
Abstract
Cadmium (Cd) is a heavy metal pollutant that adversely effects the kidney. Oxidative stress and inflammation are likely major mechanisms of Cd-induced kidney injury. Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is crucial in regulating antioxidant and inflammatory responses. To investigate the role of Nrf2 in the development of subacute Cd-induced renal injury, we utilized Nrf2 knockout (Nrf2-KO) and control mice (Nrf2-WT) which were given cadmium chloride (CdCl2, 1 or 2 mg/kg i.p.) once daily for 7 days. While subacute CdCl2 exposure induced kidney injury in a dose-dependent manner, after the higher Cd dosage exposure, Nrf2-KO mice showed elevated blood urea nitrogen (BUN) and urinary neutrophil gelatinase-associated lipocalin (NGAL) levels compared to control. In line with the findings, the renal tubule injury caused by 2 mg Cd/kg, but not lower dosage, in Nrf2-KO mice determined by Periodic acid–Schiff staining was more serious than that in control mice. Further mechanistic studies showed that Nrf2-KO mice had more apoptotic cells and severe oxidative stress and inflammation in the renal tubules in response to Cd exposures. Although there were no significant differences in Cd contents of tissues between Cd-exposed Nrf2-WT and Nrf2-KO mice, the mRNA expression of Nrf2 downstream genes, including heme oxygenase 1 and metallothionein 1, were significantly less induced by Cd exposures in the kidney of Nrf2-KO compared with Nrf2-WT mice. In conclusion, Nrf2-deficient mice are more sensitive to kidney injury induced by subacute Cd exposure due to a muted antioxidant response, as well as a likely diminished production of specific Cd detoxification metallothioneins.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Environmental cadmium (Cd) pollution has become a significant public problem worldwide. Cd is a transition metal found in the earth’s crust and occurs as a byproduct of the production of zinc, lead, cooper and other metals. In industrial applications, Cd is mainly used in battery, pigment, coating and plating manufacture. In daily life, people are exposed to cadmium mainly by eating Cd-contaminated food and smoking (Faroon et al. 2012; Jarup and Akesson 2009; Menke et al. 2009). Cd may damage multiple organs, including kidney, liver, bone, testis and lung, depending on the dose, route and duration of exposure. With acute accidental Cd exposure via inhalation, the metal causes pneumonitis, bronchitis and pulmonary edema (Boudreau 1988). Chronic Cd exposure is more common, usually through diet and/or inhalation, and renal dysfunction, anemia, osteoporosis and bone fractures are associated with chronic intake (Faroon et al. 2012; Jarup 2002). The primary target organ of chronic Cd toxicity is the kidney, which is one of the main organs where Cd deposits once it is absorbed (Edwards and Prozialeck 2009).
The mechanism of Cd-induced renal toxicity has been extensively studied. Cd has a strong affinity for, and binds to, enzymes containing sulfhydryl groups. The metal thereby inhibits many enzyme systems, affects the normal function of these systems in the kidney, especially the function of antioxidant enzymes, which results in reducing the ability to scavenge reactive oxygen species (ROS) and induces oxidative stress. Cd also affects the function of mitochondrial respiratory chain complexes, increasing ROS production which damages the structure and function of cell membrane (Jarup and Akesson 2009). Therefore, the reduction of antioxidant activity and the production of excessive ROS are likely major causes of Cd-induced renal injury (Wang 2018). In addition, Cd exposure causes an inflammatory response, and activates inflammatory cells to release a variety of cytokines, such as interleukin-6 (IL-6), tumor necrosis factor (TNF), which aggravate oxidative damage (Satarug et al. 2017). Cd also causes single-strand DNA breakage, damages DNA repair system, and leads to the imbalance of intracellular calcium homeostasis and cell apoptosis (Edwards and Prozialeck 2009; Satarug et al. 2017).
