
Abstract
Inhibition of Helicobacter pylori urease is an effective method in the treatment of several gastrointestinal diseases in humans. This bacterium plays an important role in the pathogenesis of gastritis and peptic ulceration. Considering the presence of cysteine and N-arylacetamide derivatives in potent urease inhibitors, here, we designed hybrid derivatives of these pharmacophores. Therefore, cysteine-N-arylacetamide derivatives 5a-l were synthesized through simple nucleophilic reactions with good yield. In vitro urease inhibitory activity assay of these compounds demonstrated that all newly synthesized compounds exhibited high inhibitory activity (IC50 values = 0.35–5.83 μM) when compared with standard drugs (thiourea: IC50 = 21.1 ± 0.11 μM and hydroxyurea: IC50 = 100.0 ± 0.01 μM). Representatively, compound 5e with IC50 = 0.35 μM was 60 times more potent than strong urease inhibitor thiourea. Enzyme kinetic study of this compound revealed that compound 5e is a competitive urease inhibitor. Moreover, a docking study of compound 5e was performed to explore crucial interactions at the urease active site. This study revealed that compound 5e is capable to inhibit urease by interactions with two crucial residues at the active site: Ni and CME592. Furthermore, a molecular dynamics study confirmed the stability of the 5e-urease complex and Ni chelating properties of this compound. It should be considered that, in the following study, the focus was placed on jack bean urease instead of H. pylori urease, and this was acknowledged as a limitation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Urease (EC 3.5.1.5) is a Ni-containing metalloenzyme belonging to the amidohydrolase superfamily that is ubiquitously distributed. Bacterial ureases consist of two (αβ) or three (αβγ) subunits, while plant and fungal ureases are homo-oligomers (Jabri et al. 1995; Kafarski and Talma 2018). The catalytic activity and tertiary structure of discovered ureases are similar because they have ancestral proteins (Hameed et al. 2019; Mobley et al. 1995). This ubiquitously distributed enzyme accelerates the hydrolysis of urea to ammonia and carbon dioxide and led to agriculture deterioration and pathologic conditions in humans (Amtul et al. 2002; Mobley and Hausinger 1989; Rawluk et al. 2001). In agriculture, during urea fertilization, the major issue with urease activity is plant damage caused by nutrient deprivation and increased soil pH (Rawluk et al. 2001; Ahmad 1996). Urease also has been considered the key virulence factor of many pathogens that lead to a wide range of pathologic disorders including gastric adenocarcinoma, gastric lymphoma, gastric ulcers, pyelonephritis, hepatic encephalopathy, and urolithiasis (Abbasi et al. 2019; Algood and Cover 2006). Urease has become a central issue in Helicobacter pylori (H. pylori) infection which infect half of the world’s population, especially in developing country (Rutherford 2014). The ureolytic activity of H. pylori allows bacterial survival in the gastric acid leading to gastric malignancy (Scott et al. 1998).
Many attempts have been made to inhibit urease as a pivotal virulence factor. In this regard, a vast number of natural and synthetic compounds were examined against urease including phenylurea-pyridinium, thiadiazole, thioquinazolinone, and barbituric acid, and there is an urgent need for discovering new bioavailable, safe, efficient agents (Sadat-Ebrahimi et al. 2022; Asadi et al. 2022; Sohrabi et al. 2022; Asgari et al. 2020).
Some preliminary studies showed the potential urease inhibitory activity of simple thiol-containing compounds such as cysteamine (Fig. 1) (Amtul et al. 2006). Cysteine as a thiol-containing amino acid plays a crucial role in protein synthesis, detoxification, and metabolism of co-enzyme A and biotin (Fig. 1) (Smietana et al. 2008). This amino acid has applications in organic synthesis and serves as a precursor in pharmaceutical industries (Clemente Plaza et al. 2018). Cysteine-derived compounds and their nanoparticles exhibited potent antimicrobial activities (Egbujor et al. 2022; Shahbazi-Alavi and Safaei-Ghomi 2022; Novoa et al. 2022). Furthermore, cysteine derivatives A and B have inhibitory activity against urease (Fig. 1) (Amtul et al. 2006).

