
Abstract
1,2-Dimethylhydrazine (DMH) is a plant toxicant that enters the food web through the diet. It is biotransformed into azoxymethane, a colon carcinogen, during the first hepatic passage. In mice, this study assessed the role of glutamate dehydrogenase (GDH), a key glutaminolysis enzyme in DMH-induced colorectal cancer (CRC). Colon samples were taken from mice given 6 or 15 weekly doses of 20 mg/kg DMH and serially sacrificed. Repeated DMH doses induced early aberrant crypt foci that evolved into irreversible adenocarcinomas over 24 weeks, along with an increase in GDH and lactate dehydrogenase activities (+ 122%, + 238%, P < 0.001), indicating a switch to aerobic glycolysis and glutaminolysis. Transcriptional downregulation of the endogenous GDH inhibitor, sirtuin4, and two redox regulators, mitochondrial sestrin2 and nuclear factor (erythroid derivative 2)-like 2 (− 26% and − 22%, P < 0, 05; and − 30%, P < 0.01), exacerbated mitochondrial stress by boosting mitochondrial superoxide dismutase activity (+ 240% (P < 0.001) while depressing catalase activity and GSH levels (− 57% and − 60%, P < 0.001). In vitro, allosteric GDH inhibition by 50 µM epigallocatechin gallate decreased human carcinoma (HCT-116) cells’ viability, clonogenicity, and migration (− 43% and − 57%, P < 0.001, 41%, P < 0.05), while stimulating ROS release (+ 57%, P < 0.001). Dimethylfumarate (DMF), a linear electrophile and mitochondrial fumarate analog, rebalanced ROS levels (− 34%, P < 0.05) and improved GDH activity, cell viability, and tumorogenic capacity (+ 20%, 20%, P < 0.001; and 33%, P < 0.05). Thus, the pathological remodeling of colon mucosa is supported by metabolic reprogramming bypassing uncoupled mitochondria. DMF highlights the critical role of electrophile response elements in modulating redox mithormesis and redox homeostasis during CRC.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The burst in aerobic glycolysis (Warburg effect) at the expense of mitochondrial activity is a biochemical hallmark of colorectal cancer (CRC) (Crabtree 1929; Warburg 1956; Hirayama et al. 2009; Ortmayr et al. 2009; Avolio et al. 2020). This metabolic remodeling is triggered by serial mutations and epigenetic changes such as CpG islands, microsatellite, and chromosome instability (Ogino et al. 2009; Femia et al. 2010; Sung et al. 2021).
Increased glucose uptake and consumption provides 2-carbon backbones (Acetyl-CoA) and energy (ATP, PEP, NAD(P)H) for anabolic pathways of fast-growing cells. In addition, NADPH derived from the pentose phosphate pathway and the 1- and 5-carbon backbones of the folate cycle are engaged in GSH replenishment, antioxidant shuttles, and de novo synthesis of fatty acids and nucleotides (DeBerardinis and Chandel 2016; Stincone et al. 2016). The oncogenic lactate dehydrogenase A (LDHA) reduces pyruvate into lactate, the end product of aerobic glycolysis (Baryła et al. 2022). It is actively secreted via the proton-dependent monocarboxylic transporter4 (MCT4) and promotes angiogenesis and metastasis by activating pro-inflammatory stromal cells and upregulating the bioenergetic sensor, hypoxia-inducible factor (HIF)-1α (Pollard et al. 2005; Whitaker-Menezes et al. 2011).
The crosstalk between HIF-1α and Myc upregulates glucose transporters (GLUT1/4), LDHA, and key aerobic glycolysis kinases (hexokinase2, phosfructokinase1, pyruvate kinase M2), while blocking mitochondrial pyruvate dehydrogenase kinase1 (Wise et al. 2008; Satoh et al. 2017; Belisario et al. 2020). This leads to mitochondria uncoupling and a bioenergetic shift to aerobic glycolysis (Gaude et al. 2018).
Addiction to glutaminolysis is the second metabolic switch supporting rapid proliferation (Tardito et al. 2015; Spinelli et al. 2017). It involves the glutaminase/glutamate dehydrogenase (GDH) tandem, which catalyzes the deamination of glutamine to glutamate. The latter is incorporated into GSH or further hydrolyzed into ammonia and α-ketoglutarate (α-KG) (Kovacevic 1971; Gaude et al. 2018). The human NAD(P)H-dependent GDH1 bound to the outer mitochondrial membrane catalyzes the anaplerotic insertion of glutamine into the TCA cycle via α-KG and the concomitant reduction of NAD(P)+ to NAD(P)H/H+ (Harris et al. 2015; Jin et al. 2015).
GDH is downregulated by Sirt4, a tumor-suppressive gene coding sirtuin4 (Sirt4) which is stably repressed during CRC. The mitochondrial NAD+-dependent Sirt4 inhibits the β-oxidation and pyruvate dehydrogenase activity and thus NADH/H+ and FADH2 formation for the respiratory chain (RC) (Mathias et al. 2014; Sun et al. 2018).
Uncoupled mitochondria reduced oxygen to superoxide anion (mtO2.−), readily dismutated into the stable pro-oxidant hydrogen peroxide (H2O2) by mitochondrial (MnSOD, SOD2) superoxide dismutase (Weinhouse 1956; Brand 2010; Woo et al. 2012). The subsequent crosstalk between mtO2.−, H2O2, carbonate anion (CO2−), and nitric oxide (NO) generates mixed mitochondrial reactive (oxygen and nitrogen) species (mtROS).
Sestrins (Sesn1,2,3) are ubiquitous metabolic and oxidative stress-inducible proteins (Pasha et al. 2017). Sesn2, a globin-like α-helical protein, is involved in the adaptive unfolding protein (UPR) response to endoplasmic reticulum (ER) stress. Sesn2 can restore mithormesis and metabolic homeostasis through PERK-/TFc/EBPβ signaling (Cullinan et al. 2003; Walter and Ron 2011; Park et al. 2014; Ro et al. 2016).
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a redox regulator transcription factor downstream of sesn2, is tightly anchored by low and high-affinity bonds of the DLG and ETGE domains to its endogenous inhibitor, Kelch-like ECH-associated protein 1 (Keap-1) (Hayes and Dinkova-Kostova 2014; Pasha et al. 2017). The conformational change of keap1 induced by the adduction/oxidation of specific thiol-cysteines releases a stabilized Nrf2. Its scaffolding with regulatory factors on the electrophile and antioxidant response elements (Ep/ARE) of DNA triggers adaptive responses to metabolic and oxidative stresses (Aggarwal et al. 2019; Wang et al. 2021).
This study investigated the role of mitochondrial stress and glutamate dehydrogenase activity in the oncological metabolic remodeling of the colon mucosa in vivo using a mouse model of long-term DMH-induced CRC and in vitro with human adenocarcinoma (HCT-116) cells.
Materials and methods
Materials
1,2-Dimethylhydrazine dihydrochloride (DMH) was purchased from Fluka (Buchs, Switzerland). Dimethlyfumarate (DMF) was from Alfa Aesar (Karlsruhe, Germany). 2-Aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (BCH), epigallocatechin-3-gallate (EGCG), fetal bovine serum (FBS), penicillin, streptomycin, and N-acetylcysteine (NAC) were from Sigma–Aldrich (St. Louis, USA). All other reagents are of analytical grade.
Ethics statement
All the in vivo experiments were carried out in agreement with the Ethics Committee for Animal Welfare of the University of Science and Technology Houari Boumediene (Algiers) [Algerian Law 12–235/2012; Executive Decree No. 10–90] and in accordance with the European Directive 2010/63/EU for ethics in animal experimentation.