Nuclear factor (erythroid-derived 2)-like 2 (NFE2L2, also known as Nrf2) is one of the most important regulatory factors of antioxidant response, and is also involved in the regulation of intracellular redox homeostasis, heavy metal detoxification, inflammatory response and metabolism of a variety of electrophiles (Wang 2018; Zhang 2010). In the normal physiological state, Nrf2 binds to the cytoplasmic protein Kelch-like ECH-associated protein-1 (KEAP1) and is anchored in the cytoplasm for degradation by the ubiquitin proteasome pathway. When cells are attacked by oxidative stressor, KEAP1 undergoes conformational changes and the stabilized Nrf2 translocates to the nucleus, where it heterodimerizes with small Maf proteins and binds to antioxidant response elements (AREs) initiating the transcription of its downstream target genes, including many antioxidant enzymes and phase II detoxifying enzymes, to act against oxidative damage and reduce inflammatory response (Yamamoto et al. 2018; Suzuki and Yamamoto 2015). The silencing of Nrf2 by siRNA makes rat proximal tubule-derived NRK-52E cells more sensitive to Cd-induced apoptosis, while overexpression of Nrf2 prevents apoptosis caused by Cd (Chen and Shaikh 2009). In addition, Nrf2 protects against acute Cd exposure-induced liver damage in vivo (Wu et al. 2012). Furthermore, several studies have shown that exogenous Nrf2 activators protect Cd-induced hepatotoxicity and nephrotoxicity (Zhang 2020; Akiyama 2019). However, at present, there are no studies using genetic knockout mouse models to verify the critical role of Nrf2 in the renal injury induced by Cd. Thus, the present work used Nrf2 knockout (Nrf2-KO) mice to study renal response to subacute Cd exposure. The results showed, for the first time, that constitutive deficiency of Nrf2 resulted in diminished expression of key antioxidant enzymes and phase II detoxifying enzymes in the kidney leading to aggravated renal injury induced by Cd exposure.
Materials and methods
Animals and treatment
The Nrf2-KO mice backcrossed on a C57BL/6 background were kindly provided by Dr. Masayuki Yamamoto (Tohoku University, Japan) (Itoh 1997). As needed, Nrf2-WT and Nrf2-KO littermate mice were generated at the animal facility of China Medical University as described previously (Sun 2018). Mice were housed in a specific pathogen-free environment (12 h light–dark cycle, 23 ± 1 °C and 60–70% humidity) and provided mouse chow diet and distilled water ad libitum. The genotype of mice was determined by PCR using genomic DNA which was extracted from tail snips as described previously (Sun 2018). Adult female Nrf2-WT or Nrf2-KO mice were divided into separate groups (n = 3–7 mice each) as follows: control mice that received saline (0.9% sodium chloride, NaCl) injections, i.p., at 10 ml/kg body weight once per day for 7 days (WT-Cont or KO-Cont); Nrf2-WT or Nrf2-KO mice which received cadmium chloride (#202908, Sigma, CdCl2) injections, i.p., at dose of either 1 or 2 mg Cd/kg body weight once per day for 7 days (WT-1 mg/kg, KO-1 mg/kg, WT-2 mg/kg or KO-2 mg/kg). After the last injection, a 24-h fasting urine was collected while the animals had free access to water using metabolic cages (SN-783, Ishizawa Corporation of Medical Implement, Tokyo, Japan) and the sample was frozen at –80 °C. The mice were euthanized under CO2 asphyxia and kidney, heart and spleen were collected, weighed and stored at –80 °C. Blood samples were taken and processed as outlined below for serological determination. All protocols for animal use were approved by the Institutional Animal Care and Use Committee of China Medical University (Shenyang, China) following the current guidelines for animal care and welfare.
Serum and urine biochemistry analysis
Blood samples were clotted overnight at 4 °C, and then centrifuged at 5000g for 5 min at 4 °C. The levels of blood urea nitrogen (BUN) and creatinine (SCR) in serum were determined using specific kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) according to the manufacturer’s instruction. All biochemical assessments in the serum were completed within 1 week of sample collection. Urine samples were harvested by metabolic cages, and centrifuged to clarify the urine before NGAL determination. The ELISA kits of NGAL were purchased from Cloud-Clone Corp (Wuhan, China) and performed according to the instruction.
Determination of Cd in urine, blood and tissues
The microwave digestion method was utilized to prepare the samples, and analyses of Cd in urine, blood and tissue samples were performed as previously described (Ghoochani 2019; Shi et al. 2019). Briefly, the quartz tubes for digestion were acid-washed for more than 24 h and then rinsed with ultrapure water 3 times. The samples of urine, blood or tissues were digested with the mixture of concentrated nitric acid (65%) and hydrogen peroxide (30%) at a volume ratio 5:2 in a microwave digestion apparatus according to the manufacture protocol. Then the samples were dried and diluted with ultrapure water in sterile falcon tubes.