Rational of design for compounds 5a–l as new urease inhibitors
On the other hand, several series of N-arylacetamide derivatives such as compounds C and D demonstrated high inhibitory activity against urease (Fig. 1) (Liu et al. 2022; Nazari Montazer et al. 2021). As it has been proven about the mechanism of urease, this enzyme is dependent on nickel ions of the active site pocket; therefore, compounds with chelating properties can disrupt the enzyme’s function considering these data, the amino acid head (i.e., amine group and carboxylic acid) was considered a chelator for nickels, and the compounds were designed based on this possibility. Also, the nitrogen of the pyridine group and the carbonyl of the acetamide group of the N-(2-pyridyl)acetamide derivatives also acted as a nucleus with chelating potential (an additional bidentate ligand) to strengthen the interactions of the compounds with nickel metals. Based on these above-mentioned points, using the molecular hybridization technique, we connected cysteine to N-phenylacetamide derivatives (compounds 5a–i) and N-(2-pyridyl)acetamide derivatives (compounds 5j–l) to achieve new urease inhibitors (Fig. 1). These compounds were synthesized in two steps and evaluated against jack bean (JB) urease. Furthermore, in silico molecular docking and molecular dynamics (MD) studies were also performed to further investigate the interaction, orientation, and conformation of the synthesized compounds over the active site of urease.
Experimental details
Material and methods
Chemicals, reagents, and solvents were purchased from Sigma (USA) with no need for further purifications. Pre-coated silica gel, GF-254 (Merck, Germany), was used for thin layer chromatography, and ultraviolet light at 254, 366 nm was applied to visualize spots. Melting points were determined by the means of the Kofler hot stage apparatus (Reichert, Vienna, Austria) and were uncorrected. IR studies on KBr disks were performed using Nicolet Magna FTIR 550 spectrophotometers (Nicolet Instrument Corporation, Madison, WI, USA). 1H NMR spectra were recorded on a Bruker FT-NMR spectrometer (Bruker, Darmstadt, Germany) operating at 500 MHz in DMSO-d6 using tetramethylsilane (TMS) as an internal standard. Chemical shifts are given in ppm (δ), coupling constant (J) values are determined in Hertz (Hz), and spin multiplicities are shown as s (singlet), d (doublet), t (triplet), and m (multiplet). Perkin Elmer Model 240-C apparatus (PerkinElmer, Hopkinton, USA) was used to record elemental analyses that were within ± 0.4% of theoretical values for C, H, and N.
In vitro urease assay
In an attempt to determine the urease inhibitory activity of compounds 5A–L, we used the modified Berthelot spectrophotometric method 20. Ultra-pure water (HPLC grade, Duksan, Korea) was used throughout the experiments. The Synergy H1 Hybrid multi-mode microplate reader (BioTek Instruments, Winooski, VT, USA) was used to record absorbance at 625 nm. Jack bean urease from Canavalia ensiformis has been obtained from the Sigma-Aldrich company (CAS Number: 9002-13-5). Hydroxyurea and thiourea were used as standard inhibitors and the urease inhibitory activity of compounds 5a–l was determined at a concentration of 0–100 mg/mL. The assay solution with a total volume of 985 μL was prepared by mixing urea (850 μL) with synthesized compounds (5a–l; 100 μL, 0–100 mg/mL) and potassium phosphate buffer (100 mM) adjusted to pH 7.4. Afterward, the urease enzyme (15 μL, 1 mg/mL) was added and the resulting solution was incubated at 37 °C for 60 min. Next, the concentration of ammonia was determined by adding 100 μL of the incubated solution, 500 μL of solution A (consisting of 5.0 g phenol and 25.0 mg sodium nitroprusside in 500 mL distilled water), and 500 μL of solution B (consisting of 2.5 g of sodium hydroxide and 4.2 mL of sodium hypochlorite solution containing 5% chlorine in 500 mL distilled water) to each well. The absorbance (625 nm) was measured after incubation at 37 °C for 30 min. The uninhibited urease activity was assigned 100% as the control activity. Lastly, the following equation was applied to determine the percentage of urease inhibitory activity:
In this equation, I (%) is the percentage of enzyme inhibition, and T (test) and C (control) stand for the absorbance of the evaluated synthesized compounds and the absorbance of the solvent in the presence of the enzyme, respectively. The enzyme assay was performed on three separate experiments to ensure the reliability of the results and resulting data were expressed as mean ± standard deviation (S.D.). To calculate the IC50 values, GraphPad Prism 8 has been used for statistical analysis. The inhibitor concentration values were added to the logarithmic scale and the absorbance values turned into the normalized response percentage; the IC50 calculation was conducted using nonlinear regression.
