
Abstract
Insulin resistance is a serious health condition worldwide; however, its exact mechanisms are still unclear. This study investigates agmatine (AGM; an endogenous metabolite of l-arginine) effects on insulin resistance induced by high fructose diet (HFD) in rats and the possible involved mechanisms. Sprague Dawley rats were fed 60 % HFD for 12 weeks, and AGM (10 mg/kg/day, orally) was given from week 9 to 12. AGM significantly reduced HFD-induced elevation in fasting insulin level, homeostasis model assessment of insulin resistance (HOMA-IR) index and liver glycogen content from 3.44-, 3.62- and 2.07- to 2.59-, 2.78- and 1.3-fold, respectively, compared to the control group, while it increased HFD-induced reduction in glucose tolerance. Additionally, AGM significantly decreased HFD-induced elevation in serum triglycerides, low density lipoprotein cholesterol and very low density lipoprotein cholesterol levels from 3.18-, 2.97- and 4.75- to 1.25-, 1.25- and 1.07-fold, respectively, compared to control group. Conversely, AGM had no significant effect on HFD-induced changes in fasting glucose, glycosylated hemoglobin, insulin tolerance and high density lipoprotein cholesterol. Furthermore, AGM significantly reduced HFD-induced elevation in mRNA expression of glucose transporter type-2 (GLUT-2), mammalian target of rapamycin (mTOR) and sterol regulatory element-binding protein-1c (SREBP-1c) without affecting that of peroxisome proliferator-activated receptor-alpha (PPAR-α) in the liver. Additionally, AGM enhanced ACh-induced aortic relaxation and attenuated liver steatosis induced by HFD. In conclusion, AGM may have a therapeutic potential in insulin resistance through suppressing SREBP-1c, mTOR and GLUT-2 in liver.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Insulin resistance is a serious health condition worldwide characterized by reduced insulin sensitivity in tissues including liver, skeletal muscle and adipose tissue (Virkamaki et al. 1999). It is mainly associated with obesity, type-2 diabetes mellitus, dyslipidemia and hypertension (Thomas et al. 2015). The exact mechanisms and causes of insulin resistance are still unclear; therefore, there is no definitive treatment.
Several experimental models are documented to induce insulin resistance, including high fructose diet (HFD), which is a well-established model that mimics insulin resistance observed in humans (Bezerra et al. 2000). Fructose, an isomer of glucose, is a lipogenic monosaccharide that is present in many fruits, vegetables and honey and used as a sweetener in syrups (Basciano et al. 2005; Tran et al. 2009).
HFD results in hyperinsulinemia, impaired glucose tolerance, hypertriglyceridemia and hypertension (Elliott et al. 2002). Fructose can bypass the rate-limiting reaction of phosphofructokinase (Mayes 1993), serving as unregulated source of glycerol-3-phosphate and acetyl-CoA, both of which are used for hepatic lipogenesis (Havel 2005). Additionally, HFD can increase mRNA expression of sterol regulatory element-binding protein-1c (SREBP-1c) in the liver, which is a major regulator of several key enzymes used for triglyceride (TG) and free fatty acid (FFA) production (Kazumi et al. 1997; Matsuzaka et al. 2004; Basciano et al. 2005). TG and FFA have been shown to inhibit insulin signaling (Wang et al. 2006). Furthermore, an important intermediate produced during TG synthesis is the mammalian target of rapamycin (mTOR) that act as an inhibitor for insulin signaling (Nagle et al. 2009).
Peroxisome proliferator-activated receptor-alpha (PPAR-α) is another important mediator in insulin resistance. It is responsible for fatty acid β-oxidation and ketogenesis in the liver (Lefebvre et al. 2006). Its expression has been shown to decrease in insulin resistance (Nagai et al. 2002; Shiomi et al. 2015).
In addition to SREBP-1c, mTOR and PPAR-α, GLUT-2 may be involved in insulin resistance (Narasimhan et al. 2015). GLUT-2 transports glucose in the liver across the membrane in a bi-directional manner for glycolysis and gluconeogenesis (Burcelin et al. 2000; Wood and Trayhurn 2003). Previous studies reported that the expression of GLUT-2 in liver increases in the case of insulin resistance and type-2 diabetes mellitus (Mathur et al. 2015; Liu et al. 2015).
Agmatine (AGM) is an endogenous ligand formed in mammalian tissues by decarboxylation of l-arginine by the mitochondrial enzyme arginine decarboxylase (Yang and Reis 1999). It has several pharmacological effects including potent anti-inflammatory and anti-fibrotic effects by inhibiting cytokine release (El-Agamy et al. 2014). It also possesses a neuro-protective effect due to its antagonizing action on N-methyl d-aspartate (NMDA) receptors (Yang and Reis 1999). Moreover, AGM has protective effects against dyslipidemia and associated endothelial dysfunction in rabbits (El-Awady and Suddek 2014). AGM affects hepatic metabolism including fatty acid oxidation, gluconeogenesis and glycolysis (Nissim et al. 2014). Furthermore, it increases glucose uptake (Su et al. 2009; Chang et al. 2010) and enhances glucose utilization by increasing glucose transporter-4 (GLUT-4) mRNA expression in skeletal muscle in streptozotocin-induced diabetic rat model (Hwang et al. 2005). It has a promising effect in regulating fructose-induced insulin resistance with a single injection (Ko et al. 2008). AGM possesses an insulin-like action; it increases insulin release from pancreatic islet β-cells and decreases advanced glycated end products that control many of diabetic complications (Piletz et al. 2013). AGM is considered to be safe when used for prolonged periods of time in humans (Keynan et al. 2010; Gilad and Gilad 2013, 2014).
