3390/ijms21218050
Liu D, Huang Y, Li B, Jia C, Liang F, Fu Q (2015) Carvedilol promotes neurological function, reduces bone loss and attenuates cell damage after acute spinal cord injury in rats. Clin Exp Pharmacol Physiol 42:202–212
Liu Y, Fu Y, Zhang Y, Liu F, Rose GM, He X, Yi X, Ren R, Li Y, Zhang Y, Wu H, Lv C, Zhang H (2020) Butein attenuates the cytotoxic effects of LPS-stimulated microglia on the SH-SY5Y neuronal cell line. Eur J Pharmacol 868:172858
Ludwig PE, Patil AA, Chamczuk AJ, Agrawal DK (2017) Hormonal therapy in traumatic spinal cord injury. American Journal of Translational Research 9:3881–3895
Luo YP, Jiang L, Kang K, Fei DS, Meng XL, Nan CC, Pan SH, Zhao MR, Zhao MY (2014) Hemin inhibits NLRP3 inflammasome activation in sepsis-induced acute lung injury, involving heme oxygenase-1. Int Immunopharmacol 20:24–32
Lysko PG, Lysko KA, Yue TL, Webb CL, Gu JL, Feuerstein G (1992) Neuroprotective effects of carvedilol, a new antihypertensive agent, in cultured rat cerebellar neurons and in gerbil global brain ischemia. Stroke 23(11):1630–1635. https://doi.org/10.1161/01.str.23.11.1630
Mahmoudinia S, Niapour A (2019) 2,4-D causes oxidative stress induction and apoptosis in human dental pulp stem cells (hDPSCs). Environ Sci Pollut Res Int 26(25):26170–26183. https://doi.org/10.1007/s11356-019-05837-0
Mortezaee K, Khanlarkhani N, Beyer C, Zendedel A (2018) Inflammasome: its role in traumatic brain and spinal cord injury. J Cell Physiol 233:5160–5169
Nasser S, Abdallah DM, Ahmed KA, Abdel-Mottaleb Y, El-Abhar HS (2022) The novel anti-colitic effect of β-adrenergic receptors via modulation of PS1/BACE-1/Aβ axis and NOTCH signaling in an ulcerative colitis model. Front Pharmacol 13:1008085
Niapour N, Niapour A, Sheikhkanloui Milan H, Amani M, Salehi H, Najafzadeh N, Gholami MR (2015) All trans retinoic acid modulates peripheral nerve fibroblasts viability and apoptosis. Tissue Cell 47:61–65
Niapour N, Mohammadi-Ghalehbin B, Golmohammadi MG, Gholami MR, Amani M, Niapour A (2016) An efficient system for selection and culture of Schwann cells from adult rat peripheral nerves. Cytotechnology 68:629–636
Niapour A, Ghasemi Hamidabadi H, Niapour N, Mohammadi P, Sharifi Pasandi M, Malekzadeh V (2019) Pharmacological Notch pathway inhibition leads to cell cycle arrest and stimulates ascl1 and neurogenin2 genes expression in dental pulp stem cells-derived neurospheres. Biotechnol Lett 41:873–887
Niapour A, Miran M, Seyedasli N, Norouzi F (2023) Anti-angiogenic effects of aqueous extract from Agrostemma githago L. seed in human umbilical vein endothelial cells via regulating Notch/VEGF, MMP2/9, ANG2, and VEGFR2. Environ Sci Pollut Res Int 30:22413–22429
Peng Y, Chu S, Yang Y, Zhang Z, Pang Z, Chen N (2021) Neuroinflammatory in vitro cell culture models and the potential applications for neurological disorders. Front Pharmacol 12:671734
Peng X, Li C, Yu W, Liu S, Cong Y, Fan G, Qi S (2020) Propofol attenuates hypoxia-induced inflammation in BV2 microglia by inhibiting oxidative stress and NF-κB/Hif-1α signaling. Biomed Res Int 2020:8978704. https://doi.org/10.1155/2020/8978704
Roe K (2021) An inflammation classification system using cytokine parameters. Scand J Immunol 93:e12970
Saadati H, Noroozzadeh S, Esmaeili H, Amirshahrokhi K, Shadman J, Niapour A (2021) The neuroprotective effect of mesna on cisplatin-induced neurotoxicity: behavioral, electrophysiological, and molecular studies. Neurotox Res 39(3):826–840. https://doi.org/10.1007/s12640-020-00315-9
Streit WJ, Mrak RE, Griffin WS (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation 1:14
Swanson KV, Deng M, Ting JP (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 19:477–489
