
Abstract
Asiatic acid, a triterpenoid compound derived from Centella asiatica, has been demonstrated to have antioxidant and anti-inflammatory effects. The present study evaluated the effects of asiatic acid on hemodynamic alterations, renin-angiotensin system (RAS), oxidative stress, and inflammation in 2K-1C hypertensive rats. Renovascular hypertension was induced in male Sprague-Dawley rats and treated with vehicle, asiatic acid (30 mg/kg/day), or captopril (5 mg/kg/day) for 4 weeks. We observed that 2K-1C hypertensive rats exhibited hemodynamic alterations such as high blood pressure, heart rate, hindlimb vascular resistance, and low hindlimb blood flow. Signs of RAS activation, such as increased plasma angiotensin II and serum angiotensin-converting enzyme activity, enhanced AT1R protein expression, and suppressed AT2R expression was observed in 2K-1C hypertensive rats. Overproduction of vascular superoxide, high levels of plasma MDA, low levels of plasma nitric oxide metabolites (NOx), and upregulation of gp91phox protein expression were observed in hypertensive rats. Furthermore, inflammation was observed in hypertensive rats, as evidenced by increased plasma TNF-α, NF-κB, and phospho-NF-κB protein expression. Asiatic acid or captopril alleviated hemodynamic alterations, RAS activation, oxidative stress, and inflammation in 2K-1C hypertensive rats. These findings indicate that asiatic acid is an antihypertensive agent that ameliorates hemodynamic alterations in 2K-1C hypertensive rats. This effect may involve one or both of the following mechanisms: the direct effect of asiatic acid on RAS activation, oxidative stress and inflammation, and/or asiatic acid acting as an ACE inhibitor agent to inhibit the Ang II-AT1R-NADPH oxidase-NF-κB pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The two-kidney, one clip (2K-1C) or Goldblatt hypertension model is a renin-angiotensin system dependent hypertensive model that is commonly used for the study of renovascular hypertension (Goldblatt et al. 1934). Activation of the renin-angiotensin system (RAS), including increased angiotensin converting enzyme (ACE) activity, high levels of plasma angiotensin II (Ang II), upregulation of AT1R, and downregulation of AT2R, has been shown to be an underlying mechanism in 2K-1C-induced hypertension (Boonla et al. 2015; Li et al. 2010; Park et al. 2015; Santuzzi et al. 2015). Under physiological conditions, Ang II mainly binds to AT1Rs to produce vasoconstriction, anti-natriuresis, aldosterone secretion, sympathetic nervous system activation, cellular dedifferentiation and growth, inflammation, and target organ damage (Carey 2016). However, AT2Rs can counteract the action of Ang II, since activation of AT2Rs by the Ang II metabolite (angiotensin III) induces natriuresis, blood pressure reduction, and reduction of Na+ reabsorption (Padia et al. 2008). Other abnormalities such as oxidative stress, inflammation, and cardiovascular remodeling in 2K-1C hypertension have also been reported (Boonla et al. 2014; Guo et al. 2014; Montenegro et al. 2010).
Elevation of circulating Ang II associated with oxidative stress in 2K-1C rats has been documented. Griendling et al. (1994) found that Ang II stimulates superoxide (O2 ·−) production through NADPH oxidase activation in cultured vascular smooth muscle cells (Griendling et al. 1994). It has been clearly demonstrated that NADPH oxidase is the major source of O2 ·− in vasculature (Landmesser and Harrison 2001). Jung et al. (2004) found that endothelial gp91phox-containing NADPH oxidase mediates vascular O2 ·− formation and the subsequent decrease in NO bioavailability in 2K-1C hypertensive mice (Jung et al. 2004). There is substantial evidence showing the association between reactive oxygen species (ROS) and inflammation induced by Ang II (Browatzki et al. 2005; Ortego et al. 1999). It is also clear that the transcription factor nuclear factor-kappa B (NF-κB) is the main signaling pathway involved in inflammation (Garrido and Griendling 2009; Rioja et al. 2002).
Ang II-induced NF-κB activation has been shown to be ROS dependent (Garrido and Griendling 2009). Gräfeand coworkers found that Ang II activates AT1Rs to increase the expression of adhesion molecule E-selectin and subsequent leukocyte adhesion to coronary endothelial cells (Grafe et al. 1997). Ang II enhances tumor necrosis factor (TNF)-α production in smooth muscle cells isolated from the medullary thick ascending limb (Ferreri et al. 1998). In human vascular SMCs, Ang II enhances the inflammatory response by stimulating cytokine production and activating NF-κB (Kranzhofer et al. 1999b). In addition, inflammation activated by Ang II has also been demonstrated in 2K-1C hypertensive rats (Johns 2009). Low-grade inflammation plays an important role in the development and pathophysiology of hypertension (Bautista et al. 2005). This is supported by the finding that increases in plasma inflammatory components TNF-α and IL-6, which are associated with endothelial dysfunction and cardiac hypertrophy, have been observed in 2K-1C hypertensive rats (Kalaivani et al. 2013; Moubarak et al. 2012).