The Cd content in the samples was determined by Inductively Coupled Plasma Mass Spectrometry (ICP-MS, Agilent Technologies, Waldbronn, USA) in Open Platform of Experiment Teaching Center, School of Public Health, China Medicine University as previously described (Shi et al. 2019). The reagent blanks were used for calculation, and the Cd content of the samples were expressed as μg Cd/g tissue calculated back to the original wet weight. The recovery percentages of standard samples were 86.6% (0.2 μg/L), 98.5% (2 μg/L), 101.7% (50 μg/L), respectively. The limit of detection of Cd element was 0.02 μg/L, and the regression coefficient of the calibration standard solutions was ≥ 0.99.
Histological and immunohistochemical analyses
The mice were dissected immediately after CO2 asphyxia. A slice of kidney samples was fixed in 4% of paraformaldehyde buffer overnight, and then transferred to 70% ethanol. Tissues were embedded in paraffin, sectioned at 3 μm and subjected to Periodic acid–Schiff (PAS) staining. TdT-mediated dUTP nick-end labeling (TUNEL) staining of kidney tissues was performed according to the manufacturer’s instruction (Vazyme Biotech Co., Ltd, Nanjing, China) as previously described (Kong 2018). TUNEL staining-positive cells were counted by Image J software on randomly selected kidney cortex fields. The percentage of TUNEL staining-positive cells per field was expressed as apoptotic cells.
For immunohistochemistry, the sections were mounted on poly-l-lysine slides. The levels of 4-hydroxynonenal (4-HNE) adducts were assessed using a primary rabbit anti-4-HNE adducts antibody (ab46545, 1:100, Abcam, Cambridge, UK) following the instruction of immunohistochemistry kit (IHC-DAB; Beijing Zhongshan Jinqiao Biological Technology Co., Beijing, China) (Kong 2018).
Real-time RT-PCR
Total RNA samples were extracted from a portion of kidney using Trizol reagent (Life Technologies, Carlsbad, CA, USA) using a TissueLyser II (Retsch, Newtown, PA). Extracted RNA samples were subjected to clean-up using 75% ethanol and then reverse transcribed by Prime Script RT reagent Kit (Takara, Dalian, China) as described previously (Fu 2015). Real-time fluorescence detection was performed using SYBR Premix Ex Taq mix (Takara, Dalian, China) with 5 μg cDNA and 0.1 μM primers on QuantStudio 6 Flex Real-time PCR System (Applied Biosystems, Life Technologies). Thermal cycling parameters were 95 °C for 10 min, and 40 cycles of 95 °C for 15 s and 60 °C for 1 min, followed by melting curve analysis. qPCR product was resolved by agarose gel electrophoresis to ensure the correct amplicon length. The primers were designed using Primer Express 4 (Applied Biosystems) and synthesized by Takara Co., Ltd. (Takara, Dalian, China). The following sets of primers were used: nicotinamide adenine dinucleotide (phosphate): quinine oxidoreductase 1 (Nqo1, forward 5′- TATCCTTCCGAGTCATCTCTAGCA -3′ and reverse 5′- TCTGCAGCTTCCAGCTTCTTG -3′), glutamate cysteine ligase catalytic subunit (Gclc, forward 5′- TGGCCACTATCTGCCCAATT -3′ and reverse 5′- GTCTGACACGTAGCCTCGGTAA -3′), heme oxygenase 1 (Ho-1, forward 5′- CCTCACTGGCAGGAAATCATC-3′ and reverse 5′- CCTCGTGGAGACGCTTTACATA -3′), glutamate cysteine ligase modifier subunit (Gclm, forward 5′- ACATTGAAGCCCAGGATTGG -3′ and reverse 5′- CCCCTGCTCTTCACGATGAC -3′), metallothionein 1 (Mt1, forward 5′- CTCCACCGGCGGCTC -3′ and reverse 5′- CTCCGGGAAGCAAAGGACAT -3′), metallothionein 2 (Mt2, forward 5′- GCGCTCGACCCAATACTCTC and reverse 5′- GCAGGAGCAGGATCCATCG -3′) and β-actin (forward 5′-GTATGACTCCACTCACGGCAAA-3′ and reverse 5′-GGTCTCGCTCCTGGAAGATG-3′). The mRNA levels of β-actin were used for normalization and calculations as described previously (Fu 2015).