Since AGM has several promising molecular and metabolic effects on lipid and glucose metabolism, therefore the aim of this study was to investigate the effects of AGM on HFD-induced insulin resistance in rats and the possible involved mechanisms.
Materials and methods
Drugs and chemicals
Fructose was purchased from Safety Misr (Cairo, Egypt), AGM from USPlabs (Dallas, TX, USA) and anthrone from Alpha Chemika (Mumbai, India). Acetylcholine hydrochloride (ACh), pentobarbital sodium, phenylephrine hydrochloride (PE) and sodium nitroprusside (SNP) were purchased from Sigma Aldrich chemical Co. (St. Louis, MO, USA). AGM was freshly prepared each day by dissolving in saline.
Experimental animals
Forty male Sprague Dawley rats with average age of 6–8 weeks were purchased from the “Egyptian Organization for Biological Products and Vaccines”, Giza, Egypt. The protocol of this study was approved by the Ethical Committee of the Faculty of Pharmacy, Mansoura University, Egypt, which is in accordance with the Principles of Laboratory Animal Care (NIH publication no. 85-23, revised 1985).
Experimental protocol
The rats were randomly divided into four groups, each of ten rats: group 1: control group: rats receiving regular chow for 12 weeks; group 2: HFD group: rats receiving 60 % HFD for 12 weeks (Liao et al. 2010); group 3: AGM group: rats receiving regular chow for 12 weeks and AGM (10 mg/kg /day, orally) from week 9 to 12 (Taksande et al. 2010) using a fine gastric gavage introduced directly into the stomach; group 4: HFD + AGM group: rats receiving 60 % HFD for 12 weeks and AGM (10 mg/kg /day, orally) from week 9 to 12.
The composition of regular chow was 60 % carbohydrates, 20 % protein, 4 % fats, 2 % fibers, 14 % minerals and vitamins. The HFD had the same composition, but fructose was added instead of carbohydrates. All rats had free access to drinking water and chow for 12 weeks. The food intake was determined daily, and body weight was recorded weekly.
Oral glucose tolerance test
At 48 h prior to the end day, animals were fasted for 12 h, and 40 % glucose solution (2 g/kg body weight) was given orally by gastric tube to conscious rats. Blood glucose level was measured at 0, 30, 60, 120 and 180 min after glucose administration. Blood glucose was measured via glucose dehydrogenase method using tail blood and a portable glucometer (Glucodoctor auto, All Medicus co., Gyeonggi-do, Republic of Korea).
Insulin tolerance test
At 24 h before the end day, animals were fasted for 12 h, and a dose of 0.5 IU/kg human soluble insulin (Actrapid, Novo Nordisk, Bagsvaerd, Denmark) was injected subcutaneously to conscious rats. Blood glucose level was determined at 0, 30, 60, 120 and 180 min after insulin administration using tail blood and a portable glucometer.
Sample preparation
At the end of the experiment, the rats were fasted for 12 h and euthanized using pentobarbital sodium (60 mg/kg, i.p.), and blood samples were collected to prepare serum and heparinized plasma. In addition, thoracic aortic rings were isolated for in vitro vascular reactivity measurement. Liver tissues were immediately collected and snap-frozen in liquid nitrogen, stored at −80 °C to be used later for real-time RT-PCR and glycogen content determination. Another part of liver was fixed in 10 % buffered formalin for subsequent histopathological examination.
Determination of fasting serum glucose, insulin and insulin resistance index
The fasting glucose level was measured in serum using an enzymatic colorimetric commercial kit purchased from Biodiagnostic Company (Giza, Egypt). The fasting rat insulin was measured in serum using a rat insulin ELISA kit (Mercodia Developing Diagnostics, Uppsala, Sweden) according to the manufacturer’s instructions.
Homeostasis model assessment of insulin resistance (HOMA-IR) index was calculated using HOMA2 Calculator, developed by the Diabetes Trial Unit, University of Oxford, Oxford, UK (Levy et al. 1998).
Estimation of glycosylated hemoglobin
Glycosylated hemoglobin was determined in whole blood using commercial kit containing cation-exchange resin, according to the manufacturer’s instructions (Stanbio Laboratories, TX, USA). The results were normalized to a human glycosylated hemoglobin standard.
Estimation of liver glycogen content
Liver glycogen content was assessed using the method described by Seifter et al. (1950). The method is based on the digestion of tissue in 30 % KOH and precipitating glycogen with 95 % ethanol and saturated sodium sulphate solution. The glycogen content was estimated by the use of anthrone dissolved in sulphuric acid. Sulphuric acid causes dehydration of carbohydrates that condenses in the presence of anthrone to produce a bluish green color measured at 620 nm and expressed as mg/100 g tissue.
Measurement of lipid profile in serum
Serum TG and total cholesterol (TC) were estimated in serum using commercial kits (Spinreact, Santa Coloma, Spain). High density lipoprotein cholesterol (HDL-C) was estimated in serum using a commercial kit (Vitro Scient., Hannover, Germany). Very low density lipoprotein cholesterol (VLDL-C) and low density lipoprotein cholesterol (LDL-C) were determined according to the following equations: VLDL-C = TG/5 while LDL-C = TC-[VLDL-C + HDL-C] (Yadav et al. 2007).
Determination of in vitro vascular reactivity
The descending thoracic aorta was isolated, dissected free of fat and connective tissue and cut into rings (2–4 mm) each.