Urease enzyme kinetic assay
The type of inhibition for compound 5E was determined using Michaelis–Menten kinetics. For this purpose, the initial rate of jack bean urease enzyme inhibition has been measured using (3.12, 6.25, 12.5, 25, 50, and 100 μM) concentrations of urea as the natural enzyme substrate and (0, 0.20, 0.35, and 0.70 μM) concentration of compound 5E as the inhibitor. Afterward, Lineweaver-Burk plot and secondary plot illustrated the inhibition type and the value of Ki.
Molecular docking procedure
The molecular docking investigation of synthesized compounds was performed by using the maestro molecular modelling platform (version 10.5), Schrödinger suites. X-ray crystallographic structure of jack bean urease in complex with acetohydroxamic acid by (PDB id: 4H9M) was obtained from (www.rcsb.com) (Begum et al. 2012). The protein preparation wizard was used to remove water molecules and co-crystalized atoms from the protein, and add missing loops using the prime tool and cap terminals (Sastry et al. 2013; Bell et al. 2012). Moreover, hetero atom states generated at pH 7.00 by EPIK and H-bonds were assigned using PROPKA at the same pH (Shelley et al. 2007). The 2D structure of ligands has been drawn in ChemDraw (ver.16) and exported as SDF files. The ligand preparation wizard was used to prepare the SDF files using the OPLS3e force field and defined metal binding sites using EPIK; moreover, the ligand ionization state was determined at (pH 7). The induced fit docking method was used to study the interactions of all ligands in which the AHA was considered the center of the grid (Farid et al. 2006). Van der Waals radii of receptor and ligand assigned 0.7 A and 0.5 A, respectively. The maximum number of 20 poses for each ligand has been calculated and structures beyond the prime energy levels of 30 kcal/mol were eliminated based on the sp glide docking.
Site map tool was used to investigate the active site contents of urease enzymes. Site map was tasked to report up to 5 potential binding sites with at least 15 site points per each reported site by a more restrictive definition of hydrophobicity.
Molecular dynamic simulation procedure
Molecular dynamic simulation conducted by Schrodinger maestro’s desmond interface used compound 5E-urease enzyme complex, obtained from the IFD stage as its input (Bowers et al. 2006). A system builder module was used to prepare the complex. The complex was placed at the center of a cubic cell with a 1A edge and filled by 27835 TI3P water molecules. The electrostatic charge of the system was neutralized by adding 92 Na and 78 Cl ions. The normal pressure and temperature (NPT) ensemble by Nose–Hoover chain method thermostat and Martyna–Tobias–Klein barostat was also used during the 100 ns simulation time.
General synthesis procedure for preparation of arylcarbamic chlorides 3a-l
To a mixture of aryl amines 1a–l (1mmol) and triethanolamine (1.1 mmol) in dichloromethane (10 mL), chloroacetyl chloride 2 (1.1 mmol) was added dropwise at 0 °C for 2 h. Then, a saturated bicarbonate solution was added to the reaction mixture. The dichloromethane layer was then separated and evaporated under a vacuum condition. The obtained precipitate was recrystallized with petroleum ether to get pure white products 3a–l in good yield.
General synthesis procedure for preparation of cysteine-N-arylacetamide 5a-l
A solution of compounds 3a–l (1mmol) and cysteine 4 (1.5 mmol) in methanol was prepared, and after 5 min the 0.5% sodium bicarbonate was added. Next, the solution mixture was stirred overnight at room temperature. The reaction progress was monitored by TLC. After reaction completion, water was added to achieve a white precipitate which was then washed with cold water to obtain pure compounds 5a–l in good yield. The synthesized compounds’ characterizations have been provided in a supplementary document.