Asiatic acid is a triterpenoid compound isolated from Centella asiatica. The antioxidative and anti-inflammatory activities of asiatic acid have been demonstrated in the hydrogen peroxide (H2O2)-induced injury model in human bronchial epithelial cells. Antihyperlipidemic and antidiabetic activities of asiatic acid in streptozotocin-induced diabetic rats have also been proposed (Ramachandran et al. 2014). Recent studies demonstrated that asiatic acid reduces blood pressure, improves vascular function via restoration of endothelial NO synthase (eNOS) and p47phox expression, and alleviates cardiovascular remodeling via restoration of iNOS/eNOS expression in L-NAME hypertensive rats (Bunbupha et al. 2014, 2015).
Previous studies showed that asiatic acid suppresses RAS and inflammation in rat models of HCHF diet-induced metabolic syndrome (Maneesai et al. 2016a; Pakdeechote et al. 2014). Therefore, the present study investigated whether the mechanism of antihypertensive effect of asiatic acid in 2K-1C hypertensive rats could involve reducing RAS activity, oxidative stress, and inflammation in RAS-dependent rats.
Materials and methods
Animals and experimental protocols
Chemicals
Asiatic acid (purity ≥98%, CAS NO. 464926) and captopril (purity ≥98%, CAS NO. 101296084) were purchased from ChemFaces, Hubei, China and Sigma-Aldrich Corp, MO, USA respectively.
Animals and experimental protocols
The protocols in this study were conducted in accordance with standards for the care and use of experimental animals and approved by the Animal Ethics Committee of Khon Kaen University (AEKKU-NELAC 27/2557). Rats were housed under standard conditions (25 ± 2 °C with a 12-h dark–light cycle) at the Northeast Laboratory Animal Center, Khon Kaen University, Khon Kaen, Thailand. Male Sprague-Dawley rats (160–170 g, about 5 weeks of age) were purchased from the National Laboratory Animal Center, Mahidol University, Salaya, Nakornpathom. Renovascular hypertension was induced following the 2K-1C Goldblatt model. Briefly, rats were anesthetized, and a silver clip (0.2 mm i.d.) was placed on the left renal artery. In the sham-operated group, the same surgical procedure was performed, but the clip was not applied on the left renal artery.
Sustained hypertension was observed 4 weeks after the operation. Rats that had an SP higher than 160 mmHg were included as 2K-1C hypertensive rats. Animals were then assigned into five study groups: group 1, sham-operated group (Sham; n = 12); group 2, sham-operated treated with asiatic acid group (Sham + AA; n = 6); group 3, 2K-1C group (2K-1C; n = 12); group 4, 2K-1C treated with asiatic acid group (2K-1C + AA; n = 12); and group 5, 2K-1C treated with captopril group (2K-1C + CAP; n = 12). Rats in the AA-treated groups received asiatic acid (30 mg/kg/day) and rats in the captopril-treated groups received captopril (5 mg/kg/day) for the last 4 weeks. Asiatic acid and captopril are dissolved in propylene glycol (vehicle) were intragastrically administered once daily. The dose of asiatic acid used in this study was titrated according to our previous studies (Pakdeechote et al. 2014; Umka Welbat et al. 2016). The dose of captopril was followed our previous study (Maneesai et al. 2016b).
Blood pressure measurement
Rat systolic blood pressure (SP) was measured once a week using non-invasive tail-cuff plethysmography (IITC/Life Science Instrument model 229 and model 179 amplifier; Woodland Hills, CA, USA) to monitor blood pressure changes throughout the study periods.
Hemodynamic measurement
At the end of the experimental day, rats were anesthetized with pentobarbital sodium (60 mg/kg). Polyethylene tube filled with heparinized was inserted into the femoral artery for blood pressure recording. Baseline values of systolic blood pressure (SP), diastolic blood pressure (DP), mean arterial blood pressure (MAP), and heart rate (HR) were analyzed by AcqKnowledge data acquisition and analysis software (Biopac Systems Inc., Santa Barbara, CA, USA). Thereafter, the abdominal aorta was cleaned of connective tissue and electromagnetic flow probes connected to an electromagnetic flow meter was placed around the aorta for Hindlimb blood flow (HBF) measurement (Carolina Medical Electronics, Carolina, NC, USA). Hindlimb vascular resistance (HVR) was calculated from MAP and HBF (HVR = MAP/HBF).
Assay of oxidative stress markers
Production of O2 ·−in vascular tissues was measured in carotid tissue using lucigenin-enhanced chemiluminescence as previously described (Luangaram et al. 2007). In brief, segments of the artery (1 cm in length) were cleaned of adherent fat and connective tissue and then incubated with 1 mL oxygenated Krebs-KCl buffer. The vessel was allowed to equilibrate at pH 7.4, 37 °C for 30 min before lucigenin was added to the sample tube and placed in a luminometer (Turner Biosystems, CA, USA) to count the production of O2 ·−every 30 s for 5 min. The average value was expressed as relative light unit count per minute per milligram of dried tissue weight.
Plasma MDA level was assayed spectrophotometrically using a thiobarbituric acid (TBA)–MDA assay. Briefly, a plasma sample (150 μL) was mixed with 10% TCA, 5 mmol/L EDTA, 8% SDS, and 0.5 μg/mL BHT respectively and incubated for 10 min at room temperature. Thereafter, 0.6% TBA was added to the mixture and kept in a boiling water bath for 30 min. The mixtures were cooled at room temperature and centrifuged at 10,000g for 5 min. The supernatant was collected, and the absorbance at 532 nm was read using a spectrophotometer (Amersham Bioscience, Arlington, MA, USA). MDA levels were calculated as μM concentration using standard curve at different concentrations from 0.3 to 10 μmol/L of 1,1,3,3-tetraethoxypropane.