Each aortic ring was mounted in a 10-ml organ bath at 37 °C filled with physiological salt solution of the following composition (mmol/L): NaCl 118, KCl 4.7, CaCl2 2.5 and MgSO4 · 7H2O 1.2, KH2PO4 1.2, glucose 11.1 and NaHCO3 25, pH 7.4, and aerated with a mixture of 95 % O2 and 5 % CO2.The aortic ring was allowed to equilibrate under 1 g of force for 60 min, with an exchange of bathing solution every 15 min. Vascular reactivity was measured using Riegestab K30 force transducer (Hugo Sachs electronic, D7806 march, Germany) and recorded with a Powerlab unit/400 with Chart v4.2 software (ADInstruments Pty Ltd., Australia). After equilibration, vascular responsiveness was evaluated by assessing the contractile response to 80 mmol/L of KCl.
Vascular reactivity to PE (10−10–10−5 M) was determined by constructing a cumulative concentration response curve. Additionally, the aortic ring was precontracted with 1 μM PE, and vascular relaxation to ACh (10−10–10−5 M) and SNP (10−10–10−5 M) was measured.
Measurement of mRNA expression of GLUT-2, mTOR, SREBP-1c and PPAR-α using real-time RT-PCR
RNA was isolated from liver samples using TRIZOL reagent (Life Technologies, Carlsbad, CA, USA). First-strand cDNA was synthesized from 2 μg of total RNA in 20 μl volume using Revert Aid First Strand cDNA Synthesis Kit (Thermo Scientific, Rockford, IL, USA) according to the manufacturer’s protocol. Real-time PCR was performed in Rotor Gene Q thermocycler (Qiagen, Hilden, Germany) using HOT Firepol Evagreen qPCR mix plus kit (Solis BioDyne, Tartu, Estonia).
The mRNA levels of GLUT-2, mTOR, SREBP-1c and PPAR-α were measured in duplicate PCR reactions and normalized relative to 18S ribosomal RNA (Rn18S) in the same sample. Sequences of primers (Life Technologies) were as follows:
GLUT-2: sense: TGACTGGAGCCCTCTTGATG, antisense: CCCCTGAGTGTGGTTGGAG, amplicon size =151; mTOR: sense: TATGACCCCAACCAGCCAAT, antisense: AGGAAAACAAACTCGTGCCC, amplicon size =125; SREBP-1c: sense: CTGTAGGTCACCGTTTCTTCG, antisense: CAGTTCAATGCTCGCTCCAG, amplicon size =151; PPAR-α: sense: AATCCACGAAGCCTACCTGA, antisense: CCTCTGCCTCCTTGTTTTCA, amplicon size =182 and Rn18S: sense: AGTTGGTGGAGCGATTTGTC, antisense: GAACGCCACTTGTCCCTCTA, amplicon size = 121.
The results were expressed as n-fold change of the relative expression level of target genes from control group using ∆∆C t method.
Histopathological examination
The 10 % buffered formalin fixed liver tissue was embedded in paraffin wax, sectioned (6 μm) and stained with hematoxylin–eosin (H&E), and samples were assessed for the presence of steatosis microscopically (Leica Imaging Systems, Cambridge, UK). The hepatic steatosis was evaluated by semiquantitative scale from 0 to 3: 0 = no steatosis, 1 = mild steatosis less than 33 %, 2 = moderate steatosis more than 33 % and less than 66 % and 3 = severe steatosis more than 66 %. The pathologist performing the histopathological evaluation was blinded to the study treatment assignment.
Statistical analysis
Data are expressed as mean ± standard deviation (SD). Statistical analysis and graphing were carried out using GraphPad software Prism V 5.02 (GraphPad Software Inc., San Diego, CA, USA). Vascular relaxation was calculated as percentage of the maximal contraction induced by 1 μm PE. The highest response obtained was considered as the maximum response (E max), and pEC50 (negative log of the concentration producing 50 % of maximal response) was determined from non-linear regression analysis (four-parameter curve fit). In OGTT and ITT, the change in glucose was calculated as the difference between each point and the initial baseline for each rat. One-way analysis of variance (ANOVA) followed by Tukey–Kramer’s multiple comparisons post hoc test was used to measure significant differences between groups. Statistical significance was considered at p < 0.05. Hepatic steatosis score was compared using Kruskal–Wallis test followed by Dunn's multiple comparison test.
Results
Effect of AGM on body weight gain and mean daily food intake in rats
Both body weight gain and mean food intake were not significantly different among groups after 12 weeks (Table 1).
Effect of AGM on OGTT and ITT
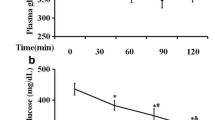
In HFD rats, the AUC of the change in blood glucose level relative to the baseline in OGTT was significantly (p < 0.05, n = 10) increased by 1.82-fold compared to the control group (Fig. 1a). AGM treatment significantly prevented the elevation in AUC induced by HFD compared to the control group. Although AGM treatment in rats receiving regular chow slightly increased AUC by 1.55-fold compared to the control group, this effect was not statistically significant.

Effect of AGM on OGTT and ITT. Rats were fed 60 % HFD for 12 weeks, and AGM (10 mg/kg/day, orally) was given from week 9 to 12; then OGTT (a) and ITT (b) were measured. Data are expressed as mean ± SD (n = 10). * and # p < 0.05, significantly different from control and HFD group, respectively, using one-way ANOVA followed by Tukey–Kramer multiple comparisons post hoc test. AGM agmatine, ANOVA analysis of variance, AUC area under the curve, HFD high fructose diet, ITT insulin tolerance test, OGTT oral glucose tolerance test, SD standard deviation
In ITT, the AUC of the change in blood glucose level relative to the baseline was significantly (p < 0.05, n = 10) decreased in HFD rats by 56.53 % compared to the control group. AGM treatment in HFD rats did not significantly affect the reduction in AUC induced by HFD compared to the control group (Fig. 1b). AGM treatment had no significant effect on ITT in normal rats.