In vivo study
Modeling colon carcinogenesis and histopathological analysis
Eight-week-old female NMRI mice (25–30 g) were acclimatized in standard cages under controlled light and temperature, with free access to food and water (animal facility of the Faculty of Biological Sciences, USTHB, Algiers, Algeria).
Colon carcinogenesis was induced in mice (n = 20) by 20 mg/kg 1,2-dimethylhydrazine (DMH) injected subcutaneously once a week for 6 or 15 weeks. Control mice (n = 10) were weekly administrated with 0.9% NaCl. Mice were sacrificed 6 (DMH6), 15 (DMH15), and 24 (DMH24) weeks post-CRC induction. Colon biopsies were collected and assessed for weight and length and macroscopic changes (Fig. 1A). Thin colon sections stained with hematoxylin and eosin (H&E) were scored for the multiplicity of aberrant crypt foci (ACF) and polyps and for histopathological changes (ACF, polyps, adenocarcinomas multiplicity, inflammatory cells infiltrate, crypt abscesses, depletion of goblet cells) (Bounaama et al. 2012; Kanehara et al. 2019).

Pathological remodeling of colon mucosa in long-term 1,2-diethylhydrazine-induced adénocarcinomas. Colon carcinogenesis was induced in NMRI (25–30 g) mice (n = 20) by 6 or 15 weekly subcutaneous doses of 1,2-dimethylhydrazine (DMH, 20 mg/kg, sc). Control mice (n = 10) received 0.9% NaCl, sc. Mice were serially sacrificed after 6 (DMH6), 15 (DMH15), and 24 weeks (DMH24) and analyzed with appropriate methods (1A). Representative macroscopic images of colon specimens of control (B1) or DMH-treated mice at weeks 6 (DMH6, B2), 15 (DMH15, B3), and 24 (DMH24, B4). Hematoxylin and eosin-stained colons sections of control (C2, D1) or DMH-treated mice at week 6 (DMH6, B3, B4), 15 (DMH15, C3, C4), and 24 weeks (DMH24, D3, D4) (100 × and 400 × magnification)
Immuno-histochemical evaluation of epithelial-to-mesenchymal transition
Thin colons sections were labeled with primary antibodies for E-cadherin (E-Cad, 1:100; Dako, Santa Clara, CA, USA) and α-smooth muscle actin (α-SMA, 1:100; Dako, Santa Clara, CA, USA) overnight at 4 °C. After application of secondary biotinylated antibodies and streptavidin-peroxidase reagent for 30 min at room temperature, the labeled sections were developed using diaminobenzidine (Dako). Immuno-labeled sections were then counterstained with Mayer’s hematoxylin and assessed for molecular alterations by light microscopy.
Reverse transcription-quantitative PCR of antioxidant genes
Total RNA was extracted from DMH24 colon biopsies with the Qiagen RNeasy Mini Kit (Carnaxide, Portugal). RNA purity and concentration were checked using a Synergy HT spectrophotometer (BioTek Instruments, VT, USA). Reverse transcription-quantitative PCR (RT-qPCR) was performed using the XpertcDNA Synthesis Mastermix kit (GRISP, Porto, Portugal), containing 1 μg of total RNA as template and designed primer sequences, and evaluated with Beacon Designer™ 7 software (PREMIER Biosoft, CA, USA). Primer specificity was assessed by NCBI BLAST analysis. Primers sequences were Glud1, forward (F) (5′-ATCCCGGACTTCAGATCCCC-3′), and reverse (R) (5′-CAACATGAAAAAGGG CTTGGG-3′), Nrf2, F (5′ CGAGATATACGCAGG AGA GG TAAG A-3′), R (5′-GCT CGACAATGTTCTCCAGCTT-3′), Sesn2, F (5′-TAGCCT GCA GC CTCACCTAT-3′), R (5′-TAT CTGATGCCAAAGACGCA-3′), Sirt4, F (5′-GAGC AT TC TT ACTAGGGATTCCA-3’), R (5′-AAC GGCTAAACAGTCGGG TT-3), Smad2, F (5′-AAG CCATCACCACTCAGAATT G-3′), R (5′-CAC TGATCTACCGTATTTGCT GT-3′), Smad3, F (5′-AGGGGCTCC CTCACGTTA TC-3′), R (5′-CAT GGCCCGTAATTCATG GTG-3′).
RT-qPCR amplifications were performed in duplicate containing 0.25 μM of each primer, 5 μl of 2X iTaq™ Universal SYBR Green Supermix, with or without (negative controls) 1 μl of template cDNA. RT-qPCR was started in a CFX384 Touch™ Real-Time PCR Detection System (Bio-Rad) (95 °C, 3 min), followed by 40 cycles of denaturation (95 °C, 10 s) and annealing (60 °C, 30 s). Melting curves of RT-qPCR amplicons were generated with temperatures ranging from 55 to 95 °C, 10 s (0.5 °C increments). β-actin and GAPDH genes were used for the normalization of quantitative RT-qPCR.
Following RT-qPCR, the dissociation curve was verified by the presence of a single peak with an observed Tm consistent with the amplicon length. Standard dilutions of cDNA were used to check the relative efficiency and quality of primers. The melting curve data were analyzed with the CFX Manager™ 2.0 (Bio-Rad, Amadora). RT-qPCR were run for 40 cycles, and all detected genes had Ct values below 30. Ct values from duplicate measurements were averaged, and relative expression levels were determined by the 2−ΔΔCt method (Pfaffl 2001).
In vitro study
Glutamate dehydrogenase implication in HCT-116 cells’ migration and invasion
HCT-116 cells’ viability assay
Human colorectal carcinoma (HCT-116) cells (American Type Culture Collection, ATCC) were grown in RPMI-1640 medium containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin, 5% CO2 at 37 °C. Cell viability was assessed with the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tetrazolium reduction assay (Denizot and Lang 1986). Briefly, HCT-116 cells (7.5 × 103 cells/well) were incubated for 24 h and 48 h in 100 µl of fresh medium containing the appropriate drugs (2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (BCH) (Zaleski et al. 1986), an activator of glutamate dehydrogenase (GDH, EC: 1.4.1.3), epigallocatechin-3-gallate (EGCG), an allosteric inhibitor of GDH (Liao et al. 1995), dimethylfumarate (DMF), a cell-permeable form of fumarate, N-acetylcysteine (NAC), a standard antioxidant, or their respective solvents. Formazan crystals formed after 3 h incubation of HCT-116 cells in 0.5 mg/ml MTT were dissolved in DMSO. Absorbance was measured at 570 nm using an automated microplate reader (Sinergy HT, BioTek). Results were expressed as the relative percentage of control.
Wound healing assay
HCT-116 cells were seeded in a 24-well plate (1.5 × 105 cells/well) and allowed to attach overnight in 5% CO2 at 37 °C. Single scratches were made across the confluent cell monolayer (Liang et al. 2007). After washing, attached cells were treated with appropriate drugs or the respective solvents. Images of the cells’ migration front were taken at 0 h and 24 h with a Lionheart FX automated microscope (BioTek) and analyzed using Gen5 3.0 software (BioTek).
Clonogenic assay
HCT-116 cells were seeded in a 12-well plate (500 cells/well) and allowed to attach for 24 h. They were cultured for 7 additional days in a medium containing the appropriate drugs or solvents, renewed every 72 h. On day 7, the newly formed colonies were fixed with 4% paraformaldehyde for 10 min, stained with 0.5% (v/v) crystal violet, and counted on representative images taken with a digital camera using ImageJ software (NIH, Bethesda, MD). Results were expressed as a percentage of the respective controls.