Statistical analysis
Data analysis was performed using GraphPad Prism 5 software (San Diego, CA), and a p < 0.05 was considered as showing a significant difference. All the results were expressed as mean ± standard deviation. For comparisons between two groups, a Student’s t test was performed. For comparisons among multiple groups, two-way ANOVA followed by a Bonferroni multiple comparison test was used.
Results
Cd exposure induces more serious renal injury in Nrf2-KO mice with more proximal tubule epithelial cell apoptosis
During Cd treatment, body weight was closely monitored and the body weight of all six groups did not change significantly during the whole experimental period (Fig. 1a). The organ masses of kidney, heart and spleen measured at the end of treatment were unaltered by Cd exposure (Figs. 1b and S1). This suggests that the dose of Cd selected for use in this experiment did not cause overt systemic toxicity. To help evaluate the specific renal function after Cd exposure, we measured serum BUN, SCR and urinary NGAL. As shown in Fig. 1c–e, the BUN levels were significantly increased in 2 mg/kg of Cd-treated Nrf2-KO mice, but not in 1 mg/kg group of Nrf2-KO mice and all groups of Nrf2-WT mice (Fig. 1d). There were no significant increases in serum SCR levels in Cd-treated Nrf2-WT and Nrf2-KO mice (Fig. 1c). In agreement with the results of serum BUN measurements, urinary NGAL levels were also significantly increased in 2 mg/kg Cd-exposed Nrf2-KO mice compared to either Nrf2-WT mice with the same treatment or the control mice (Fig. 1e).

Physiological and biochemical measurements of Nrf2-KO and Nrf2-WT mice exposed subacutely to Cd. The mice were treated as controls (0.9% NaCl, 10 ml/kg/day) or with Cd (1 or 2 mg/kg/day) by i.p. injection every day for seven consecutive days. Body weight (a). Kidney mass expressed as ratios to body weight (b). c–e Serum levels of SCR (c), BUN (d) and urinary NGAL (e) determined by ELISA. WT represents Nrf2-WT, KO represents Nrf2-KO. Cont represents mice exposed to saline (0.9% NaCl) i.p. injection, n = 5. 1 mg/kg or 2 mg/kg represents mice treated with 1 mg Cd/kg (n = 3) or 2 mg Cd/kg (n = 7) i.p. once per day for 7 consecutive days. Values are expressed as mean ± S.D. *, p < 0.05 vs Control of the same genotype; #, p < 0.05 vs Nrf2-WT given the same treatment
The renal proximal tubules were normal in Nrf2-WT and Nrf2-KO mice treated with saline (Figs. 2a, b, S2 and S3). In groups receiving 1 mg Cd/kg (Figs. 2c, d, S2 and S3), the brush border of renal proximal tubules was particularly thin or absent in Nrf2-WT mice. In contrast, in Nrf2-KO mice, the proximal tubules were dilated, and most of the brush border was lost. In 2 mg Cd/kg treatment groups (Figs. 2e, f, S2 and S3), a small number of renal tubular epithelial cells shed into the lumen in the Nrf2-WT mice. In Nrf2-KO mice, the epithelial cells of proximal tubules disappeared completely, and the normal structure of renal tubules had often disappeared. These histochemical staining results are highly consistent with the results of serum BUN and urinary NGAL measurements, indicating that Nrf2-KO mice are prone to high dose Cd-induced renal toxicity.

PAS staining of kidney tissues of Nrf2-KO and Nrf2-WT mice. For treatment details, see the legend for Fig. 1. Left panels in each genotype, low magnification (10 × , scale bar = 1 mm). Right panels in each genotype, high magnification (200 × , scale bar = 100 μm). The proximal tubules showed a regular and well-stained brush border in the control mice (a, b). In the mice given 1 mg Cd/kg, the brush border is either particularly thin or absent (arrowhead) in WT mice (c), while in KO mice, the proximal tubules are dilated and most of the brush border is lost (arrow, d). In 2 mg Cd/kg Cd groups, a small number of renal tubular epithelial cells are shed into the lumen in WT mice (+ , e), but in the KO mice, the epithelial cells of proximal tubules have disappeared completely, and the structure of renal tubules is essentially lost (*, f). Thus, Cd induced more serious kidney injury in Nrf2-KO mice
To investigate the nature of damage in renal tubules, we performed TUNEL staining to look for proximal tubule epithelial cell apoptosis after Cd exposure (Fig. 3). There were no TUNEL positive nuclei in Controls of either Nrf2-WT or Nrf2-KO mice. In contrast, there were substantial increases of TUNEL staining-positive renal tubular epithelial cells in both Nrf2-WT and Nrf2-KO mice exposed to Cd. Importantly, Nrf2-KO mice showed much more TUNEL staining-positive renal tubular epithelial cells than those of Nrf2-WT mice with the same high dose of Cd exposure (Fig. 3a, b).