Results and discussion
Chemistry
Cysteine-N-arylacetamide derivatives 5a–l were synthesized through facile nucleophilic reactions (Sch. 1). In the first step, amine derivatives 1a–l were reacted with chloroacetyl chloride 2 in the presence of N(Et)3 in CH2Cl2 to give N-arylacetamide derivatives 3a–l. In the second step, the latter compounds and cysteine 4 in the presence of sodium bicarbonate were converted to target compounds 5a–l. All target compounds were obtained in good yield. Furthermore, their structures were confirmed by NMR and IR spectroscopies and elemental analysis.

The synthesis procedure for cysteine-N-arylacetamides 5a-l
In vitro urease inhibitory activity
Cysteine-N-arylacetamide derivatives 5a–l were evaluated against urease. The in vitro anti-urease assay demonstrated that all title compounds showed high inhibitory activity (IC50 values = 0.35–5.83 μM) in comparison to positive controls thiourea (IC50 = 21.1 ± 0.11 μM) and hydroxyurea (IC50 = 100.0 ± 0.01 μM) (Table 1). Structurally, the synthesized compounds are divided into two series: N-phenylacetamide derivatives 5a–i and N-(2-pyridyl)acetamide derivatives 5j–l. The most active compounds were 4-chloro and unsubstituted analogs of the N-phenylacetamide series (compounds 5e and 5a) with IC50 values of 0.35 and 0.43, respectively.
The inhibitory activity of N-phenylacetamide derivatives 5a–i against urease demonstrated that 4-chloro derivative 5e showed the most potent activity, while 3-fluoro derivative 5d was the less active compound. Removing the 4-chloro substituent of compound 5e, as in compound 5a (the second potent compound), led to a slight decrease in inhibitory activity, while replacement of 4-chloro substituent with methoxy or bromo, as in compounds 5c and 5f, inhibitory activity reduced by more than 2- and 6-fold. Moreover, the introduction of 2-methoxy, 2-fluoro-4-nitro, 2,5-dichloro, or 2,4-dibromo on the phenyl ring of unsubstituted compound 5a did not improve anti-urease potency as observed in compounds 5b, 5g, 5h, and 5i.
In the N-(2-pyridyl)acetamide series (compounds 5j–l), the 5-substituted compounds 5j and 5l, respectively, with methyl and chloro substituents showed approximately the same inhibitory activity against urease, while 6-methyl derivative 5k shows less inhibitory activity compared to compounds 5j and 5l.
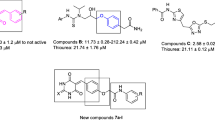
The comparison of anti-urease activities of the new cysteine-N-arylacetamide derivatives 5a–l with cysteine derivatives A–B and N-arylacetamide derivatives C–D demonstrated that our new compounds inhibit the target enzyme urease much more potently than the template compounds A–D (Amtul et al. 2006; Liu et al. 2022; Nazari Montazer et al. 2021). For example, as can be seen in Sch. 2, the comparison of IC50 values of the new cysteine-N-arylacetamide derivatives 5 with their corresponding analogs of 2-((5-amino-1,3,4-thiadiazol-2-yl)thio)-N-arylacetamide derivatives D revealed that replacement of 2-((5-amino-1,3,4-thiadiazol-2-yl)thio) moiety of these compounds with cysteine unit improved anti-urease activity (Nazari Montazer et al. 2021).

Anti-urease activities of 2-((5-amino-1,3,4-thiadiazol-2-yl)thio)-N-arylacetamide derivatives D and their corresponding analogs of new compounds 5
Urease kinetic assay
To understand the mechanism of inhibition of newly synthesized compounds, kinetic studies of the most potent urease inhibitor 5e were performed. The analysis of the Lineweaver-Burk plot and the secondary plot indicates a competitive inhibition for this compound with a ki value of 0.53 μM (Fig. 2).