Biochemical analyses
Redox status assessment
The level of cellular hydrogen peroxide (H2O2) released by growing HCT-116 cells was measured with the 2′,7′-dichlorodihydrofluorescein diacetate (H2-DCFDA) fluorescence kit (Sigma Aldrich). HCT-116 cells (7.5 × 103 cells/well) were seeded in a 96-well plate and treated for 24 h with appropriate drugs. The level of oxidized DCFDA was measured according to the manufacturer’s instructions using a microplate reader (Sinergy HT).
Colon supernatants were assayed for antioxidant capacity. Manganese superoxide dismutase (MnSOD) was measured by the rate of pyrogallol oxidation (10 mM in 50 mM Tris buffer pH 8.2, 5 mM KCN) at 420 nm for 3 min (Marklund and Marklund 1974). Catalase (CAT) activity was quantified by the rate of H2O2 consumption (10 mM in 50 mM phosphate buffer pH 7) at 240 nm for 1 min (Aebi 1984).
The level of reduced glutathione (GSH) was measured in colon supernatants and cancer HCT-116 cells at 405 nm by Ellman’s reagent (DTNB, 10 mM in 0.2 mM phosphate buffer pH 8) (Ellman 1959).
Determination of lactate dehydrogenase and glutamate dehydrogenase activities
Colon tissues were homogenized at low speed in ice-cold mitochondria isolation buffer pH 7.2 (200 mM mannitol, 50 mM sucrose, 5 mM 3-morpholino-1-propane sulfonic acid, 5 mM KH2PO4, 1 mM EDTA, and 0.1% BSA) and centrifuged at 1000 × g, 20 min at 4 °C. Supernatants containing mitochondria were centrifuged at 12,000 × g, 20 min at 4 °C (Holmuhamedov et al. 1998). Lactate dehydrogenase (LDH) activity was assayed in the mitochondria-free supernatant by monitoring the rate of NADH oxidation with an LDH estimation kit (Spinreact, Bio Elite).
Glutamate dehydrogenase (GDH) activity was assayed with a GDH assay kit (Sigma Aldrich, St. Louis, USA) by monitoring the rate of NAD+ reduction in mitochondria fraction and in HCT-116 cells’ homogenates using a non-denaturing cell lysis buffer (25 mM Tris–HCl pH 7.4, 100 mM NaCl, and 1% Nonidet P-40) and a precellys homogenizer.
Statistical analysis
Graphs and statistical analysis were performed using GraphPad Prism 8 software. Data were tested for normality by the Shapiro–Wilk test. Values are presented as mean ± SD. P value < 0.05 was considered statistically significant (*P < 0.05; **P < 0.05; ***P < 0.05). The statistical differences between the two groups were determined by Student’s t-test. One-way ANOVA, followed by Tukey’s post hoc, was applied for multiple comparisons.
Results
Long-term DMH triggers the multistep process of colon carcinogenesis in mice
Low-grade dysplastic areas represent early oncological changes in DMH-treated mice. They gradually evolved into highly invasive adenocarcinomas (ADC) (Fig. 1B-D).
They were characterized by increased histopathological scores (+ 38%, P < 0.05), multiplicity (+ 73%, P < 0.05), and tumor size (+ 130%, P < 0.01) (Fig. 2).

Disease activity index. Long-term DMH-induced colon carcinogenesis was assessed by scoring the number of tumors per mouse (A), mean tumor size (B), and histological score (C). Data are mean ± SD of five different samples with *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. Statistical differences between different conditions were evaluated using one-way ANOVA, followed by Tukey’s post hoc test (GraphPad Prism 8)
Colon epithelial-to-mesenchymal transition is independent of Smad2/3 transcription factors
Long-term DMH resulted in a sustained colon epithelial-to-mesenchymal transition (EMT) as indicated by the threefold (P < 0.05) decrease in E-cadherin expression (E-Cad, epithelial phenotype), compared with a twofold (P < 0.001) increase in that of α-smooth muscle actin (α-SMA, mesenchymal phenotype) (Fig. 3A).

Long-term DMH-induced epithelial-to-mesenchymal transition (EMT). E-cadherin (E-cad, epithelial marker) and alpha-smooth muscle actin (α-SMA, mesenchymal marker) immunostaining of control and DMH24 colon sections (A). Smad2 (B) and Smad3 (C) relative mRNA expression were quantified by RT-qPCR in control and DMH24 colon biopsies. Data are mean ± SD of three different samples with *P < 0.05 vs. control; NS: non-significant. Statistical differences between different conditions were evaluated using the Student t-test (GraphPad Prism 8)
Additionally, reduced mRNA levels of Smad2 (− 36%, P < 0.05) and Smad3 (− 34%, P > 0.05) suggested that DMH-induced EMT occurred independently of TGF-β-SMAD2/3 pathway (Fig. 3B, C).
DMH sustains redox imbalance by downregulating Sesn2 and Nrf2
DMH depressed redox regulators as evidenced by reduced Sens2 and Nrf2 mRNA levels (− 22%, P < 0.05 and − 30%, P < 0.01, respectively) in DMH 24 colon biopsies (Fig. 4A, B).

Modulation of antioxidant response by long-term DMH-induced colon adenocarcinomas. Sesn2 (A) and Nrf2 (B) relative mRNA expression were quantified by RT-qPCR in control and DMH24 colon biopsies. Reduced glutathione (GSH) levels (C), superoxide dismutase 2 (MnSOD) (D), and catalase (E) activities were evaluated in control and in DMH24 colon tissues using appropriate methods. Data are mean ± SD of five different samples with *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. Statistical differences between different conditions were evaluated using Student’s t-test (GraphPad Prism 8)
Downstream, the antioxidant capacity is drastically altered. The decrease in GSH levels (− 60%, P < 0.001) and catalase activity (− 57%, P < 0.001) associated with a burst in MnSOD matrix activity (+ 240%, P < 0.001) resulted in persistent mitochondrial stress (Fig. 4C−E).
DMH sustains glutamate dehydrogenase and lactate dehydrogenase activities
GDH activity, an index of glutaminolysis, increased time dependently, reaching + 122% (P < 0.001) increase in DMH24 colon biopsies. Similarly, lactate dehydrogenase (LDH) activity assessed as a marker of aerobic glycolysis reached + 238% (P < 0.001) compared to control (Fig. 5A, B).

Glutamate dehydrogenase and lactate dehydrogenase expression and activity in long-term DMH-induced colon adenocarcinomas. Glutamate dehydrogenase (GDH) (A) and lactate dehydrogenase (LDH) (B) activities were evaluated in control and in DMH6, DMH15, and DMH24 colon biopsies using appropriate methods. Glud1 (C) and Sirt4 (D) mRNA expression were quantified by RT-qPCR in control and in DMH24 colon biopsies. Results are mean ± SD of five different samples, with *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; NS: non-significant. Statistical differences between different conditions were evaluated using Student’s t-test when comparing two groups and one-way ANOVA, followed by Tukey’s post hoc test when comparing more than two groups (GraphPad Prism 8)
DMH sustains GDH activity through sirtuin4 downregulation
Next, we examined the expression of Sirt4 and Glud1 genes encoding Sirt4 (endogenous GDH inhibitor) and GDH in DMH24 colon biopsies. Sirt4 mRNA levels were reduced (− 26%, P < 0.05), while those of Glud1 mRNA increased (+ 18%, P > 0.05), suggesting sustained GDH activity in CCR progression (Fig. 5C, D).
GDH contributes to the survival, migration, and invasion of HCT-116 CRC cells
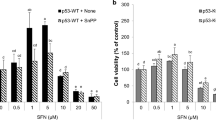
The role of GDH in CRC progression was explored pharmacologically using HCT-116 cells. Epigallocatechin gallate (EGCG), a green tea-derived polyphenol and allosteric inhibitor of GDH activity (Liao et al. 1995), reduced HCT-116 cell viability in a time and concentration-dependent manner. The 50% inhibitory concentration (IC50) was reached with approximately 50 µM EGCG at 24 h (− 43%, P < 0.001) (Fig. 6).