The level of plasma NO x was assayed using an enzymatic conversion method as previous described (Luangaram et al. 2007). Briefly, plasma samples were deproteinized and then the supernatant was mixed with 1.2 μmol/L NADPH, 4 mmol/L glucose-6-phosphate disodium, 1.28 unit/mL glucose-6-phosphate dehydrogenase, and 0.2 U/mL nitrate reductase. Thereafter, the mixture was incubated at 30 °C for 30 min and reacted with Griess solution (4% sulfanilamide in 0.3% NED) for 15 min. The absorbance of the sample was read at a wavelength of 540 nm using a microplate reader (Tecan GmbH., Grödig, Austria).
Assay of serum ACE activity
ACE activity in serum was measured using a fluorescence assay following a previously described method (Friedland and Silverstein 1976) with some modifications. Briefly, 25 μl of serum was mixed with 3.5 mMhippuryl-l-histidyl-l-leucine(HHL) in assay buffer containing 20 mM sodium borate and 0.3 M NaCl, pH 8.3 in a final volume of 35 μl. The mixtures were then incubated at 37 °C for 30 min, and the reaction was stopped by adding 150 μl of 0.34 M NaOH. The product of the reaction was fluorogenically labeled with 10 mg/mL o-phthaldialdehyde (OPA) and read on a fluorescent plate reader at 355 nm excitation; 535 nm emission. ACE activity is reported as mU/mL.
Assay of plasma Ang II and plasma TNF-α concentration
The concentrations of plasma Ang II and plasma TNF-α was measured using an angiotensin II EIA kit (St. Louis, MO, USA) and enzyme-immunoassay (ELISA) kit (eBioscienc, Inc., CA, USA), respectively.
Western blot analysis
The expression of AT1R, AT2R, gp91phox, Basal-NF-κB, and Phospho-NF-κB proteins were measured in mesenteric homogenates (superior mesenteric artery and its first-order and the second-order branches). The tissue samples were electrophoresed through a sodium dodecyl sulfate polyacrylamide gel and transferred onto a polyvinylidenedifluoride (PVDF) membrane. Non-specific binding to the membrane was blocked by treatment with 5% skimmed milk in phosphate buffer saline with 0.1% Tween-20 (PBST) for 2 h at room temperature and then washed with PBST. Thereafter, the membrane were incubated overnight (4 °C) with mouse monoclonal antibodies to gp91phox (BD Biosciences, San Jose, CA, USA) or rabbit polyclonal antibodies to AT1R, AT2R (Santa Cruz Biotechnology, Indian Gulch, CA, USA) or Basal-NF-κB (Santa Cruz Biotechnology, Indian Gulch, CA, USA), or Phospho-NF-κB (Cell Signaling Technology, Danvers, MA, USA) and goat polyclonal IgG to β-actin (Santa Cruz Biotechnology, Indian Gulch, CA, USA). After washing with PBST, membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature. Antigen–antibody reaction was detected using Amersham™ ECL™ Prime solution (Amersham Biosciences Corp., Piscataway, NJ, USA), and densitometric analysis was performed using an ImageQuant™400 imager (GE Healthcare Life Science, Piscataway, NJ, USA). β-Actin was used for protein loading control in the same gel.
Statistical analysis
Data are present as mean ± SEM. The differences among treatment groups were analyzed by one-way analysis of variance (ANOVA). Where appropriate, a post hoc Bonferroni correction was used to assess differences among groups. A p value less than 0.05 was considered statistically significant.
Results
Effects of asiatic acid and captopril on blood pressure
At baseline (week 0), there was no significant difference in SP among experimental groups. After clipping the left renal artery, systolic blood pressure progressively increased in 2K-1C hypertensive rats compared to sham-operated rats (SP at 8 weeks of study period, 217.4 ± 10 vs. 136.5 ± 2.7 mmHg, p < 0.01). Daily administration of asiatic acid (30 mg/kg/day) or captopril (5 mg/kg/day) for 4 weeks significantly reduced SP in 2K-1C hypertensive rats compared with untreated rats (SP, 177.8 ± 7.6 and 166.6 ± 8.5 mmHg respectively, p < 0.05). However, supplementation with asiatic acid had no effect on SP in sham-operated rats (131.1 ± 1.6 mmHg, Fig. 1).

Effects of asiatic acid and captopril on blood pressure. The chemical structure of asiatic acid (a). Effects of asiatic acid and captopril on systolic blood pressure in conscious rats of all experimental groups (b). Data are expressed as mean ± SEM (n = 6–12 in each group). *p < 0.05 vs. sham group, # p < 0.05 vs. 2K-1C group
Effects of asiatic acid and captopril on hemodynamic status
After the week study period, marked increases in SP, DP, MAP, and HR were found in 2K-1C hypertensive rats compared to sham-operated rats (p < 0.05). Moreover, an increase in HVR and decrease in HBF were also observed. Treatment with asiatic acid (30 mg/kg/day) caused significant decreases in SP, DP, and MAP compared to the untreated group (p < 0.05). Improvement of HBF and HVR was also observed in the asiatic acid treated group compared to the untreated group (p < 0.05). In addition, asiatic acid did not affect hemodynamic status in sham-operated rats (Table 1). Hypertensive rats that received captopril also showed improvement in all hemodynamic parameters except HR compared to untreated hypertensive rats (p < 0.05) (Table 1).