Lineweaver-Burk plot (left) and secondary plot (right) of compound 5e inhibition of jack bean urease
Docking study
Through an analysis comparing the H. pylori urease (PDB: 1E9Z) (Ha et al. 2001) and the active sites of jack bean urease using the sitemap tool (Halgren 2009), notable similarities have been observed. Specifically, significant resemblances have been identified in the h-bond donor/acceptor and hydrophobic regions of the active sites (Fig. 3a); the surface content of each active site pocked is shown in Fig. 3b. Considering the similarities of the active sites and since the enzyme urease assay has been conducted on the jack bean urease, further molecular simulations also used the jack bean urease by the PDB ID of 4H9M as the receptor.

a The surface site map of jack bean urease (4H9M) and H. pylori urease (1E9Z). Each active site contains H-bond donor (blue), h-bond acceptor (red), and hydrophobic (yellow) surface areas. b The percentage of H-bond donor (blue), h-bond acceptor (red), and hydrophobic (yellow) surface areas of enzymes from the whole accessible surface area of the enzyme active site pocket
In addition to the similarity of interactive surfaces in the active sites of JBU and HPU enzymes, compound 5e was also docked in the active site pocket of HPU enzyme. The detailed 2D interaction of compound 5e with both enzymes is shown in Fig. 4. The interactions are notably similar: in both enzymes, Ni ions are chelated by the ligand’s C-terminal, and the amide group’s oxygen and nitrogen atoms form hydrogen bonds with the corresponding HIS and ALA residues in both active sites. Furthermore, in both active sites, the quaternary nitrogen exhibits a salt bridge interaction with the ASP residue.

The detailed 2D interactions of compound 5e with the active site pocket of HPU and JBU enzymes
The tertiary structure of the JB urease includes four domains: two αβ domains that form the handle and one head of this hammer shape enzyme and the other head of the enzyme is an (αβ)8 TIM barrel domain which contains the active site of the enzyme (Fig. 5) (Balasubramanian and Ponnuraj 2010). These head sides are connected through a middle β domain.

A The tertiary structure of jack bean urease. B The 2D interactions of compound 5e and the active site of JB urease. C 3D interaction of compound 5e and the active site of jack bean urease
The docking study was performed to elucidate the binding mode of the synthesized compounds in the active site of the JB urease. All synthesized compounds (5a–l) were capable of efficiently fitting into the active site of the enzyme with glide scores ranging from − 10.475 to − 7.45 kcal/mol. As can be seen in Table 1, the most potent derivatives were compound 5e (IC50 = 0.35 μM) and compound 5a (IC50 = 0.43 μM) which also exhibited the best in silico results with docking score values of − 10.475 and − 10.015 kcal/mol, respectively. Similarly, the least active derivatives 5d (IC50 = 5.83 μM) and 5k (IC50 = 3.01 μM) demonstrated the worse results with dock values of − 8.03 and − 7.48 kcal/mol, respectively.
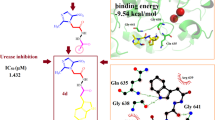
Figure 5 depicts the interactions of compound 5e within the urease active site. Docking studies revealed efficient inhibition through electrostatic, halogen, hydrophobic, and H-bond interactions. It is important to note that in silico ligand optimization, considering physiological pH, would result in ionization of the compound’s acid and amine moieties, as demonstrated.
In Fig. 5, the carboxylate group of compound 5e interacted with both Ni ions by salt bridge interaction and H-bond interaction with His492. Another salt bridge and H-bond interaction were established between the ammonium group and Asp633 and Ala440, respectively. The amide group of the ligand exhibited two H-bond interactions with His593 and Ala636. The chlorine atom of the phenyl ring created a halogen bond with Arg439. Moreover, several hydrophobic interactions were observed with CME592, KCX490, Met637, and His409. The vital unusual amino acid CME592 is a critical residue in the mobile flap of the active site in the open-closed-open procedure, and restriction of this flexibility leads to the destruction of reaction and enzyme activity. It can be inferred that numerous hydrogen bonds and hydrophobic interaction of compound 5e with the active site and CME592 could prevent the mobility of the flap and, eventually, reduce the activity of the urease enzyme.