Tozaki-Saitoh H, Sasaki I, Yamashita T, Hosoi M, Kato TA, Tsuda M (2020) Involvement of exchange protein directly activated by cAMP and tumor progression locus 2 in IL-1beta production in microglial cells following activation of beta-adrenergic receptors. J Pharmacol Sci 143:133–140
Wang J, Ono K, Dickstein DL, Arrieta-Cruz I, Zhao W, Qian X, Lamparello A, Subnani R, Ferruzzi M, Pavlides C, Ho L, Hof PR, Teplow DB, Pasinetti GM (2011) Carvedilol as a potential novel agent for the treatment of Alzheimer’s disease. Neurobiol Aging 32:2321.e2321–2312
Wang L, Wang R, Jin M, Huang Y, Liu A, Qin J, Chen M, Wen S, Pi R, Shen W (2014) Carvedilol attenuates 6-hydroxydopamine-induced cell death in PC12 cells: involvement of Akt and Nrf2/ARE pathways. Neurochem Res 39:1733–1740
Wong WT, Li LH, Rao YK, Yang SP, Cheng SM, Lin WY, Cheng CC, Chen A, Hua KF (2018) Repositioning of the β-blocker carvedilol as a novel autophagy inducer that inhibits the NLRP3 inflammasome. Front Immunol 9:1920
Wu F, Luo T, Mei Y, Liu H, Dong J, Fang Y, Peng J, Guo Y (2018) Simvastatin alters M1/M2 polarization of murine BV2 microglia via Notch signaling. J Neuroimmunol 316:56–64
Wu X, Gong L, Xie L, Gu W, Wang X, Liu Z, Li S (2021) NLRP3 deficiency protects against intermittent hypoxia-induced neuroinflammation and mitochondrial ROS by promoting the PINK1-parkin pathway of mitophagy in a murine model of sleep apnea. Front Immunol 12:628168
Xu Y, Chen S, Cao Y, Zhou P, Chen Z, Cheng K (2018) Discovery of novel small molecule TLR4 inhibitors as potent anti-inflammatory agents. Eur J Med Chem 154:253–266
Xu J, Zhao S, Zhao L, Sun M (2022) Carvedilol alleviates lipopolysaccharide (LPS)-induced acute lung injury by inhibiting Ras homolog family member A (RhoA)/ROCK activities. Bioengineered 13:4137–4145
Yang Y, Wang H, Kouadir M, Song H, Shi F (2019) Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 10:128
Yang CC, Wu CH, Lin TC, Cheng YN, Chang CS, Lee KT, Tsai PJ, Tsai YS (2021) Inhibitory effect of PPARgamma on NLRP3 inflammasome activation. Theranostics 11:2424–2441
Zendedel A, Johann S, Mehrabi S, Joghataei MT, Hassanzadeh G, Kipp M, Beyer C (2016) Activation and regulation of NLRP3 inflammasome by intrathecal application of SDF-1a in a spinal cord injury model. Mol Neurobiol 53:3063–3075
Zhuang JC, Wogan GN (1997) Growth and viability of macrophages continuously stimulated to produce nitric oxide. Proc Natl Acad Sci U S A 94:11875–11880
Funding
This study was financially supported by the Ardabil University of Medical Sciences (grant no. IR.ARUMS.REC.1400.203). Author Ali Niapour has received research support from the Ardabil University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
MJ.KH. performed cell culture and MTT assay. K.A. performed the ELISA assay. A.N. performed ROS and conditioned media assays, Western blotting, and data analysis. A.N. and Z.N. designed the study and prepared the manuscript and revisions. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding authors
Ethics declarations
Ethics approval
This study was approved by the ethical committee of Ardabil University of Medical Sciences (IR.ARUMS.REC.1400.203).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khoshnavay Foumani, M., Amirshahrokhi, K., Namjoo, Z. et al. Carvedilol attenuates inflammatory reactions of lipopolysaccharide-stimulated BV2 cells and modulates M1/M2 polarization of microglia via regulating NLRP3, Notch, and PPAR-γ signaling pathways. Naunyn-Schmiedeberg's Arch Pharmacol 397, 4727–4736 (2024). https://doi.org/10.1007/s00210-023-02914-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-023-02914-7