Effects of asiatic acid and captopril on serum ACE activity and plasma Ang II concentration, AT1R, and AT2R protein expression
Serum ACE activity in 2K-1C hypertensive rats was elevated compared to that of sham-operated rats (245.11 ± 10.6 vs. 187.8 ± 7.1 mU/mL, p < 0.01). However, the activity of ACE was attenuated in hypertensive rats that were treated with asiatic acid or captopril (208.8 ± 10.2 and 206.4 ± 9.5 mU/mL respectively, p < 0.05, Fig. 2a). Similarly, a high level of plasma Ang II was observed in 2K-1C hypertensive rats compared to that of sham-operated rats (57.9 ± 1.9 vs. 19.5 ± 2.1 pg/mL, p < 0.01). The level of plasma Ang II in hypertensive rats was markedly reduced after treatment with asiatic acid or captopril rats compared to untreated rats (28.2 ± 3.1 and 38.7 ± 7.6 pg/mL respectively, p < 0.05, Fig. 2b).

Effects of asiatic acid and captopril supplementation on serum angiotensin converting enzyme (ACE) activity (a) and angiotensin II (Ang II) level (b) in sham-operated, 2K-1C, 2K-1C + AA, and 2K-1C + Cap groups. Data are expressed as mean ± SEM (n = 8–10 in each group), *p < 0.05 vs. sham-operated group, # p < 0.05 vs. 2K-1C group
Upregulation of AT1R protein expression in mesenteric arteries was observed in 2K-1C hypertensive rats (p < 0.05). Asiatic acid partially decreased the expression of AT1R protein level in mesenteric arteries (Fig. 3a). Interestingly, treatment with captopril completely restored AT1R protein expression in mesenteric arteries of hypertensive rats (p < 0.05). In addition, downregulation of AT2R was found in mesenteric tissue of hypertensive rats (p < 0.05). An upregulation of AT2R expression was observed in the asiatic acid or captopril-treated groups (p < 0.05) (Fig. 3b).

Mesenteric protein expression of angiotensin II type I receptor (AT1R) (a) and type II receptor (AT2R) (b) collected from sham-operated, 2K-1C, 2K-1C + AA, and 2K-1C + Cap groups. Data are expressed as mean ± SEM (n = 5 in each group). Top: representative bands for AT1R. *p < 0.05 vs. sham-operated group, # p < 0.05 vs. 2K-1C group
Effects of asiatic acid and captopril supplementation on oxidative stress markers and gp91phox protein expression
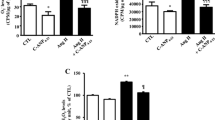
There was a significant increase in vascular O2 ·− production in 2K-1C hypertensive group compared to sham-operated control (160.3 ± 20.4 vs. 67.8 ± 7.4 count/mg dry wt/min, p < 0.001). O2 ·− production was significantly reduced in hypertensive rats treated with asiatic acid and captopril compared to untreated rats (97.2 ± 17.2 and 101.9 ± 15.2 count/mg dry wt/min respectively, p < 0.05) (Fig. 4a). Plasma MDA level in the 2K-1C hypertensive group was significantly higher than that of the sham-operated group (11 ± 0.9 vs. 7.2 ± 0.3 μM, p < 0.05). This elevation of plasma MDA was markedly attenuated in hypertensive rats treated with asiatic acid or captopril compared to that of untreated rats (8.1 ± 0.9 and 7 ± 1.1 μM, respectively, <0.05) (Fig. 4b). In contrast, a significant decrease in plasma NO x concentration was found in 2K-1C hypertensive rats compared to sham-operated rats (8.3 ± 1.6 vs. 12.7 ± 0.8 μM, p < 0.01). This low level of NO x was normalized by asiatic acid or captopril treatment (12.8 ± 0.5 and 11.8 ± 0.9 μM, respectively, p < 0.05) (Fig. 5a).

Effects of asiatic acid or captopril supplementation on vascular O2 ·− production (a) and plasma malondialdehyde (MAD) (b) levels in all experimental groups. Data are expressed as mean ± SEM (n = 6–10 in each group). *p < 0.05 vs. sham-operated group, # p < 0.05vs. 2K-1C group

Effects of asiatic acid and captopril supplementation on plasma nitric oxide metabolite (NOx) level (n = 7–10 in each group) (a) and protein expression of gp91phox expression (n = 4) (b) in mesenteric arteries in sham-operated, 2K-1C, 2K-1C + AA, and 2K-1C + Cap groups. Top: representative bands showing gp91phox subunit expression. Data are expressed as mean ± SEM, *p < 0.05 vs. sham-operated group, # p < 0.05 vs. 2K-1C group
Protein expression of gp91phox was upregulated in mesenteric arteries of 2K-1C hypertensive rats compared to those of sham-operated rats (p < 0.01). The elevation of gp91phox in mesenteric arteries of 2K-1C hypertensive rats was reduced by the administration of asiatic acid or captopril (p < 0.05) (Fig. 5b).