Effect of AGM on liver glycogen content
Liver glycogen content was significantly (p < 0.05, n = 10) elevated by HFD by 2.07-fold compared to the control group (Table 1).
Treatment with AGM in HFD rats from week 9 to 12 managed to decrease liver glycogen content significantly (p < 0.05, n = 10), which was reduced to 1.3-fold compared to control group. AGM treatment had no significant effect on liver glycogen content in control animals.
Effect of AGM on fasting serum glucose, insulin, HOMA-IR index and glycosylated hemoglobin
The HFD caused a significant state of insulin resistance indicated by a significant (p < 0.05, n = 10) increase in fasting insulin level and HOMA-IR index by 3.44- and 3.62-fold respectively compared to the control group (Table 1). Moreover, HFD caused a significant state of hyperglycemia as elucidated by an increase in fasting glucose level and glycosylated hemoglobin by 1.09- and 1.37-fold respectively compared to the control group.
Treatment with AGM decreased the level of insulin resistance induced by HFD by significantly (p < 0.05, n = 10) decreasing both fasting insulin level and HOMA-IR index to 2.59- and 2.78-fold, respectively, compared to the control group. AGM did not significantly affect fasting glucose and glycosylated hemoglobin levels in HFD rats (Table 1). AGM treatment had no significant effect on fasting glucose, insulin, HOMA-IR index or glycosylated hemoglobin in normal rats.
Effect of AGM on lipid profile in serum
The HFD caused a significant (p < 0.05, n = 10) increase in serum TG, VLDL-C and LDL-C levels by 3.18-, 2.97- and 4.75-fold, respectively (Fig. 2), and it caused a significant decrease in serum HDL-C level by 19 % compared to the control group, while serum TC level was not significantly affected by HFD.

Effect of AGM on lipid profile in serum. Rats were fed 60 % HFD for 12 weeks, and AGM (10 mg/kg/day, orally) was given from week 9 to 12; then TC (a), TG (b), HDL (c), VLDL (d) and LDL (e) were measured. Data are expressed as mean ± SD (n = 10). * and # p < 0.05, significantly different from control and HFD group, respectively, using one-way ANOVA followed by Tukey–Kramer multiple comparisons post hoc test. AGM agmatine, ANOVA analysis of variance, HFD high fructose diet, TC total cholesterol, TG triglycerides, HDL-C high density lipoprotein cholesterol, VLDL-C very low density lipoprotein cholesterol, LDL-C low density lipoprotein cholesterol, SD standard deviation
Treatment with AGM significantly (p < 0.05, n = 10) decreased the HFD-induced elevated levels of serum TG, VLDL-C and LDL-C levels to 1.25-, 1.25- and 1.07-fold, respectively, compared to the control group. AGM treatment did not attenuate the HFD-induced decrease in serum HDL-C. AGM treatment had no significant effect on lipid profile in normal rats.
Effect of AGM on vascular reactivity in vitro
The HFD had no significant effect on contraction induced by either KCl or PE or the relaxation induced by SNP in aortic rings (Fig. 3) when compared to the control group. Conversely, HFD caused a significant (p < 0.05, n = 6) decrease in endothelium-dependent ACh-induced maximal relaxation, where E max decreased significantly by 57 % compared to the control group. However, HFD had no significant effect on pEC50.

Effect of AGM on vascular reactivity in vitro. Rats were fed 60 % HFD for 12 weeks, and AGM (10 mg/kg/day, orally) was given from week 9 to 12; then aortic rings were isolated and tested for vascular reactivity by evaluating the contractile response to 80 mmol/L of KCl (a), vascular reactivity to cumulative concentration response curve of PE (b) and vascular relaxation to ACh (c) and SNP (d) after precontracting the aortic ring with PE; e a table showing E max and pEC50 values. Data are expressed as mean ± SD (n = 6). * and # p < 0.05, significantly different from control and HFD group, respectively, using one-way ANOVA followed by Tukey–Kramer multiple comparisons post hoc test. ACh acetylcholine, AGM agmatine, ANOVA analysis of variance, HFD high fructose diet, KCl potassium chloride, PE phenylephrine, SD standard deviation, SNP sodium nitroprusside
AGM in HFD rats significantly (p < 0.05, n = 6) enhanced vascular reactivity by increasing ACh-induced maximal relaxation (E max), which returned to 69 % compared to the control group. AGM treatment had no significant effect on vascular reactivity in normal rats.
Effect of AGM on HFD-induced changes in mRNA expression of GLUT-2, mTOR, SREBP-1c and PPAR-α
The HFD caused a significant (p < 0.05, n = 4) elevation in mRNA expression of GLUT-2, mTOR and SREBP-1c by 2.42-, 3.59- and 6.78-fold, respectively, and a significant decrease in PPAR-α mRNA expression by 68 % when compared to the control group (Fig. 4).

Effect of AGM on HFD-induced changes in mRNA expression of GLUT-2, mTOR, SREBP-1c and PPAR-α. Rats were fed 60 % HFD for 12 weeks, and AGM (10 mg/kg/day, orally) was given from week 9 to 12; then mRNA expression of GLUT-2 (a), mTOR (b), SREBP-1c (c) and PPAR-α (d) were measured using real-time RT-PCR. Data are expressed as mean ± SD (n = 4). * and # p < 0.05, significantly different from control and HFD group, respectively, using one-way ANOVA followed by Tukey–Kramer multiple comparisons post hoc test. AGM agmatine, ANOVA analysis of variance, HFD high fructose diet, GLUT-2 glucose transporter type-2, mTOR the mammalian target of rapamycin, SREBP-1c sterol regulatory element-binding protein-1, PPAR-α peroxisome proliferator-activated receptor-alpha, SD standard deviation
Treatment with AGM significantly (p < 0.05, n = 4) prevented HFD-induced elevation in mRNA expression of GLUT-2, mTOR and SREBP-1c, which returned to 1.17-, 1.09- and 1.19-fold, respectively, compared to the control group. AGM treatment in HFD rats attenuated the PPAR-α mRNA expression by 47 % compared to the control group; however, it was not statistically significant. AGM treatment had no significant effect on mRNA expression of GLUT-2, mTOR, SREBP-1c and PPAR-α in normal rats.