Influence of GDH inhibition on HCT-116 cells’ viability and ability to form colonies. HCT-116 cells (7.5 × 10.3 cells/well) were treated with 25 µM, 50 µM, or 100 µM epigallocatechin-3-gallate (EGCG), an allosteric inhibitor of GDH activity for 24 h and 48 h, and their viability was measured by MTT reduction assay (A). Their ability to form colonies was evaluated by the clonogenic assay (C). HCT-116 cells (500 cells/well) were treated with 25 µM, 50 µM, or 100 µM EGCG in a 12-well plate for 7 days. Violet crystal-stained viable new colonies (B) were counted on representative images. The results were expressed as a percentage of control. Data are mean ± SD of three independent experiments with *P < 0.05, **P < 0.01, ***P < 0.001 vs. control. Statistical differences between different conditions were evaluated using one-way ANOVA, followed by Tukey’s post hoc test (GraphPad Prism 8)
GDH inhibition by 50 µM of EGCG also reduced cell migration (− 41%, P < 0.05) and the number of newly formed colonies (− 57%, P < 0.001).
The GDH activator, 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid (Zaleski et al. 1986), restored in stride HCT-116 cells’ GDH activity, viability, ability to form colonies, and migrate close to control level (P < 0.01) (Fig. 7), suggesting a crucial involvement of GDH in the metastatic phenotype of HCT-116 cells.

Effect of pharmacological modulation of GDH on HCT-116 cell viability, wound healing, and colony formation. Influence of 24 h treatment by 50 μM EGCG on cell viability (A), GDH activity (B), colony formation (C), and wound healing (D) in the absence or the presence of 5 mM BCH and 10 mM BCH. Data were presented as mean ± SD values from three independent experiments with *P < 0.05, ***P < 0.001 vs. control. #P < 0.05, ##P < 0.01, and.###P < 0.001 vs. EGCG; NS: non-significant. Statistical differences between different conditions were evaluated using one-way ANOVA, followed by Tukey’s post hoc test (GraphPad Prism 8)
DMF facilitates the tumorigenic capacity of GDH
To assess which GDH by-products contribute to its oncogenic capacity, HCT-116 cells were treated with EGCG alone or in the presence of NH4Cl or dimethylfumarate (DMF).
NH4Cl and DMF alone did not alter cell viability, wound healing, or colony formation (Data not shown). DMF can restore cell viability and colony formation, previously inhibited by 50 µM EGCG (+ 20%, P < 0.001; and + 33%, P < 0.05, respectively), without improving wound healing. However, NH4Cl slightly restored cell viability (+ 9%, P < 0.05) (Fig. 8).

Influence of NH4Cl and DMF on cell viability, migration, and colony formation by EGCG-treated HCT-116 cells
Effect of 0.5 mM NH4Cl or 10 µM dimethylfumarate (DMF) on 50 µM EGCG-treated HCT-116 cells’ cell viability (A), colony formation (B), and wound healing assay (C). Results are expressed as a percentage of the respective control. Data were presented as mean ± SD of three independent experiments with *P < 0.05, ***P < 0.001 vs. control. #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. EGCG; NS: non-significant. Statistical differences between different conditions were evaluated using one-way ANOVA, followed by Tukey’s post hoc test (GraphPad Prism 8).
DMF rebalances redox status in HCT-116 cells
To gain further insights into the oncogenic capacity of GDH, we assessed its involvement in ROS production by HCT-116 cancer cells. ROS levels increased in the presence of EGCG (+ 57%, P < 0.001), an effect reversed by DMF (− 34%, P < 0.05), similar to N-acetylcysteine, a standard antioxidant (NAC, − 40%, P < 0.01). Interestingly, DMF increased GSH levels (1.34-fold, P < 0.05) and MnSOD activity (P > 0.05) without improving catalase activity (Fig. 9A−D).

Effect of GDH modulation on ROS scavenging in HCT-116. Influence of 50 µM EGCG in the absence or in the presence of 5 µM BCH and 10 µM BCH, 10 µM DMF, or 1 mM NAC on ROS levels (A). GSH levels (B) and on the activity of MnSOD (C) or catalase (D). Data were presented as mean ± SD of three independent experiments with *P < 0.05, ***P < 0.001 vs. control. #P < 0.05, ##P < 0.01, and.###P < 0.001 vs. EGCG; NS: non-significant. Statistical differences between different conditions were evaluated using one-way ANOVA, followed by Tukey’s post hoc test (GraphPad Prism 8)
Discussion
This study examined the relationship between the pathological remodeling of colon mucosa and GDH activity in a mice model of 1,2-dimethyhydrazine (DMH)-induced adenocarcinomas.
The early-onset ACF differentiated into irreversible adenocarcinomas by 15 weekly doses of colon carcinogen (Fig. 1), indicating that long-term DMH closely mimics the ACF-adenocarcinomas sequence occurring in human CRC (Herron and Shank 1981; Bird and Good 2000; Femia et al. 2010). However, azoxymethane-induced sporadic colon cancer in mice shows some differences in the genetic background compared to human CRC (Perse and Cerar 2011; Pan et al. 2017).
DMH is a secondary metabolite, a toxic contaminant of the seed starch of Japanese Salco palm (Cycas revoluta Thunb., Cycadaceae). During the first hepatic passage, DMH is biotransformed into azoxymethane (AOM) by the superoxide-generating CYP2E1, which is transported as AOM-β-glucuronide (Cycasin) via the bile to the intestine.
Colonic β-glucuronidase releases the carcinogenic aglycone which is sequentially degraded into the alkylated derivatives methyldiazonium, methylcarbabonium, and methyl cation by CYP2E1 and gut microbiota enzymes (Fig. 10).

Oxidative effects of 1,2-dimethylhydrazine metabolites
These free radicals can alkylate and oxidize proteins and DNA (Matsumoto and Higa 1966; Nagasawa et al. 1972; Fiala 1975; Cavanna et al. 1979). The subsequent epigenetic changes and oncogenic activation trigger sporadic colon cancer in humans and adenomas in various target organs in animal models (Sohn et al. 2001; Femia et al. 2010).
In addition, DMH, by disrupting tight junctions, enhances the permeability of colonic epithelium to gut microbiota, which sustains intestinal oxidative inflammation (Bekusova et al. 2018).
1,2-Dimethylhydrazine (DMH) is a naturally occurring plant secondary metabolite accumulated in the seed starch of Japanese Salco palm (Cycas revoluta Thunb., Cycadaceae). DMH is toxic to animals and humans upon chronic exposure. DMH is sequentially bioactivated into azoxymethane (AOM) and various alkylated methyldiazonium, methylcarbabonium, and methyl cation by the phase I hepatic superoxide-generating cytochrome CYP2E1 and gut microbiota enzymes.
Repeated doses of DMH induced a long-lasting epithelial-to-mesenchymal transition (EMT), glutamate dehydrogenase (GDH, glutaminolysis) and lactate dehydrogenase (LDH, Warburg effect, aerobic glycolysis) activation, and mitochondrial uncoupling supporting the progression of early aberrant crypt foci to invasive adenocarcinomas.
The parallel between drastic LDH activity and the severity of colonic lesions (Figs. 1, 2, and 5) highlights the critical role of aerobic glycolysis in experimental carcinogenesis and in CRC patients (Hirayama et al. 2009; Hanna et al. 2014; Grazziano et al. 2017; Mishra and Banerjee 2019).