Effects of asiatic acid and captopril on tumor necrosis factor alpha (TNF-α) and basal NF-κB and phospo-NF-κB protein expression
A marked increase in plasma TNF-α was observed in 2K-1C hypertensive rats compared to those of sham-operated rats (1159.8 ± 46 vs. 810.6 ± 40.2 pg/mL, p < 0.01). Asiatic acid or captopril reversed these high levels of plasma TNF-α in hypertensive rats (855.5 ± 92.9 and 857 ± 70.4 pg/mL, respectively, p < 0.05) (Fig. 6a). Overexpression of basal and phospho-NF-κB levels was found in the mesenteric arteries of 2K-1C hypertensive rats compared to those of sham-operated rats (p < 0.01). This overexpression was attenuated by asiatic acid or captopril supplementation (p < 0.05) (Fig. 6b, c).

Effects of asiatic acid and captopril supplementation on plasma TNF-α level (n = 7–10 in each group) (a), protein expression of basal-NF-κB (NF-κB) (n = 3) (b), and phospho-NF-κB (P-NF-κB) (n = 3) (c) in mesenteric artery collected from sham-operated, 2K-1C, 2K-1C + AA, and 2K-1C + Cap groups. Top: representative bands showing basal-NF-κB and phospho-NF-κB expression. Data are expressed as mean ± SEM, *p < 0.05 vs. sham-operated group, # p < 0.05 vs. 2K-1C group
Discussion
The present study examined the mechanism of the antihypertensive effect of asiatic acid in renovascular hypertensive rats. After reducing renal blood flow by clipping the left renal artery, we observed hemodynamic alterations such as high blood pressure, increased HR, increased HVR, and decreased HBF. Activation of RAS was also observed in these 2K-1C hypertensive rats, as evidenced by increased ACE activity, high levels of plasma Ang II, upregulation of AT1R, and downregulation of AT2R. In addition, we observed increases in oxidative stress markers, upregulation of gp91phox, and low levels of NO metabolites in 2K-1C hypertensive rats. We also observed inflammation in 2K-1C hypertensive rats, as evidenced by high levels of plasma TNF-α as well as upregulation of basal NF-κB and phosphor-NF-κB expression. Asiatic acid and captopril significantly alleviated hemodynamic alterations associated with reducing RAS activation, oxidative stress, and inflammation in 2K-1C hypertensive rats.
In 2 K-1C hypertensive rats, we observed increases in BP, HR, and HVR and a decrease in HBF. Hemodynamic alterations found in this study were associated with increased ACE activity, increased plasma Ang II and AT1R expression, and decreased AT2R expression. It has been well documented that high blood pressure in renovascular hypertension is mediated by RAS activation (Goldblatt et al. 1934). Our data are consistent with several previous studies on hypertension. For example, Boonla and coworkers reported that 2K-1C hypertensive rats showed hemodynamic alterations and increased plasma ACE activity (Boonla et al. 2015). Increased plasma renin activity and increased circulating Ang II have been observed in 2K-1C hypertensive rats (Goldblatt et al. 1934). Ang II binds to AT1R in vascular smooth muscle cells of blood vessels and subsequently mediates vasoconstriction, high vascular resistance, and high blood pressure (Higuchi et al. 2007). Overexpression of AT1R in aortic and renal tissues (Santuzzi et al. 2015) and the suppression of AT2R expression in renal and heart tissue (Li et al. 2010; Santuzzi et al. 2015) have been reported. Thus, our results confirm that overactivation RAS leads to the pathophysiology observed in renovascular hypertension. Furthermore, we found that asiatic acid alleviated all hemodynamic alterations present in hypertensive rats. These effects involved suppression of RAS activation by asiatic acid. Our data are consistent with a recent study that showed that the antihypertensive effect of asiatic acid in metabolic syndrome-induced hypertensive rats was due to its ability to reduce RAS overactivation (Maneesai et al. 2016a). The precise mechanism by which asiatic acid alleviates RAS activation is unclear. However, it is possible the ACE inhibitor activity of asiatic acid mediates this effect, since captopril, an ACE inhibitor, also ameliorates hemodynamic disturbance and RAS activation in 2K-1C hypertensive rats. The mechanisms of ACE inhibition may be a direct interaction between asiatic acid and ACE and/or asiatic acid reduced ACE protein levels as reported by our recent report (Maneesai et al. 2016a).
Ang II activates AT1R, leading to production of oxidative stress mainly via NADPH oxidase (Garrido and Griendling 2009; Rincon et al. 2015). In addition, upregulation of NADPH oxidase subunits such as p47phox, p22phox, and gp91phox together with increases in vascular O2 ·− and plasma MDA level and a reduction of NO metabolite have been observed in a 2K-1C hypertensive model (Boonla et al. 2014; da Costa et al. 2012; Jung et al. 2004). Our study showed high levels of oxidative stress markers, plasma MDA, and vascular O2 ·− production in 2K-1C hypertensive rats. It is possible that the main source of vascular O2 ·− production found in this study is the NADPH oxidase subunit gp91phox, since we found upregulation of gp91phox expression in mesenteric arteries of 2K-1C hypertensive rats. O2 ·− can react with NO to produce ONOO−, a powerful oxidant (Squadrito and Pryor 1995). This reaction causes a reduction of NO bioavailability, which then increases vascular tone and blood pressure. Thus, high production of O2 ·− could decrease NO bioavailability, as evidenced by the low levels of plasma NO x we observed in 2K-1C hypertensive rats. Treatment with asiatic acid significantly reduced oxidative stress markers, suppressed gp91phox expression, and increased plasma NO x in hypertensive rats. This beneficial effect of asiatic acid in decreasing oxidative stress markers in the present study may involve at least two mechanisms. Firstly, asiatic acid has an ACE inhibitor activity and reduces circulating Ang II. Secondly, asiatic acid has antioxidant properties (Huang et al. 2011) and directly quenches O2 ·−. There is direct evidence that the antioxidant property of asiatic acid is mediated via several hydroxyl groups, an olefin group, and a carboxylic acid group (Jew et al. 2000). Additionally, triterpenes that contains asiatic acid had greater antioxidative effects against glucose-induced glutathione loss and malondialdehyde and oxidized glutathione production in renal homogenates isolated from mice (Yin et al. 2012). A previous study demonstrated that asiatic acid improves oxidative stress status in L-NAME-induced hypertensive rats by reducing the expression of NADPH oxidase subunit p47phox in the thoracic aorta (Bunbupha et al. 2014). Similarly, captopril reduces blood pressure and RAS activation in 2K-1C hypertensive rats. These effects of captopril are associated with its ACE inhibitor (Lewis et al. 1985) and antioxidant properties (Mak et al. 1990).