Histopathological examination
Liver specimen (n = 5) from the control group (score = 0.33) showed normal hepatic architecture with normal hepatocytes arranged in 1–2 thickness plates with no evidence of steatosis, while HFD rats (score = 2.83) showed severe steatosis affecting >66 % of hepatocytes mainly periportal (microvesicular and macrovesicular steatosis) (Fig. 5). Treatment with AGM in HFD rats (score = 1) caused a dramatic regression of steatosis with only remaining microfoci of macrovesicular steatosis (mild steatosis). AGM treatment in normal rats (score = 0.33) showed normal hepatocytes radiating from central veins with intervening normal sinuses in between with no evidence of steatosis.

Effect of AGM on HFD-induced changes in histopathology of rat liver. Rats were fed 60 % HFD for 12 weeks and AGM (10 mg/kg/day, orally) was given from week 9 to 12; then liver specimens (n = 5) were collected and stained with H&E (×200). a Control showing normal hepatocytes, b HFD showing severe steatosis, c AGM showing normal hepatocytes, d HFD+AGM showing mild steatosis and e scoring values of steatosis. Data are expressed as mean (n = 5). *p < 0.05, significantly different from control group using Kruskal–Wallis followed by Dunn's multiple comparison test. AGM agmatine, HFD high fructose diet
Discussion
In this study, HFD for 12 weeks induced impaired glucose and insulin tolerance in addition to elevated levels of fasting insulin, glucose, glycosylated hemoglobin and HOMA-IR index, confirming the state of insulin resistance. Furthermore, liver glycogen content was increased, and dyslipidemia was induced by HFD. It managed to induce endothelial dysfunction. Additionally, the mRNA expression of GLUT-2, mTOR and SREBP-1c was increased, while that of PPAR-α was decreased as a result of HFD. AGM treatment significantly alleviated HFD-induced insulin resistance. In addition, AGM treatment decreased liver glycogen content and improved lipid profile and endothelial-dependent relaxation. Additionally, the elevated mRNA expression of GLUT-2, mTOR and SREBP-1c was decreased as a result of the AGM treatment without affecting PPAR-α expression.
Several studies showed that feeding rats with HFD induced insulin resistance. The duration of HFD feeding varies mostly from 4 up to 12 weeks (Shawky et al. 2014; Liao et al. 2010; Ko et al. 2008). In this study, by the end of 8 weeks of HFD feeding, AGM treatment was started after confirming insulin resistance by the elevated levels of fasting glucose and insulin and the alteration of lipid profile.
Feeding rats with HFD has been shown to induce insulin resistance, which is elucidated by hyperinsulinemia, hyperglycemia, increase in HOMA-IR index, increase in glycosylated hemoglobin and increase in liver glycogen content with a decrease in both glucose and insulin tolerance. In this study, AGM increased glucose tolerance and decreased fasting insulin levels, which is consistent with a previous study (Ko et al. 2008). This effect could be attributed to an indirect action through the secretion of β-endorphin, in addition to the direct activation of imidazoline-I2 binding sites in peripheral tissues (Su et al. 2009) increasing insulin sensitivity.
Previous studies have shown that a single intravenous dose of AGM activated I2-imidazoline receptors and increased β-endorphin secretion, increasing insulin action in a 4-week model of HFD (Ko et al. 2008; Su et al. 2009). However, the link between the continuous use of AGM daily for 4 weeks in HFD-induced insulin resistance model and dyslipidemia has not been thoroughly investigated.
Since GLUT-2 is involved in insulin resistance (Narasimhan et al. 2015), its mRNA expression was therefore investigated. The expression of GLUT-2 in liver was reported to increase in insulin resistance and type-2 diabetes mellitus (Mathur et al. 2015; Liu et al. 2015). In this study, HFD increased GLUT-2 mRNA expression in liver, and AGM ameliorated this elevation in GLUT-2 expression, which could contribute to AGM effect on insulin resistance.
In this study, HFD-induced insulin resistance caused an increase in both liver glycogen content and glycosylated hemoglobin as reported in previous studies (Yadav et al. 2007; Madani et al. 2012). Although in this study ion exchange resin method was used to estimate glycosylated hemoglobin, affinity boronate chromatography method is more suitable for rats (Nagisa et al. 2003). Fructose metabolism in the liver mainly forms glycogen and TG by stimulating their synthesis (Koo et al. 2008). Additionally, fructose 1-phosphate, an intermediate of fructose metabolism in liver, inhibits glycogen phosphorylase activity (Mayes 1993), decreasing glycogen breakdown. Elevated level of glycosylated hemoglobin correlates with increased fasting blood glucose level in insulin resistance model. AGM treatment decreased liver glycogen content, but it did not attenuate the slightly elevated glycosylated hemoglobin, suggesting that modifying glycogen content is another pathway through which AGM ameliorates insulin resistance.
In this study, HFD managed to cause dyslipidemia, which was shown mainly by the elevation of TG. These findings were consistent with previous studies (Yadav et al. 2007, 2009; Shawky et al. 2014).