By accelerating the uptake and metabolism of glucose up to 100-fold, aerobic glycolysis straightens ATP formation relative to mitochondrial oxidative phosphorylation [4 ATP plus 2 NADH (basal glycolysis) vs. 38 ATP (OxPhos)], thereby limiting high-dose mtROS discharge and mitochondrial stress (Warburg 1956; Wise et al. 2008; Woo et al. 2012; Leone et al. 2017).
The antioxidant and uncoupling protein2 (UCP2) is a protonophore channel crossing the inner mitochondrial membrane. UCP2 is overexpressed in various cancers and functionally engaged in adaptation to mitochondrial nitro-oxidative stress (Weinhouse 1956; Orrenius et al. 2007; Li et al. 2013; Avolio et al. 2020). UCP2 blocks both β-oxidation and the TCA cycle, generating substrates for the respiratory chain (RC), thereby bypassing uncoupled mitochondria (Vozza et al. 2014; Brandi et al. 2016; Aguilar et al. 2019).
As a thermogenin, UCP2 allows the re-entry of protons ejected through the redox loops of RC. The parallel flow back of electrons at respirasomes I and III induces the monoelectronic reduction of oxygen to superoxide anion, which in turn sustains UCP2 activation (Porporato et al. 2014; Gaude et al. 2018). This collapses the mitochondrial pH gradient and electrochemical potential (mt∆pH/∆ψ, Michaelis proton power) necessary for ATP synthesis (Weinhouse 1956). Succinate dehydrogenase (SDH, respirasome II) is a critical redox checkpoint linking the TCA cycle to the coenzyme Q shuttle. Its uncoupling prevents FAD reduction to FADH2 and exacerbates ROS superoxide formation.
Otherwise, the active oxidative metabolism of glutamine through transforming growth factor β (TGFβ)-Smad pathway supports EMT and cancer progression. In contrast, the antitumoral effect of TGF-β relies on inducing cell cycle arrest and apoptosis (Valcourt et al. 2006).
Long-term DMH lowers Smad2 and/or Smad3 transcripts in DMH24 biopsies with a mesenchymal phenotype (Fig. 3B, C). Loss of Smad2 and/or Smad3 expression has been reported in 10% of metastatic CRC patients (Sung et al. 2021). DMH-carcinogenesis reproduces this poor prognosis, and the spontaneous development of deadly adenocarcinomas is related to Smad3 loss (Zhu et al. 1998). The sixfold increase in α-SMA expression at the expense of E-cadherin in DMH24 colon biopsies (Fig. 3A) refers to long-lasting EMT of malignant mucosal glands (Aspuria et al. 2014; Colvin et al. 2016; Daniel et al. 2021).
Oncogenic mutations of fumarate hydratase (FH), succinate (SDH), and isocitrate (IDH) of the TCA cycle generate analogs of α-KG, fumarate, succinate, and D-2-hydroxyglutarate (D(R)-2HG and L(S)-2HG) that cause epigenomic and transcriptional remodeling (Pollard et al. 2005; Xiao et al. 2012; Baryła et al. 2022). These mitochondrial oncometabolites and GDH have been implicated in EMT (Aspuria et al. 2014; Colvin et al. 2016; Brabletz et al. 2021; Jia et al. 2021).
The 1.6-fold increase in Glut1/Sirt4 ratio and a twofold increase in GDH activity suggest that its reactivation following Sirt4 slowdown can sustain the pathological remodeling of colon mucosa (Fig. 5) (Haigis et al. 2006; Mathias et al. 2014; Miyo et al. 2015; Sun et al. 2018). By replenishing the NADPH pool, GDH is critically involved in redox shuttles evolving between cytosol, peroxisomes, mitochondria, and nucleus (Spinelli et al. 2017; Gaude et al. 2018). GDH has been suggested as a prognostic marker for CRC metastasis (Jin et al. 2015).
In vitro, pharmacological modulation of GDH activity by EGCG, BCH, and DMF demonstrated its functional role in carcinoma-derived HCT116 cells’ invasiveness by modulating their ability to migrate and form colonies (Figs. 6, 7, and 8).
DMH-induced colon mucosa remodeling is associated with GSH depletion and with an
8.3-fold increase in MnSOD/catalase ratio. Chronic H2O2 levels (mtROS) rely on a burst of MnSOD activity, leading to oxidative paracrine changes to mitochondria and uncoupling. The tandem catalase and MnSOD can adjust ROS levels for CRC progression (Sarsour et al. 2014; Zelko et al. 2022).
Besides, high levels of volatile oxidized lipids such as 4-hydroxy-2-nonenal (4-HNE) and malondialdehyde (MDA) alter proteins and DNA through covalent adduction (Fig. 3) (Ayala et al. 2014).
Low ROS flow promotes cell survival and proliferation through oncogenic PI3K1A/Akt-PKB/PKB/mTORC2 and ERK1/2 pathways and evades apoptosis through resistance to TRAIL-mediated cell death pathways (Steelman et al. 2008). Two cellular lipid phosphatase and tensin homolog isoforms coexist in the nucleus (PTEN-β) and/or mitochondria (PTEN-Long, PTEN-L, mtPTEN). Succinylation by the onco fumarate or the H2O2-dependent oxidation of the redox-sensitive Cys71 and Cys124 ties an intramolecular thiol bond in the active site of PTEN, locking down both PI3K antagonist activity of the tumor suppressor PTEN and p53-dependent cell cycle arrest and reactivating carcinogenesis.
Upstream, H2O2 oxidation of thioredoxin-1 resulted in an intermolecular disulfur bond between
Trx-1-Cys32-122Cys-PTEN, which triggers the tumor-suppressive pathway (Lee et al. 2002; Zhang et al. 2020).
Sen2 can buffer electrophilic and oxidant stresses through activation of nuclear electrophilic and/or antioxidant responses (E/ARE), leading to de novo synthesis of phase II enzymes, NADPH, and GSH (Cullinan et al. 2003; Park et al. 2014; Ro et al. 2016).
Dimethylfumarate (DMF), a permeable form of fumarate, is a US Food and Drug Administration (FDA)-approved drug for relapsing multiple sclerosis (Altmeyer et al. 1994; Arnold et al. 2014). DMF and its metabolites monomethylfumarate (MMF) and monoethylfumarate (MEF) are hydrophilic linear electrophiles, which inhibit oxidative inflammation by blocking NF-ĸB and NLRP3 inflammasome-dependent pro-inflammatory cytokines. Their α, β-unsaturated carbonyl common backbone can adduct to and inactivate Keap-1 by succinylation at Cys151, Cys277, or Cys288, each carrying selective pharmacological sensing. Succinylated or ROS-oxidized Keap-1 undergoes a conformational change, releasing a stable functional Nrf2 (Ashrafian et al. 2012; Scannevin et al. 2012). The blockade of Nrf2 and Sen2 transcripts further confirms chronic oxidative stress in DMH24 colon biopsies (Fig. 4).
DMF, a mitochondrial fumarate analog, rescued HCT-116 cells from allosteric blockade induced by EGCG, thus restoring GDH-dependent NADPH(H+) and GSH recycling through redox shuttles and HCT-116 cells’ proliferation and oncogenicity (Figs. 6, 7, and 8). The oncometabolite fumarate can bind to and activate glutathione peroxidase 1 (GPx1), which increases GSH consumption, through GSH recycling by glutathione reductase or by promoting Nrf2 activation by succinylation and blockade of Keap-1 (Satoh and Lipton 2007; Saito et al. 2016; Sciacovelli and Frezza 2017).