Assessment of apoptotic cells in the kidneys by TUNEL staining in control or Cd-treated Nrf2-WT or Nrf2-KO mice. For treatment details, see the legend for Fig. 1. Representative images of TUNEL staining (a). Magnification 200 × , scale bar = 100 μm. Quantitative analysis of TUNEL staining (b). Values are expressed as mean ± S.D. *, p < 0.05 vs Control of the same genotype; #, p < 0.05 vs Nrf2-WT given the same treatment
Taken together, the key findings in the evaluation of renal function, histochemical analysis and immunostaining of kidney tissues reveal that Nrf2-KO mice are sensitive to subacute Cd exposure-induced kidney damage.
Nrf2-KO mice show more severe renal oxidative stress and inflammation in kidney after Cd treatment
To characterize the protective role of Nrf2 against Cd-induced renal toxicity, we measured multiple biomarkers of oxidative stress and inflammation in kidney tissues. Using immunofluorescence staining, 4-HNE, a product of lipid peroxidation and a marker of oxidative/electrophilic stress, was determined in kidney tissues. As shown in Fig. 4a, b, Cd exposure dose-dependently increased levels of 4-HNE adducts in the kidney tissues of Nrf2-WT and Nrf2-KO mice. Importantly, the fluorescence intensity of 4-HNE adducts were significantly higher in the kidney of Nrf2-KO mice treated with 2 mg Cd/kg than Nrf2-WT mice with the same treatment. In line with the results above, the Nrf2-KO mice with 2 mg Cd/kg treatment showed significantly augmented transcript levels of renal cluster of differentiation 68 (Cd68) and Tnf genes in their kidney tissues (Fig. 4c, d). These findings indicate that high dose of Cd induces more severe oxidative damage and subsequent inflammation in the kidney of Nrf2-KO mice compared to Nrf2-WT mice.

Assessment of oxidative damage and inflammation in kidney by immunofluorescent staining for 4-HNE adducts and mRNA expression of cytokines. For treatment details, see the legend for Fig. 1. a, b Representative images (a) and quantitation (b) of immunofluorescent stainings for 4-HNE adducts. Magnification 400 × , scale bar = 50 μm in panel A. (c, d) mRNA expression of Cd68 (c) and Tnf (d). Values were expressed as mean ± S.D in (b–d). n = 3–7. *, p < 0.05 vs Control of the same genotype. #, p < 0.05 vs Nrf2-WT given the same treatment
Nrf2 deficiency does not affect the distribution of Cd in mice
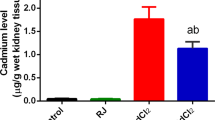
Typically, after the administration of CdCl2 by i.p. injection, Cd is absorbed into the blood and deposits in the liver and elsewhere, where it is conjugated with proteins and small molecules, often containing thiol groups, and stimulates formation of Cd–metallothioneins (Cd–MTs) and other Cd complexes. During Cd excretion, the metal will reach the kidney, where Cd is normally re-absorbed by renal tubular epithelial cells, and will thereby accumulate in the kidney. A small amount of Cd can be excreted into the urine. To investigate the effect of Nrf2 deficiency on the absorption, disposition, accumulation and excretion of Cd, the content of Cd in blood, liver, kidney and urine samples was determined. After the administration of Cd for seven consecutive days, the tissue content of Cd increased dramatically in both Nrf2-WT and Nrf2-KO mice (Fig. 5a–c), and correlated with the dose of Cd administered. However, Nrf2 deficiency had no impact on Cd levels in blood, liver and kidney after Cd treatment, indicating Nrf2 plays marginal role in the biodistribution of subacute Cd exposure. As expected, 24-h excretion of Cd from urine was also dramatically increased in both Nrf2-WT and Nrf2-KO mice, which was correlated with Cd exposure dose-dependently (Fig. 5d). There was no significant difference in 24-h excretion of Cd between genotypes, partially due to the big variations among mice (Fig. 5d).