Hepatic steatosis due to TG accumulation is considered a major contributor to insulin resistance (Flannery et al. 2012), which was confirmed in our rat liver specimens by histopathological examination. The intermediates produced during TG synthesis such as mTOR, protein kinase-C (PKC) and ceramide can act as inhibitors for insulin signaling (Nagle et al. 2009). Both PKC and mTOR affect insulin signaling by phosphorylation of insulin receptor substrate-1, leading to inhibition of phosphoinositide-3 kinase (PI3K) activation (Moeschel et al. 2004; Gual et al. 2005). In this study, HFD-induced insulin resistance increased mRNA gene expression of mTOR which was attenuated by AGM treatment.
The decrease in the mRNA expression of PPAR-α in insulin resistance was reported in previous studies (Nagai et al. 2002; Shiomi et al. 2015), suggesting that fructose or its metabolites can directly reduce lipid β-oxidation(Basciano et al. 2005), leading to cellular lipid accumulation and a remarked hepatic steatosis as observed in this study. AGM treatment decreased hepatic steatosis; however, it did not affect PPAR-α mRNA expression, suggesting that lipid β-oxidation is not involved in AGM beneficial effects on insulin resistance in our model.
The mRNA expression of SREBP-1c has been shown to increase in insulin resistance (Kazumi et al. 1997; Matsuzaka et al. 2004; Basciano et al. 2005). The increase in VLDL-C upregulates SREBP-1c mRNA gene expression and subsequently downregulates PPAR-α mRNA gene expression (Lucero et al. 2015). In this study, HFD increased mRNA expression of SREBP-1c, and AGM treatment inhibited this increase.
AGM showed a beneficial effect on lipid profile in this study, which is consistent with a previous study (El-Awady and Suddek 2014). The decrease in the mRNA gene expression of mTOR and SREBP-1c could be the reason for these beneficial effects on lipid profile and on insulin sensitivity. Unlike what was reported in Nissim et al. (2014) that AGM consumption can increase the mRNA gene expression of PPAR-α in high-fat-diet-fed rats, PPAR-α was not increased in our study. This discrepancy could be attributed to the difference in AGM treatment duration and dose.
In insulin resistance, the increase in GLUT-2 expression and the decrease in PPAR-α expression are regulated by the overexpression of SREBP-1c (Im et al. 2005; Narasimhan et al. 2015), so our results suggest that AGM managed to increase insulin sensitivity, improve lipid profile and interrupt de novo lipogenesis by decreasing SREBP-1c and mTOR expression, followed by decreasing the expression of GLUT-2. A scheme representing the possible protective mechanisms of AGM is outlined in Fig. 6.

Suggested scheme of the possible protective mechanism of AGM in HFD-induced insulin resistance in rat liver. AGM agmatine, AKt protein kinase B, GLUT-2 glucose transporter type-2, HFD high fructose diet, IRS insulin receptor substrate, mTOR the mammalian target of rapamycin, PI3K phosphoinositide-3 kinase, PKC protein kinase-C, PPAR-α peroxisome proliferator-activated receptor-alpha, SREBP-1c sterol regulatory element-binding protein-1, TG triglycerides
Endothelial dysfunction is widely reported in HFD-induced insulin resistance model (Shawky et al. 2014; Vasudevan et al. 2006). PI3K and protein kinase B (Akt) are compromised in HFD-induced insulin resistance model (Teruel et al. 2001), leading to a decrease in endothelial nitric oxide synthase (eNOS) phosphorylation and a decrease in nitric oxide (NO) release. The imbalance between the decreased NO release and secretion of the powerful vasoconstrictor endothelin-1 can contribute to endothelial dysfunction in insulin resistance (Muniyappa and Sowers 2013).
In this study, relaxation response to Ach but not to SNP was affected by HFD. This suggests that NO production, through eNOS, but not NO signaling, is affected by HFD in this model and that vasodilatory function is normally maintained (Akar et al. 2012). AGM treatment improved endothelial-dependent relaxation impaired by HFD. AGM has been shown to stimulate eNOS enzyme to release NO (Ozyazgan et al. 2003; El-Awady and Suddek 2014); in addition to possessing antioxidant and anti-inflammatory effects (El-Agamy et al. 2014), these actions could explain its endothelial protective effects.
In conclusion, AGM may attenuate insulin resistance through inhibiting SREBP-1c, mTOR and GLUT-2 in liver without affecting PPAR-α. Taking into consideration its safety as an endogenous compound, AGM may present a promising therapeutic compound in insulin resistance.