Conclusion
Long-term DMH through its alkylated endometabolites (1) differentiates preneoplastic ACF into invasive adenocarcinomas supported by a long-lasting EMT of malignant mucous glands, (2) induces metabolic reprogramming switching bioenergetic supply to aerobic glycolysis and glutaminolysis, (3) drives subtle transcriptional downregulation of redox sensors Sesn2 and Nrf2, which boosts MnSOD activity and generates mitochondrial stress, and (4) blocks Sirt4, thus reactivating oncogenic GDH activity and its critical antioxidant effect. Ex vivo, the pharmacological modulation of HCT-166 cells by DMF, a mitochondrial fumarate analog and Ep/ARE inducer, could reactivate the oncogenic activity of GDH and its antioxidant capacity. Fumarate has been suggested as a signaling molecule for survival pathways. DMF highlights the oncogenic role of GDH and the critical role of electrophilic and antioxidant response elements in modulating mithormesis and redox homeostasis during CRC progression.
Together, our results suggest long-term DMH as a translational model for the multistep process of human CRC.
Data availability
All histological, immunochemical, biochemical, and RT-PCR data are available upon request from the first author.
Abbreviations
- AOM:
-
Azoxymethane
- BCH:
-
2-Aminobicyclo-(2,2,1)-heptane-2-carboxylic acid
- DMF:
-
Dimethylfumarate
- DMH:
-
1,2-Dimethylhydrazine
- E-Cad:
-
E-cadherin
- EGCG:
-
Epigallocatechin gallate
- Ep/ARE:
-
Electrophile/antioxidants response elements
- GDH:
-
Glutamate dehydrogenase
- GSH:
-
Reduced glutathione
- GLUT:
-
Glucose transporter
- MDZ:
-
Methyldiazonium
- MAM:
-
Methylcarbabonium
- CH3+ :
-
Methyl cation
- MnSOD:
-
Manganese superoxide dismutase
- mtROS:
-
Mitochondrial reactive oxygen species
- mtO2 − :
-
Mitochondrial superoxide anion
- NAC:
-
N-acetylcysteine
- Nrf2:
-
Nuclear factor (erythroid-derived 2)-like 2
- PTEN:
-
Phosphatase and tensin homolog
- ROS:
-
Reactive oxygen species
- Sesn2:
-
Sestrin2
- Sirt4:
-
Sirtuin4
- α-SMA:
-
α-Smooth muscle actin
References
Aebi H (1984) Catalase in vitro Methods. Enzymol 105(121):26
Aggarwal V, Tuli HS, Varol A, Thakral F, Yerer MB, Sak K, Varol M, Jain A, Md A, Sethi G (2019) Role of reactive oxygen species in cancer progression: molecular mechanisms and recent advancements. Biomolecules 9(11):735
Aguilar E, Esteves P, Sancerni T, Lenoir V, Aparicio T, Bouillaud F, Dentin R, Prip-Buus C, Ricquier D, Pecqueur C, Guilmeau S, Alves-Guerra MC (2019) UCP2 deficiency increases colon tumorigenesis by promoting lipid synthesis and depleting NADPH for antioxidant defenses. Cell Rep 28(9):2306–2316
Altmeyer P, Matthes U, Pawlak F, Hoffmann K, Frosch P, Ruppert P (1994) Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. J Am Acad Dermatol 30:977–981
Arnold P, Mojumder D, Detoledo J, Lucius R, Wilms H (2014) Pathophysiological processes in multiple sclerosis: focus on nuclear factor erythroid-2-related factor 2 and emerging pathways. Clin Pharmacol 6:35–42
Ashrafian H, Czibik G, Bellahcene M, Aksentijevic D, Smith AC, Mitchel SJ, Dodd MS, Kirwan J, Byrne JJ, Ludwig C, Isackson H, Yavari A, Støttrup NB, Contractor H, Thomas Cahill TJ, Sahgal N, Ball DR, Birkler RID, Hargreaves I, Tennant DA, Land J, Lygate CA, Johannsen M, Kharbanda RK, Neubauer S, Redwood C, de Cabo R, Ahmet I, Talan M, Günther UL, Robinson AJ, Viant MR, Pollard PJ, Tyler DJ, Watkins H (2012) Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab 15:361–371
Aspuria PJP, Lunt SY, Väremo L, Vergnes L, Gozo M, Beach JA, Salumbides B, Reue K, Wiedemeyer WR, Nielsen J, Karlan BY, Orsulic S (2014) Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab 2:21
Avolio R, Matassa DS, Criscuolo D, Landriscina M, Esposito F (2020) Modulation of mitochondrial metabolic reprogramming and oxidative stress to overcome chemoresistance in cancer. Biomolecules 10(1):135
Ayala A, Muñoz MF, Sandro Argüelles S (2014) Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Medic Cell Longev 2014:360438
Baryła M, Semeniuk-Wojta´s A, Róg L, Kraj L, Małyszko M, Stec R, (2022) Oncometabolites—a link between cancer cells and tumor microenvironment. Biology (basel) 11(2):270
Bekusova V, Falchuk EL, Okorokova LS, Kruglova NM, Nozdrachev AD, Markov AG (2018) Increased paracellular permeability of tumor-adjacent areas in 1,2-dimethyhydrazine-induced colon carcinogenesis in rats. Cancer Biol Med 15:251–259
Belisario DC, Kopecka J, Pasino M, Akman M, De Smaele E, Donadelli M, Riganti C (2020) Hypoxia dictates metabolic rewiring of tumors: implications for chemoresistance. Cells 9:2598
Bird RP, Good CK (2000) The significance of aberrant crypt foci in understanding the pathogenesis of colon cancer. Toxicol Lett 112:395–402
Bounaama A, Djerdjouri B, Laroche-Clary A, Le Morvan V, Robert J (2012) Short curcumin treatment modulates oxidative stress, arginase activity, aberrant crypt foci, and TGF- 1 and HES-1 transcripts in 1,2-dimethylhydrazine-colon carcinogenesis in mice. Toxicology 302(308):17
Brabletz S, Schuhwerk H, Brabletz T, Stemmler MP (2021) Dynamic EMT: a multi-tool for tumor progression. EMBO J 40:e108647
Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45(7–8):466–472
Brandi J, Cecconi D, Cordani M, Torrens-Mas M, Pacchiana R, DallaPozza E, Butera G, Manfredi M, Marengo E, Oliver J, Roca P, Dando I, Donadelli M (2016) The antioxidant uncoupling protein 2 stimulates hnRNPA2/B1, GLUT1 and PKM2 expression and sensitizes pancreas cancer cells to glycolysis inhibition. Free Radic Biol Med 101:305–316
Cavanna M, Parodi S, Taningher M, Bolognesi C, Sciaba L, Brambilla G (1979) DNA fragmentation in some organs of rats and mice treated with cycasin. Br J Cancer 39:383
Colvin H, Nishida N, Konno M, Haraguchi N, Takahashi H, Nishimura J, Hata T, Kawamoto K, Asai A, Tsunekuni K, Koseki J, Mizushima T, Satoh T, Doki Y, Mori M, Ishii H (2016) Oncometabolite D-2-Hydroxyglurate directly induces epithelial-mesenchymal transition and is associated with distant metastasis in colorectal cancer. Sci Rep 6:36289
Crabtree HG (1929) Observations on the carbohydrate metabolism of tumours. Biochem J 23:536–545
Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 20:7198–7209
Daniel Y, Lelou E, Aninat C, Corlu A, Cabillic F (2021) Interplay between metabolism reprogramming and epithelial-to-mesenchymal transition in cancer stem cells. Cancers (basel) 13:1973
DeBerardinis RJ, Chandel NS (2016) Non-essential amino acids serve as precursors to purine and pyrimidine synthesis and methyl groups are obtained from the carbon (1C)/folate pool. Fundamentals of Cancer Metabolism Sci Adv 2:e1600200
Denizot F, Lang R (1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 89:271–277
Ellman G (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82(1):70–77
Femia AP, Luceri C, Toti S, Giannini A, Dolara P, Caderni G (2010) Gene expression profile and genomic alterations in colonic tumours induced by 1,2-dimethylhydrazine (DMH) in rats. BMC Cancer 10:194
Fiala E (1975) Carcinogen 1,2-dimehylhydrazine: mode of action of the colon. Cancer 36:2407–2412