Molecular dynamics simulations
To study the stability of the protein-ligand complex, the root mean square deviation (RMSD) of both complexed backbones of urease with thiourea and compound 5e was investigated through MD simulation. For small, globular protein normal variance of change is considered to be 1–3 A. Higher deviations from these amounts indicate a major conformational change of the protein tertiary structure. Based on the ligand-complex RMSD result, the simulation period was enough for both complexes to reach steadiness, after almost 30 ns (Fig. 6). The RMSD plot of urease backbone in complex with the compound 5e and complex with the thiourea is demonstrated in Fig. 6. As can be seen, fluctuation of compound 5e (average of 2 Å) seemed to be significantly lower than the thiourea complex (average of 3.1 Å).

RMSD plots of the urease backbone complexed with compound 5e (blue) and thiourea (red)
The active site of JB urease has a mobile flap consisting of Met590 to His607 residues. This mobile flap can physically restrict the active site pocket of the enzyme. As is shown in Fig. 7, compound 5e stabilized the closed-flap conformation of the enzyme’s active site which can be explained by a transition state inhibition of the enzyme. Distance between Ile599 and Ala440 is considered an indicator of mobile flap conformation toward the active site. The distance was found to be 21 Å and 32 Å in thiourea-urease and 5e-urease complexes, respectively. This explains the competitive inhibition of compound 5e.

Distance of Ile599 and Ala440 residues in the thiourea complex as the open flap state (left) and compound 5e complex as the closed flap state (right)
As is shown in Fig. 8a, compound 5e in an ionized state chelated with both nickel ions for almost all the duration of the 100-ns simulation. Unusual residue KCX and other important residues of the enzyme’s active site pocket including Gly500, His545, His519, His407, His409, and Asp633 made their interactions with the compound 5e through Ni ions in the whole simulation. Moreover, the Ala440 residue formed a hydrogen bond with the ionized amine group of the ligand. As shown in Fig. 8b, other interacting residues of the enzyme which had less interaction in 100 ns simulation time are CME592, Arg439, Ala549, Ala636, Gln635, and Arg609.

a Interactions of compound 5e and urease enzyme represented by two-dimensional and b timeline plots
In silico drug-likeness, ADME, and toxicity studies
The druglikeness, ADME (absorption, distribution, metabolism, and excretion), and toxicity properties of the most potent compounds 5e and 5a were predicted using PreADMET software. (Bioinformatics and Molecular Design Research Center 2014). Results are presented in Table 2, indicating that both compounds comply with Lipinski’s “rule of five.” They exhibit poor Caco-2 cell permeability but fall within acceptable ranges for blood-brain barrier (BBB) and skin permeability. Additionally, these compounds demonstrate high human oral absorption (HIA). Toxicity predictions reveal mutagenicity and low risk of cardiotoxicity (hERG inhibition). Compounds 5e and 5a showed carcinogenicity in mice but not in rats.
Conclusion
In this study, a novel series of cysteine-N-arylacetamide derivatives (5a–l) were successfully synthesized via a cost-effective and straightforward nucleophilic reaction, yielding promising urease inhibitors. It is worth noting that the study focused on jack bean urease instead of H. pylori urease, which should be considered a limitation. Nevertheless, the synthesized compounds exhibited remarkable inhibitory activity, outperforming standard inhibitors. Compound 5e, in particular, demonstrated exceptional potency, being approximately 60-fold more potent than the positive control, thiourea. In silico studies further elucidated the chelation of compound 5e with the Ni cofactor at the active site of urease, along with significant hydrophobic and H-bond interactions involving key residues such as CME592 and His593. Molecular dynamics simulations confirmed the stability of the 5e-urease complex and highlighted the interaction with dynamic flap residues. Despite the use of jack bean urease, these findings contribute valuable insights into urease inhibition.
Data Availability
The online version contains supplementary material and data.