Abbreviations
- AGM:
-
Agmatine
- GLUT-2:
-
Glucose transporter type-2
- HFD:
-
High fructose diet
- HOMA-IR:
-
Homeostasis model assessment of insulin resistance
- mTOR:
-
The mammalian target of rapamycin
- PPAR-α:
-
Peroxisome proliferator-activated receptor-alpha
- SREBP-1c:
-
Sterol regulatory element-binding protein-1c
References
Akar F, Uludag O, Aydin A, Aytekin YA, Elbeg S, Tuzcu M, Sahin K (2012) High-fructose corn syrup causes vascular dysfunction associated with metabolic disturbance in rats: protective effect of resveratrol. Food Chem Toxicol 50:2135–2141
Basciano H, Federico L, Adeli K (2005) Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond) 2:5
Bezerra RM, Ueno M, Silva MS, Tavares DQ, Carvalho CR, Saad MJ (2000) A high fructose diet affects the early steps of insulin action in muscle and liver of rats. J Nutr 130:1531–1535
Burcelin R, Dolci W, Thorens B (2000) Glucose sensing by the hepatoportal sensor is GLUT2-dependent: in vivo analysis in GLUT2-null mice. Diabetes 49:1643–1648
Chang CH, Wu HT, Cheng KC, Lin HJ, Cheng JT (2010) Increase of beta-endorphin secretion by agmatine is induced by activation of imidazoline I(2A) receptors in adrenal gland of rats. Neurosci Lett 468:297–299
El-Agamy DS, Sharawy MH, Ammar EM (2014) Agmatine attenuates silica-induced pulmonary fibrosis. Hum Exp Toxicol 33:650–660
El-Awady MS, Suddek GM (2014) Agmatine ameliorates atherosclerosis progression and endothelial dysfunction in high cholesterol-fed rabbits. J Pharm Pharmacol 66:835–843
Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ (2002) Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr 76:911–922
Flannery C, Dufour S, Rabol R, Shulman GI, Petersen KF (2012) Skeletal muscle insulin resistance promotes increased hepatic de novo lipogenesis, hyperlipidemia, and hepatic steatosis in the elderly. Diabetes 61:2711–2717
Gilad GM, Gilad VH (2013) Evidence for oral agmatine sulfate safety—a 95-day high dosage pilot study with rats. Food Chem Toxicol 62:758–762
Gilad GM, Gilad VH (2014) Long-term (5 years), high daily dosage of dietary agmatine—evidence of safety: a case report. J Med Food 17:1256–1259
Gual P, Le Marchand-Brustel Y, Tanti JF (2005) Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 87:99–109
Havel PJ (2005) Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr Rev 63:133–157
Hwang SL, Liu IM, Tzeng TF, Cheng JT (2005) Activation of imidazoline receptors in adrenal gland to lower plasma glucose in streptozotocin-induced diabetic rats. Diabetologia 48:767–775
Im SS, Kang SY, Kim SY, Kim HI, Kim JW, Kim KS, Ahn YH (2005) Glucose-stimulated upregulation of GLUT2 gene is mediated by sterol response element-binding protein-1c in the hepatocytes. Diabetes 54:1684–1691
Kazumi T, Odaka H, Hozumi T, Ishida Y, Amano N, Yoshino G (1997) Effects of dietary fructose or glucose on triglyceride production and lipogenic enzyme activities in the liver of Wistar fatty rats, an animal model of NIDDM. Endocr J 44:239–245
Keynan O, Mirovsky Y, Dekel S, Gilad VH, Gilad GM (2010) Safety and efficacy of dietary agmatine sulfate in lumbar disc-associated radiculopathy. An open-label, dose-escalating study followed by a randomized, double-blind, placebo-controlled trial. Pain Med 11:356–368
Ko WC, Liu IM, Chung HH, Cheng JT (2008) Activation of I(2)-imidazoline receptors may ameliorate insulin resistance in fructose-rich chow-fed rats. Neurosci Lett 448:90–93
Koo HY, Wallig MA, Chung BH, Nara TY, Cho BH, Nakamura MT (2008) Dietary fructose induces a wide range of genes with distinct shift in carbohydrate and lipid metabolism in fed and fasted rat liver. Biochim Biophys Acta 1782:341–348
Lefebvre P, Chinetti G, Fruchart JC, Staels B (2006) Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest 116:571–580
Levy JC, Matthews DR, Hermans MP (1998) Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care 21:2191–2192
Liao Q, Shi DH, Zheng W, Xu XJ, Yu YH (2010) Antiproliferation of cardamonin is involved in mTOR on aortic smooth muscle cells in high fructose-induced insulin resistance rats. Eur J Pharmacol 641:179–186
Liu Y, Song A, Zang S, Wang C, Song G, Li X, Zhu Y, Yu X, Li L, Wang Y, Duan L (2015) Jinlida reduces insulin resistance and ameliorates liver oxidative stress in high-fat fed rats. J Ethnopharmacol 162:244–252
Lucero D, Miksztowicz V, Macri V, Lopez GH, Friedman S, Berg G, Zago V, Schreier L (2015) Overproduction of altered VLDL in an insulin-resistance rat model: influence of SREBP-1c and PPAR-alpha. Clin Investig Arterioscler.