Gaude E, Schmidt C, Gammage PA, Dugourf A, Blacker T, Chew SP, Saez Rodriguez J, O’Neill JS, Szabadkai G, Minczuk M, Frezza C (2018) NADH shuttling couples cytosolic reductive carboxylation of glutamine with glycolysis in cells with mitochondrial dysfunction. Mol Cell 69(4):581–593
Grazziano F, Ruzzo A, Giacomini E, Ricciardi T, Aprile G, Loupakis F, Lorenzini P, Ongaro E, Zoratto F, Catalano V, Sari D, Rulli E, Cremolini C, De Nictolis M, De Maglio G, Falcone A, Fiorentini G, Magnani M (2017) Glycolysis gene expression analysis and selective metabolic advantage in the clinical progression of colorectal cancer. Pharmacogenomics J 17(3):258–264
Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ, Valenzuela DM, Yancopoulos GD, Karow M, Blandre G, Wolberger C, Prolla TA, Weindruch R, Alt FW, Guarente L (2006) SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126(5):941–945
Hanna N, Woods C, Zheng Z, Onukwugha E, Seal BS, Mullins CD (2014) Survival benefit associated with the number of chemotherapy/biologic treatment lines in 5,129 metastatic colon cancer patients. J Clin Oncol 32(3):559–559. https://doi.org/10.1200/jco.2014.32.3_suppl.559
Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, Elia E, Berger T, Cescon DW, Adeoye A, Brustle A, Molyneux SD, Mason JM, Li WY, Yamamoto K, Wakeman A, Berman HK, Khokha SJ, Done SJ, Kavanagh TJ, Lam CW, Mak TW (2015) Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27(2):211–222
Hayes JD, Dinkova-Kostova AT (2014) The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci 39(4):199–218
Herron DC, Shank RC (1981) In Vivo Kinetics of O6-methylguanine and 7-methylguanine formation and persistence in DMA of rat treated with symmetrical dimethylhydrazine. Cancer Res 41(3967):72
Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, Esumi H (2009) Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res 69(11):4918–4925
Holmuhamedov EL, Jovanovic S, Dzeja PP, Jovanovic A, Terzic A (1998) Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondrial function. Am J Physiol 275:H1567–H1576
Huang G, Cheng J, Yu F, Liu X, Chen X, Peng Z (2016) Clinical and therapeutic significance of sirtuin 4 expression in colorectal cancer. Oncol. Rep. 35(5):2801–2810
Jia D, Park JH, Kaur H, Jung KH, Yang S, Tripathi S, Galbraith M, Deng Y, Jolly MK, Kaipparettu BA, Onuchic JN, Levine H (2021) Towards decoding the coupled decision-making of metabolism and epithelial-to-mesenchymal transition in cancer. Br J Can 124:1902–1911
Jin L, Li D, Alesi GN, Fan J, Kang HB, Lu Z, Boggon TJ, Jin P, Yi H, Wright ER, Duong D, Seyfried NT, gnatchik R, Deberardinis RJ, Magliocca KR, He C, Arellano ML, Khoury HJ, Shin DM, Khuri FR, Kang S, (2015) Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 27(2):257–270
Kanehara K, Ohnuma S, Kanazawa Y, Sato K, Kokubo S, Suzuki H, Kaeasawa H, Suzuki T, Suzuki C, Naitoh T, Unno M, Abe T (2019) The indole compound MA-35 attenuates tumorigenesis in an inflammation-induced colon cancer model. Sci Rep 9:12739
Kovacevic Z (1971) The pathway of glutamine and glutamate oxidation in isolated mitochondria from mammalian cells. Biochem J 125(3):757–763
Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG (2002) Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 277(23):20336–20342
Leone A, Roca MS, Ciardiello C, Costantini S, Budillon A (2017) Oxidative stress gene expression profile correlates with cancer patient poor prognosis: identification of crucial pathways might select novel therapeutic approaches. Oxid Med Cell Longev 2017:2597581
Li W, Nichols K, Nathan CA, Zhao Y (2013) Mitochondrial uncoupling protein 2 is up-regulated in human head and neck, skin, pancreatic, and prostate tumors. Cancer Biomark 13(5):377–383
Liang CC, Park AY, Guan JL (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2:329–333
Liao S, Umekita Y, Guo J, Kokontis JM, Hiipakka RA (1995) Growth inhibition and regression of human prostate and breast tumors in athymic mice by tea epigallocatechin gallate. Cancer Lett 96(2):239–243
Manna SK, Tanaka N, Krausz KW, Haznadar M, Xue X, Matsubara T, Bowman ED, Fearon ER, Harris CC, Shah YM, Gonzalez FJ (2014) Biomarkers of coordinate metabolic reprogramming in colorectal tumors in mice and humans. Gastroenterology 146(5):1313–1324
Marklund S, Marklund G (1974) Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47:469–474
Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, Kang Y, Shenk T, Cristea I (2014) Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity. Cell 159(7):1615–1625
Matsumoto H, Higa HH (1966) Studies on methylazoxymethanol, the aglycone of cycasin: methylation of mucleic acids in vitro. Biochem J 98(20c):22c
Mishra D, Banerjee D (2019) Lactate dehydrogenases as metabolic links between tumor and stroma in the tumor microenvironment. Cancers (basel) 11(6):750
Miyo M, Yamamoto H, Konno M, Colvin H, Nishida N, Koseki J, Kawamoto K, Ogawa H, Hamabe A, Uemura M, Nishimura J, Hata T, Takemasa I, Mizushima T, Doki Y, Mori M, Sshii H (2015) Tumour-suppressive function of SIRT4 in human colorectal cancer. Br J Can 113(3):49299
Mouzaoui S, Banerjee S, Djerdjouri B (2020) Low-dose curcumin reduced TNBS-associated mucin depleted foci in mice by scavenging superoxide anion and lipid peroxides, rebalancing matrix NO synthase and aconitase activities, and recoupling mitochondria. Inflammopharm. 28(4):949–965
Nagasawa HT, Shirota FN, Matsumota H (1972) Decomposition of methylazoxymethanol. The aglycone of cycasin, in DO. Nature 236:234–235
Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M, Giovannucci EL, Fuchs CS (2009) CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 58(1):90–96
Orrenius S, Gogvadze V, Zhivotovsky B (2007) Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol 47:143–183
Ortmayr K, Dubuis S, Zampieri M (2009) Metabolic profiling of cancer cells reveals genome wide crosstalk between transcriptional regulators and metabolism. Nature Commun 10:1841
Pan Q, Lou X, Zhang J, Zhu Y, Li F, Shan Q, Chen X, Xie Y, Su S, Wei H, Lin L, Wu L, Liu S (2017) Genomic variants in mouse model induced by azoxymethane and dextran sodium sulfate improperly mimic human colorectal cancer. Sci Rep 1:25
Park HW, Park H, Ro SH, Semple JA, Kim DN, Kim M, Nam M, Yin L, Lee JH (2014) Hepatoprotective role of Sestrin2 against chronic ER stress. Nat Commun 5:4233
Pasha M, Eid AH, Eid AA, Gorin Y, Munusamy S (2017) Sestrin2 as a novel biomarker and therapeutic target for various diseases. Oxid Med Cell, Longev, pp 3296294–3296294
Perse P, Cerar A (2011) Morphological and molecular alterations in 1,2 dimethylhydrazine and azoxymethane induced colon carcinogenesis in rats. J Biomed Biotechnol 2011:473964
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath IA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IPM (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14(15):2231–2239
Porporato PE, Payen VL, Perez-Escudo J, De Saedeleer CJ, Danhier P, Copetti T, Dhup S, Tardy M, Vazeille T, Bouzin C, Feron O, Michiels C, Gallez B, Sonveaux P (2014) Mitochondrial switch promotes tumor metastasis. Cell Rep 8:754–766
Ro SH, Xue X, Ramakrishnan SK, Cho CS, Namkoong S, Jang I, Semple IA, Ho A, Park HW, Shah YMn Lee JH (2016) Tumor suppressive role of Sestrin2 during colitis and colon carcinogenesis. Elife 5:e12204