Inflammation is involved in the development and pathophysiology of hypertension, since it plays an important role in atherosclerosis and vascular remodeling (Li and Chen 2005). We found overexpression of the basal and phosphorylated forms of NF-κB in mesenteric arteries and high levels of plasma TNF-α in 2K-1C hypertensive rats. Kranzhofer et al. (1999a, b) found Ang II-induced NF-κB activation via AT1R in human monocytes (Kranzhofer et al. 1999a). Furthermore, expression of NF-κB was enhanced in 2K-1C hypertensive rats with left ventricular remodeling (Cau et al. 2015). Significant increases in inflammatory markers that were associated with cardiac hypertrophy in 2K-1C hypertensive rats have also been observed (Moubarak et al. 2012). Our results may indicate that the inflammation in 2K-1C hypertensive rats is a consequence of Ang II stimulating AT1R to produce O2 ·− production via NADPH oxidase, which then induces the expression of NF-κB. In our study, asiatic acid attenuated the expression of NF-κB and the elevation of plasma TNF-α in hypertensive rats. This result was consistent with previous studies in which asiatic acid exhibited anti-inflammatory properties by reducing iNOS protein expression and reducing the level of plasma TNF-α in L-NAME-induced hypertensive rats (Bunbupha et al. 2015). Another mechanism by which asiatic acid might alleviate oxidative stress is through its ACE inhibitor activity, which could subsequently suppress NF-κB expression, since Ang II-induced NF-κB activation is ROS dependent (Garrido and Griendling 2009). Similarly, treatment with captopril also decreased inflammation induced by activation of RAS in hypertensive rats. It is possible that captopril reduces Ang II production through its ACE inhibitor and antioxidant activity and/or through its anti-inflammatory effects (Miguel-Carrasco et al. 2010).
The present study provides evidence that asiatic acid normalizes hemodynamic alterations in 2K-1C hypertensive rats. At least two mechanisms by which asiatic acid reduces blood pressure have been proposed. Firstly, asiatic acid might directly reduce ACE activity, Ang II levels, oxidative stress, and inflammation in 2K-1C hypertensive rats. Secondly, ACE inhibitor activity of asiatic acid might restore the Ang II-AT1R-gp91phox-NF-κB pathway (Fig. 7).

Schematic representation the effect of asiatic acid (AA) on RAS-induced hypertension and Ang II-AT1R-ROS-NF-κB pathway in 2K-1C hypertensive rats. TPR represents total peripheral resistance
References
Bautista LE, Vera LM, Arenas IA, Gamarra G (2005) Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens 19:149–154. doi:10.1038/sj.jhh.1001785
Boonla O, Kukongviriyapan U, Pakdeechote P, Kukongviriyapan V, Pannangpetch P, Prachaney P, Greenwald SE (2014) Curcumin improves endothelial dysfunction and vascular remodeling in 2K-1C hypertensive rats by raising nitric oxide availability and reducing oxidative stress. Nitric Oxide 42:44–53. doi:10.1016/j.niox.2014.09.001
Boonla O, Kukongviriyapan U, Pakdeechote P, Kukongviriyapan V, Pannangpetch P, Thawornchinsombut S (2015) Peptides-derived from Thai Rice bran improves endothelial function in 2K-1C renovascular hypertensive rats. Nutrients 7:5783–5799. doi:10.3390/nu7075252nu7075252
Browatzki M et al (2005) Angiotensin II stimulates matrix metalloproteinase secretion in human vascular smooth muscle cells via nuclear factor-kappaB and activator protein 1 in a redox-sensitive manner. J Vasc Res 42:415–423. doi:10.1159/000087451
Bunbupha S, Pakdeechote P, Kukongviriyapan U, Prachaney P, Kukongviriyapan V (2014) Asiatic acid reduces blood pressure by enhancing nitric oxide bioavailability with modulation of eNOS and p47phox expression in L-NAME-induced hypertensive rats. Phytother Res 28:1506–1512. doi:10.1002/ptr.5156
Bunbupha S, Prachaney P, Kukongviriyapan U, Kukongviriyapan V, Welbat JU, Pakdeechote P (2015) Asiatic acid alleviates cardiovascular remodeling in rats with L-NAME-induced hypertension. Clin Exp Pharmacol Physiol. doi:10.1111/1440-1681.12472
Carey RM (2016) Update on angiotensin AT2 receptors. Curr Opin Nephrol Hypertens. doi:10.1097/MNH.0000000000000304
Cau SB, Guimaraes DA, Rizzi E, Ceron CS, Gerlach RF, Tanus-Santos JE (2015) The nuclear factor kappaB inhibitor pyrrolidine dithiocarbamate prevents cardiac remodelling and matrix metalloproteinase-2 up-regulation in renovascular hypertension basic. Clin Pharmacol Toxicol 117:234–241. doi:10.1111/bcpt.12400