References
Abbasi MA, Raza H, ur Rehman A, Siddiqui SZ, Nazir M, Mumtaz A, Shah SA, Seo SY, Hassan M (2019) Synthesis, antioxidant and in-silico studies of potent urease inhibitors: N-(4-{[(4-methoxyphenethyl)-(substituted) amino] sulfonyl} phenyl) acetamides. Drug Res 69:111–120
Ahmad N (1996) Nitrogen economy in tropical soils. Kluwer Academic Publishers, Dordrech, The Netherlands
Algood HMS, Cover TL (2006) Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin Microbiol Rev 19:597–613
Amtul Z, Kausar N, Follmer C, Rozmahel RF, Kazmi SA, Shekhani MS, Eriksen JL, Khan KM, Choudhary MI (2006) Cysteine based novel noncompetitive inhibitors of urease (s)—distinctive inhibition susceptibility of microbial and plant ureases. Bioorg Med Chem 14:6737–6744
Amtul Z, Siddiqui RA, Choudhary MI (2002) Chemistry and mechanism of urease inhibition. Curr Med Chem 9:1323–1348
Asadi M, Iraji A, Sherafati M, Montazer MN, Ansari S, Khanaposhtani MM, Tanideh N, Dianatpour M, Biglar M, Larijani B, Foroumadi A (2022) Synthesis and in vitro urease inhibitory activity of 5-nitrofuran-2-yl-thiadiazole linked to different cyclohexyl-2-(phenylamino) acetamides, in silico and kinetic studies. Bioorg Chem 120:105592
Asgari MS, Azizian H, Nazari Montazer M, Mohammadi-Khanaposhtani M, Asadi M, Sepehri S, Ranjbar PR, Rahimi R, Biglar M, Larijani B, Amanlou M (2020) New 1, 2, 3-triazole–(thio) barbituric acid hybrids as urease inhibitors: design, synthesis, in vitro urease inhibition, docking study, and molecular dynamic simulation. Arch Pharm 353:2000023
Balasubramanian A, Ponnuraj K (2010) Crystal structure of the first plant urease from jack bean: 83 years of journey from its first crystal to molecular structure. J Mol Biol 400:274–283
Begum A, Choudhary MI, Betzel C (2012) The first jack bean urease (Canavalia ensiformis) complex obtained at 1.52 resolution
Bell JA, Cao Y, Gunn JR, Day T, Gallicchio E, Zhou Z, Levy R, Farid R (2012) PrimeX and the Schrödinger computational chemistry suite of programs
Bioinformatics and Molecular Design Research Center (2014) Pre-ADMET program. Bioinformatics and Molecular Design Research Center, Seoul, South Korea http://preadmet.bmdrc.org
Bowers KJ, Chow DE, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK (2006) Scalable algorithms for molecular dynamics simulations on commodity clusters. InSC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing. IEEE 43-43
Clemente Plaza N, Reig García-Galbis M, Martínez-Espinosa RM (2018) Effects of the usage of l-cysteine (l-Cys) on human health. Molecules 23:575
Egbujor MC, Okoro UC, Okafor SN, Egu SA, Amasiatu IS, Egwuatu PI, Umeh OR, Ibo EM (2022) Design, synthesis, and molecular docking of cysteine-based sulphonamide derivatives as antimicrobial agents. Res Pharm Sci 17:99
Farid R, Day T, Friesner RA, Pearlstein RA (2006) New insights about HERG blockade obtained from protein modeling, potential energy mapping, and docking studies. Bioorg Med Chem 14:3160–3173
Ha NC, Oh ST, Sung JY, Cha KA, Lee MH, Oh BH (2001) Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat Struct Mol Biol Jun 8(6):505–509
Halgren TA (2009) Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 49(2):377–389
Hameed A, Al-Rashida M, Uroos M, Qazi SU, Naz S, Ishtiaq M, Khan KM (2019) A patent update on therapeutic applications of urease inhibitors (2012-2018). Expert Opin Ther Pat 29:181–189
Jabri E, Carr MB, Hausinger RP, Karplus PA (1995) The crystal structure of urease from Klebsiella aerogenes. Sci 268:998–1004
Kafarski P, Talma M (2018) Recent advances in design of new urease inhibitors: a review. J Adv Res 13:101–112