Madani Z, Louchami K, Sener A, Malaisse WJ, Ait YD (2012) Dietary sardine protein lowers insulin resistance, leptin and TNF-alpha and beneficially affects adipose tissue oxidative stress in rats with fructose-induced metabolic syndrome. Int J Mol Med 29:311–318
Mathur R, Dutta S, Velpandian T, Mathur SR (2015) Psidium guajava Linn. leaf extract affects hepatic glucose transporter-2 to attenuate early onset of insulin resistance consequent to high fructose intake: an experimental study. Pharmacognosy Res 7:166–175
Matsuzaka T, Shimano H, Yahagi N, Amemiya-Kudo M, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Tomita S, Sekiya M, Hasty A, Nakagawa Y, Sone H, Toyoshima H, Ishibashi S, Osuga J, Yamada N (2004) Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes 53:560–569
Mayes PA (1993) Intermediary metabolism of fructose. Am J Clin Nutr 58:754S–765S
Moeschel K, Beck A, Weigert C, Lammers R, Kalbacher H, Voelter W, Schleicher ED, Haring HU, Lehmann R (2004) Protein kinase C-zeta-induced phosphorylation of Ser318 in insulin receptor substrate-1 (IRS-1) attenuates the interaction with the insulin receptor and the tyrosine phosphorylation of IRS-1. J Biol Chem 279:25157–25163
Muniyappa R, Sowers JR (2013) Role of insulin resistance in endothelial dysfunction. Rev Endocr Metab Disord 14:5–12
Nagai Y, Nishio Y, Nakamura T, Maegawa H, Kikkawa R, Kashiwagi A (2002) Amelioration of high fructose-induced metabolic derangements by activation of PPARalpha. Am J Physiol Endocrinol Metab 282:E1180–E1190
Nagisa Y, Kato K, Watanabe K, Murakoshi H, Odaka H, Yoshikawa K, Sugiyama Y (2003) Changes in glycated haemoglobin levels in diabetic rats measured with an automatic affinity HPLC. Clin Exp Pharmacol Physiol 30:752–758
Nagle CA, Klett EL, Coleman RA (2009) Hepatic triacylglycerol accumulation and insulin resistance. J Lipid Res 50(Suppl):S74–S79
Narasimhan A, Chinnaiyan M, Karundevi B (2015) Ferulic acid regulates hepatic GLUT2 gene expression in high fat and fructose-induced type-2 diabetic adult male rat. Eur J Pharmacol 761:391–397
Nissim I, Horyn O, Daikhin Y, Chen P, Li C, Wehrli SL, Nissim I, Yudkoff M (2014) The molecular and metabolic influence of long term agmatine consumption. J Biol Chem 289:9710–9729
Ozyazgan S, Bicakci B, Ozaydin A, Denizbasi A, Unluer EE, Akkan AG (2003) The effect of agmatine on the vascular reactivity in streptozotocin-diabetic rats. Pharmacol Res 48:133–138
Piletz JE, Aricioglu F, Cheng JT, Fairbanks CA, Gilad VH, Haenisch B, Halaris A, Hong S, Lee JE, Li J, Liu P, Molderings GJ, Rodrigues AL, Satriano J, Seong GJ, Wilcox G, Wu N, Gilad GM (2013) Agmatine: clinical applications after 100 years in translation. Drug Discov Today 18:880–893
Seifter S, Dayton S, Novic B, Muntwyler E (1950) The estimation of glycogen with the anthrone reagent. Arch Biochem 25:191–200
Shawky NM, Shehatou GS, Abdel RM, Suddek GM, Gameil NM (2014) Levocetirizine ameliorates high fructose diet-induced insulin resistance, vascular dysfunction and hepatic steatosis in rats. Eur J Pharmacol 740:353–363
Shiomi Y, Yamauchi T, Iwabu M, Okada-Iwabu M, Nakayama R, Orikawa Y, Yoshioka Y, Tanaka K, Ueki K, Kadowaki T (2015) A novel peroxisome proliferator-activated receptor (PPAR)alpha agonist and PPARgamma antagonist, Z-551, ameliorates high-fat diet-induced obesity and metabolic disorders in mice. J Biol Chem 290:14567–14581
Su CH, Liu IM, Chung HH, Cheng JT (2009) Activation of I2-imidazoline receptors by agmatine improved insulin sensitivity through two mechanisms in type-2 diabetic rats. Neurosci Lett 457:125–128
Taksande BG, Kotagale NR, Patel MR, Shelkar GP, Ugale RR, Chopde CT (2010) Agmatine, an endogenous imidazoline receptor ligand modulates ethanol anxiolysis and withdrawal anxiety in rats. Eur J Pharmacol 637:89–101
Teruel T, Hernandez R, Lorenzo M (2001) Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 50:2563–2571
Thomas S, Senthilkumar GP, Sivaraman K, Bobby Z, Paneerselvam S, Harichandrakumar KT (2015) Effect of s-methyl-L-cysteine on oxidative stress, inflammation and insulin resistance in male Wistar rats fed with high fructose diet. Iran J Med Sci 40:45–50
Tran LT, Yuen VG, McNeill JH (2009) The fructose-fed rat: a review on the mechanisms of fructose-induced insulin resistance and hypertension. Mol Cell Biochem 332:145–159
Vasudevan H, Nagareddy PR, McNeill JH (2006) Gonadectomy prevents endothelial dysfunction in fructose-fed male rats, a factor contributing to the development of hypertension. Am J Physiol Heart Circ Physiol 291:H3058–H3064
Virkamaki A, Ueki K, Kahn CR (1999) Protein–protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest 103:931–943
Wang XL, Zhang L, Youker K, Zhang MX, Wang J, LeMaire SA, Coselli JS, Shen YH (2006) Free fatty acids inhibit insulin signaling-stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes 55:2301–2310
Wood IS, Trayhurn P (2003) Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr 89:3–9
Yadav H, Jain S, Sinha PR (2007) Antidiabetic effect of probiotic dahi containing Lactobacillus acidophilus and Lactobacillus casei in high fructose fed rats. Nutrition 23:62–68
Yadav H, Jain S, Yadav M, Sinha PR, Prasad GB, Marotta F (2009) Epigenomic derangement of hepatic glucose metabolism by feeding of high fructose diet and its prevention by rosiglitazone in rats. Dig Liver Dis 41:500–508
Yang XC, Reis DJ (1999) Agmatine selectively blocks the N-methyl-d-aspartate subclass of glutamate receptor channels in rat hippocampal neurons. J Pharmacol Exp Ther 288:544–549
Acknowledgments
The authors acknowledge Dr. Mahmoud Gabr and the staff members of the Research Department, Urology and Nephrology Center, Mansoura University, Egypt, for kindly sharing the facilities of their laboratory and their helpful support.
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sharawy, M.H., El-Awady, M.S., Megahed, N. et al. Attenuation of insulin resistance in rats by agmatine: role of SREBP-1c, mTOR and GLUT-2. Naunyn-Schmiedeberg's Arch Pharmacol 389, 45–56 (2016). https://doi.org/10.1007/s00210-015-1174-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00210-015-1174-6