da Costa CA et al (2012) Euterpe Oleracea Mart.-derived polyphenols prevent endothelial dysfunction and vascular structural changes in renovascular hypertensive rats: role of oxidative stress. Naunyn Schmiedeberg's Arch Pharmacol 385:1199–1209. doi:10.1007/s00210-012-0798-z
Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC (1998) Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Phys 274:F148–F155
Friedland J, Silverstein E (1976) A sensitive fluorimetric assay for serum angiotensin-converting enzyme. Am J Clin Pathol 66:416–424
Garrido AM, Griendling KK (2009) NADPH oxidases and angiotensin II receptor signaling. Mol Cell Endocrinol 302:148–158. doi:10.1016/j.mce.2008.11.003
Goldblatt H, Lynch J, Hanzal RF, Summerville WW (1934) Studies on experimental hypertension : I. The Production of Persistent Elevation of Systolic Blood Pressure by Means of Renal Ischemia. J Exp Med 59:347–379
Grafe M et al (1997) Angiotensin II-induced leukocyte adhesion on human coronary endothelial cells is mediated by E-selectin. Circ Res 81:804–811
Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW (1994) Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 74:1141–1148
Guo K, Lan CZ, Yu TT, Huang LL, Wang XH, Pan C, Gao S (2014) Effects of Xin-Ji-Er-Kang formula on 2K1C-induced hypertension and cardiovascular remodeling in rats. J Ethnopharmacol 155:1227–1235. doi:10.1016/j.jep.2014.07.006
Higuchi S, Ohtsu H, Suzuki H, Shirai H, Frank GD, Eguchi S (2007) Angiotensin II signal transduction through the AT1 receptor: novel insights into mechanisms and pathophysiology. Clin Sci (Lond) 112:417–428. doi:10.1042/CS20060342
Huang SS et al (2011) Antinociceptive activities and the mechanisms of anti-inflammation of asiatic Acid in mice. Evid Based Complement Alternat Med 2011:895857. doi:10.1155/2011/895857
Jew SS et al (2000) Structure-activity relationship study of asiatic acid derivatives against beta amyloid (a beta)-induced neurotoxicity. Bioorg Med Chem Lett 10:119–121
Johns EJ (2009) Inflammation: the underlying foe in renovascular hypertension? J Hypertens 27:1964–1965. doi:10.1097/HJH.0b013e328331a881
Jung O, Schreiber JG, Geiger H, Pedrazzini T, Busse R, Brandes RP (2004) gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation 109:1795–1801. doi:10.1161/01.CIR.0000124223.00113.A4
Kalaivani P et al (2013) Cuminum cyminum, a dietary spice, attenuates hypertension via endothelial nitric oxide synthase and NO pathway in renovascular hypertensive rats. Clin Exp Hypertens 35:534–542. doi:10.3109/10641963.2013.764887
Kranzhofer R, Browatzki M, Schmidt J, Kubler W (1999a) Angiotensin II activates the proinflammatory transcription factor nuclear factor-kappaB in human monocytes. Biochem Biophys Res Commun 257:826–828. doi:10.1006/bbrc.1999.0543
Kranzhofer R, Schmidt J, Pfeiffer CA, Hagl S, Libby P, Kubler W (1999b) Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 19:1623–1629
Landmesser U, Harrison DG (2001) Oxidative stress and vascular damage in hypertension. Coron Artery Dis 12:455–461
Lewis RA, Baker KM, Ayers CR, Weaver BA, Lehman MR (1985) Captopril versus enalapril maleate: a comparison of antihypertensive and hormonal effects. J Cardiovasc Pharmacol 7(Suppl 1):S12–S15
Li JJ, Chen JL (2005) Inflammation may be a bridge connecting hypertension and atherosclerosis. Med Hypotheses 64:925–929. doi:10.1016/j.mehy.2004.10.016
Li J, Xie ZZ, Tang YB (2010) Genistein prevents myocardial hypertrophy in 2-kidney 1-clip renal hypertensive rats by restoring eNOS pathway. Pharmacology 86:240–248. doi:10.1159/000320457000320457
Luangaram S, Kukongviriyapan U, Pakdeechote P, Kukongviriyapan V, Pannangpetch P (2007) Protective effects of quercetin against phenylhydrazine-induced vascular dysfunction and oxidative stress in rats. Food Chem Toxicol 45:448–455. doi:10.1016/j.fct.2006.09.008
Mak IT, Freedman AM, Dickens BF, Weglicki WB (1990) Protective effects of sulfhydryl-containing angiotensin converting enzyme inhibitors against free radical injury in endothelial cells. Biochem Pharmacol 40:2169–2175
Maneesai P, Bunbupha S, Kukongviriyapan U, Prachaney P, Tangsucharit P, Kukongviriyapan V, Pakdeechote P (2016a) Asiatic acid attenuates renin-angiotensin system activation and improves vascular function in high-carbohydrate, high-fat diet fed rats. BMC Complement Altern Med 16:123. doi:10.1186/s12906-016-1100-6