Saito R, Suzuki T, Hiramoto K, Asami S, Naganuma E, Suda H, Iso T, Yamamoto H, Morita M, Baird L, Furusawa Y, Negishi T, Ichinose M, Yamamoto M (2016) Characterizations of three major cysteine sensors of keap1 in stress response. Mol Cell Biol 36(2):271–284
Sarsour EH, Kalen AL, Goswami PC (2014) Manganese superoxide dismutase regulates a redox cycle within the cell cycle. Antioxid Redox Signal 20(10):1618–1627
Satoh T, Lipton SA (2007) Redox regulation of neuronal survival by electrophilic compounds. Trends Neurosci 30:38–45
Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, Akamoto S, Tabata S, Saitoh K, Kato K, Sato S, Igarachi K, Aizawa Y, Kajino-Sakamoto R, Kojima Y, Fujishita T, Enomoto A, Hirayama A, Ishakawa T, Taketo MM, Kushida Y, Haba R, Okano K, Tomita M, Susuki Y, Fukuda S, Aoki M, Soga T (2017) Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci USA 114:E7697-e7706
Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, Rhodes KJ (2012) Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J Pharmacol Exp Ther 341(1):274–284
Sciacovelli M, Frezza C (2017) Fumarate drives EMT in renal cancer. Cell Death Differ 24:1–2
Snezhkina AV, Kudryavtseva AV, Kardymon OL, Savvateeva MV, Melnikova NV, Krasnov GS, Dmitriev AA (2019) ROS generation and antioxidant defense systems in normal and malignant cells oxid. Med Cell Longev 5:2019
Sohn OS, Fiala ES, Requeijo SO, Weisburger JH, Gonzalez FJ (2001) Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res 61(23):8435–8440
Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, Haigis MC (2017) Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Sci 358:941–946
Steelman LS, Abrams SL, Whelan J, Libra M, Stivala F, Milella M, Tafuri A, Lunghi P, Bonati A, Martelli AM, McCubrey AJ (2008) Contributions of the Raf/MEK/ ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 22:686–707
Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, Cheung E, Olin-Sandoval V, Grüning NM, Krüger A, Alam MT, Keller MA, Breitenbach M, Brindle KM, Rabinowitz JD, Ralser M (2016) The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc 90:927–963
Sun H, Huang D, Liu G, Jian F, Zhu J, Zhang L (2018) SIRT4 acts as a tumor suppressor in gastric cancer by inhibiting cell proliferation, migration, and invasion. Onco Targets Ther 11:3959–3968
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71:209–249
Tardito S, Oudin A, Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen PO, Weinstock A, Wagner A, LindsaySL HAK, Barett SC, Ruppin E, Mørkve SH, LundJohansen M, Chalmers AJ, Bjerkvig R, Niclou SP, Gottlieb E (2015) Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nature Cell Biol 17:1556–1568
Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A (2006) TGF and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 16:1987–2002
Vozza A, Parisi G, de Leonardis F, Lasorsa FM, Castegna A, Amorese D, Marmo R, Calcagnile VM, Palmieri L, Ricquier D, Paradies E, Scarcia P, Palmieri F, Bouillaud F, Fiermonte G (2014) UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci USA 111:960–965
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Sci 334:1081–1086
Wang Y, Qi H, Liu Y, Duan C, Liu X, Xia T, Chen D, Piao HL, Liu HX (2021) The double-edged roles of ROS in cancer prevention and therapy. Theranostics 11(10):4839–4857
Warburg O (1956) On the origin of cancer cells. Sci 123:309–314
Weinhouse S (1956) On respiratory impairment in cancer cells. Science 124:267–269
Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, Birbe RC, Howell A, Pavlides S, Gandara R, Pestell RG, Sotgia FS, Philp NJ, Lisanti MP (2011) Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 10:1772–1783
Wise DR, DeBrardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, Thompson CB (2008) Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA 105(48):18782–18787
Woo DK, Green PD, Santos JH, D’Souza AD, Walther Z, Martin WD, Christian BE, Chandel NS, Shadel GS (2012) Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APCMin/+ mice. Am J Pathol 180:24–31
Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Ying Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL (2012) Inhibition of alpha-KG dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26:1326–3138
Zaleski J, Wilson DF, Erecinska M (1986) 2-Aminobicyclo-(2.2.1)-heptane-2-carboxylic acid, a new activator of glutaminase in intact rat liver mitochondria. J Biol Chem 261(30):14091–10494
Zelko IN, Mariani TJ, Folz RJ (2022) Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) Gene Structures, Evolution, and Expression. Free Radic Biol Med 33:337–349
Zhang Y, Park J, Han SJ, Yang SY, Yoon HJ, Park I, Woo HA, Lee SR (2020) Redox regulation of tumor suppressor PTEN in cell signaling. Red Biol 34:101553
Zhu Y, Richardson JA, Parada LF, Graff JM (1998) Smad3 mutant mice develop metastatic colorectal cancer. Cell 94:703–714
Acknowledgements
We acknowledge all the staff of the Laboratory of Pharmacology (University of Porto, Portugal) for their valuable scientific contribution to this study.
This study was supported in part by the “Direction Générale de la Recherche Scientifique et du Development Technique (DG-RSDT),” Algiers, Algeria.
Author information
Authors and Affiliations
Contributions
BD, JG, and BA designed the research. AB contributed to the experimental tumorigenesis protocol. BA, DS, and CQ conducted experiments. SYD made the histological and immunohistological evaluations. BD, JG, and BA analyzed data and wrote the manuscript. BD revised the manuscript.
Corresponding author
Ethics declarations
Ethical approval
All the in vivo experiments were carried out in agreement with the Ethics Committee for Animal Welfare of the University of Science and Technology Houari Boumediene (Algiers) [Algerian Law 12–235/2012; Executive Decree No. 10–90] and in accordance with the European Directive 2010/63/EU for ethics in animal experimentation.
Consent to participate and for publication
All authors consent to participate in this study. They read and approved the manuscript. The authors declare that all data were generated in-house and that no paper mill was used.
Competing interests
Not applicable.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Highlights
• 1,2-Dimethylhydrazine (DMH) depresses Sesn2 and Nrf2 and redox homeostasis.
• Long-term DMH supports glutamate dehydrogenase (GDH) activity by blocking sirtuin4.
• the burst in matrix superoxide dismutase activity induced mitochondrial stress.
• Dimethylfumarate (DMF) rescued GDH activity inhibited by epigallocatechin gallate.
• DMF triggers GDH-mediated cancer cell's viability, motility and clogenicity.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Allal, BE., Bounaama, A., Silva, D. et al. Long-term 1,2-dimethylhydrazine triggers pathological remodeling of colon mucosa through repression of sestrin2, nuclear factor (erythroid-derived 2)-like 2, and sirtuin4 stimulating mitochondrial stress and metabolic reprogramming. Naunyn-Schmiedeberg's Arch Pharmacol 396, 1291–1307 (2023). https://doi.org/10.1007/s00210-023-02403-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-023-02403-x