Liu ML, Li WY, Fang HL, Ye YX, Li SY, Song WQ, Xiao ZP, Ouyang H, Zhu HL (2022) Synthesis and biological evaluation of dithiobisacetamides as novel urease inhibitors. Chem Med Chem 17:e202100618
Mobley HL, Hausinger RP (1989) Microbial ureases: significance, regulation, and molecular characterization. Microbiol Rev 53:85–108
Mobley HL, Island MD, Hausinger RP (1995) Molecular biology of microbial ureases. Microbiol Rev 59:451–480
Nazari Montazer M, Asadi M, Bahadorikhalili S, Hosseini FS, Amanlou A, Biglar M, Amanlou M (2021) Design, synthesis, docking study and urease inhibitory activity evaluation of novel 2-((5-amino-1, 3, 4-thiadiazol-2-yl) thio)-N-arylacetamide derivatives. Med Chem Res 30:729–742
Novoa CC, Tortella G, Seabra AB, Diez MC, Rubilar O (2022) Cotton textile with antimicrobial activity and enhanced durability produced by L-cysteine-capped silver nanoparticles. Processes 10:958. https://doi.org/10.3390/pr10050958
Rawluk CDL, Grant CA, Racz GJ (2001) Ammonia volatilization from soils fertilized with urea and varying rates of urease inhibitor NBPT. Can J Soil Sci 81:239–246
Rutherford JC (2014) The emerging role of urease as a general microbial virulence factor. PLoS Pathog 10:e1004062. https://doi.org/10.1371/journal.ppat.1004062
Sadat-Ebrahimi SE, Bigdelou A, Sooreshjani RH, Montazer MN, Zomorodian K, Irajie C, Yahya-Meymandi A, Biglar M, Larijani B, Amanlou M, Iraji A (2022) Novel phenylurea-pyridinium derivatives as potent urease inhibitors: synthesis, in vitro, and in silico studies. J Mol Struct 1263:133078
Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27:221–234
Scott DR, Weeks D, Hong C, Postius S, Melchers K, Sachs G (1998) The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterology 114:58–70
Shahbazi-Alavi H, Safaei-Ghomi J (2022) Sonosynthesis of pyrimidines as antimicrobial agents using nano-Fe3O4–L-cysteine. Nano Res 7:28–35
Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M (2007) Epik: a software program for pK(a ) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 21:681–691
Smietana M, Clayette P, Mialocq P, Vasseur JJ, Oiry J (2008) Synthesis of new N-isobutyryl-L-cysteine/MEA conjugates: evaluation of their free radical-scavenging activities and anti-HIV properties in human macrophages. Bioorg Chem 36:133–140
Sohrabi M, Nazari Montazer M, Farid SM, Tanideh N, Dianatpour M, Moazzam A, Zomorodian K, Yazdanpanah S, Asadi M, Hosseini S, Biglar M (2022) Design and synthesis of novel nitrothiazolacetamide conjugated to different thioquinazolinone derivatives as anti-urease agents. Sci Rep 12:1–4
Acknowledgements
We are indebted and grateful to acknowledge the Research Council of Tehran University of Medical Sciences.
Funding
M. Amanlou was founded by the Research Council of Tehran University of Medical Sciences, Tehran, Iran under grant agreement Number 46127-104-3-98.
Author information
Authors and Affiliations
Contributions
Conceptualization and idea, M.N. Montazer, M. Asadi, and M. Amanlou. Synthesis and enzyme assay, M.N. Montazer and M. Asadi. In silico methodology and software, M.N. Data analysis, M.N. Montazer and M. Asadi. Original draft preparation, Z.B. Omrany and F. Moradkhani. Supervision and administration, M. Amanlou. The authors declare that all data were generated in-house and that no paper mill was used.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
ESM 1
(DOCX 2601 kb)
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Montazer, M.N., Asadi, M., Moradkhani, F. et al. Design, synthesis, and biological studies of the new cysteine-N-arylacetamide derivatives as a potent urease inhibitor. Naunyn-Schmiedeberg's Arch Pharmacol 397, 305–315 (2024). https://doi.org/10.1007/s00210-023-02596-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-023-02596-1