Maneesai P et al (2016b) Synergistic antihypertensive effect of Carthamus tinctorius L. extract and captopril in L-NAME-induced hypertensive rats via restoration of eNOS and AT(1)R expression. Nutrients 8:122. doi:10.3390/nu8030122
Miguel-Carrasco JL, Zambrano S, Blanca AJ, Mate A, Vazquez CM (2010) Captopril reduces cardiac inflammatory markers in spontaneously hypertensive rats by inactivation of NF-kB. J Inflamm (Lond) 7:21. doi:10.1186/1476-9255-7-211476-9255-7-21
Montenegro MF et al (2010) Quercetin restores plasma nitrite and nitroso species levels in renovascular hypertension. Naunyn Schmiedeberg's Arch Pharmacol 382:293–301. doi:10.1007/s00210-010-0546-1
Moubarak M, Jabbour H, Smayra V, Chouery E, Saliba Y, Jebara V, Fares N (2012) Cardiorenal syndrome in hypertensive rats: microalbuminuria, inflammation and ventricular hypertrophy. Physiol Res 61:13–24
Ortego M, Bustos C, Hernandez-Presa MA, Tunon J, Diaz C, Hernandez G, Egido J (1999) Atorvastatin reduces NF-kappaB activation and chemokine expression in vascular smooth muscle cells and mononuclear cells. Atherosclerosis 147:253–261
Padia SH, Kemp BA, Howell NL, Fournie-Zaluski MC, Roques BP, Carey RM (2008) Conversion of renal angiotensin II to angiotensin III is critical for AT2 receptor-mediated natriuresis in rats. Hypertension 51:460–465. doi:10.1161/HYPERTENSIONAHA.107.103242
Pakdeechote P, Bunbupha S, Kukongviriyapan U, Prachaney P, Khrisanapant W, Kukongviriyapan V (2014) Asiatic acid alleviates hemodynamic and metabolic alterations via restoring eNOS/iNOS expression, oxidative stress, and inflammation in diet-induced metabolic syndrome rats. Nutrients 6:355–370. doi:10.3390/nu6010355
Park BM, Gao S, Cha SA, Kim SH (2015) Attenuation of renovascular hypertension by cyclooxygenase-2 inhibitor partly through ANP release. Peptides 69:1–8. doi:10.1016/j.peptides.2015.03.022
Ramachandran V, Saravanan R, Senthilraja P (2014) Antidiabetic and antihyperlipidemic activity of asiatic acid in diabetic rats, role of HMG CoA: in vivo and in silico approaches. Phytomedicine 21:–225, 232. doi:10.1016/j.phymed.2013.08.027
Rincon J et al (2015) Role of angiotensin II type 1 receptor on renal NAD(P)H oxidase, oxidative stress and inflammation in nitric oxide inhibition induced-hypertension. Life Sci 124:81–90. doi:10.1016/j.lfs.2015.01.005
Rioja I, Terencio MC, Ubeda A, Riguera R, Quintela JM, Alcaraz MJ (2002) A new ditriazine inhibitor of NF-kappaB modulates chronic inflammation and angiogenesis. Naunyn Schmiedeberg's Arch Pharmacol 365:357–364. doi:10.1007/s00210-002-0544-z
Santuzzi CH, Tiradentes RV, Mengal V, Claudio ER, Mauad H, Gouvea SA, Abreu GR (2015) Combined aliskiren and L-arginine treatment has antihypertensive effects and prevents vascular endothelial dysfunction in a model of renovascular hypertension Braz. J Med Biol Res 48:65–76
Squadrito GL, Pryor WA (1995) The formation of peroxynitrite in vivo from nitric oxide and superoxide. Chem Biol Interact 96:203–206
Umka Welbat J et al (2016) Asiatic acid prevents the deleterious effects of valproic acid on cognition and hippocampal cell proliferation and survival. Nutrients 18:8(5). doi:10.3390/nu8050303
Yin MC, Lin MC, Mong MC, Lin CY (2012) Bioavailability, distribution, and antioxidative effects of selected triterpenes in mice. J Agric Food Chem 60:7697–7701. doi:10.1021/jf302529x
Acknowledgments
This study was funded by grants from the Invitation Research Fund (IN58342, IN59145), Faculty of Medicine, Khon Kaen University and from Khon Kaen University (590050), Thailand and the Thailand Research Fund (RSA6080005). Putcharawipa Maneesai is financially supported by the Cardiovascular Research Group, Khon Kaen University, Thailand. We would like to acknowledge Dr. Justin Thomas Reese for editing this manuscript via the Publication Clinic, KKU, Thailand.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Rights and permissions
About this article

Cite this article
Maneesai, P., Bunbupha, S., Kukongviriyapan, U. et al. Effect of asiatic acid on the Ang II-AT1R-NADPH oxidase-NF-κB pathway in renovascular hypertensive rats. Naunyn-Schmiedeberg's Arch Pharmacol 390, 1073–1083 (2017). https://doi.org/10.1007/s00210-017-1408-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-017-1408-x