Biodistribution of Cd in control or Cd-treated Nrf2-WT and Nrf2-KO mice. For treatment details, see the legend for Fig. 1. a–d Cd levels were measured in blood (a), liver (b), kidney (c) and urine (d). n = 3–7. Values were expressed as mean ± S.D. *, p < 0.05 vs Control of the same genotype
The induction of mRNA expression of antioxidant genes and Cd disposition-related enzymes was inhibited in the kidney of Nrf2-KO mice
Blood Cd, Cd–MT, Cd–protein and Cd–thiol complexes undergo glomerular filtration and are often reabsorbed into proximal tubules, with subsequent breakdown releasing the toxic metal thereby inducing local renal toxicity. To determine the mechanism by which Nrf2 protected against subacute Cd-induced kidney injury, the mRNA levels of Nrf2 downstream genes, as well as the Mts, were measured in the kidney of Nrf2-WT and Nrf2-KO mice treated with 1 or 2 mg Cd/kg for 7 days (Fig. 6a). In Nrf2-WT mice, the induction of the antioxidant genes by 2 mg/kg Cd exposure was lower than 1 mg/kg Cd, suggesting that the mice with consecutive 7-day injections of 2 mg/kg Cd have severe kidney injury. Although basal Gclc, Gclm, Ho1, Mt1 and Mt2 mRNA levels were comparable between genotypes, Cd-induced expression of these genes was substantially reduced in Nrf2-KO mice. The expression of Nqo1 under both basal and Cd-exposed conditions was substantially decreased in Nrf2-deficient groups (Fig. 6a). In line with mRNA levels, the protein levels of GCLC and GCLM induced by Cd exposure was also attenuated in Nrf2-KO kidneys (Fig. 6b, c).

Renal expression of antioxidant and detoxification-related enzymes in mice after Cd exposure. The transcript levels of Nrf2 downstream genes were assessed (a). For treatment details, see the legend for Fig. 1. n = 3–7. Values were expressed as mean ± S.D. *, p < 0.05 vs Control of the same genotype. #, p < 0.05 vs Nrf2-WT given the same treatment. b, c Representative images (b) and quantitation (c) of immunoblots of Nrf2 downstream targets in the kidneys. TUBULIN was as loading control. The transcript levels of Nrf2 downstream genes in acute Cd-treated mice (d). The mice received a single Cd dose (2 mg Cd/kg; i.p.) and samples were prepared 12 h later. n = 6. Values were expressed as mean ± S.D. *, p < 0.05 vs Control in the same genotype; #, p < 0.05 vs Nrf2-WT given the same treatment
To exclude the interfering effect of kidney injury on the expression of the antioxidant and Cd detoxification genes, the tests were run on mice at early time point (12 h) post injection of 2 mg Cd/kg, i.p. and the same panel of mRNA were assessed. As shown in Fig. 6d, the 12 h of Cd exposure significantly upregulated the mRNA expression of Nqo1, Ho1 and Mt1 in the kidney of Nrf2-WT mice, whereas Nrf2-KO mice showed significantly lowered induction of the genes. The Cd treatment showed limited effect on the mRNA expression of Gclc, Gclm and Mt2. Taken the findings from subacute and acute experiments together, our results indicate that Nrf2-mediated antioxidant and detoxification responses are involved in the prevention of Cd-induced kidney injury.
Discussion
Cd pollution is a major public health problem, as it is widely dispersed in the human environment and can damage multiple organs, including the kidney, bone, lung, liver, and testes (Jarup and Akesson 2009; Menke et al. 2009; Jarup 2002). The kidney is a primary target organ of both acute and chronic cadmium toxicity (Edwards and Prozialeck 2009; Satarug et al. 2017). The present study investigated the critical protective role of Nrf2 in Cd-induced renal toxicity using a Nrf2-KO mouse model. The data showed that constitutive Nrf2 deficiency in mice greatly exacerbated the impaired renal tubular function induced by subacute Cd exposure (2 mg Cd/kg, 7 days) as evaluated by levels of serum BUN and urinary NGAL. Furthermore, we found that the Nrf2-KO mice given subacute Cd treatment had more apoptotic cells, increased oxidative stress and inflammation specifically in the renal tubules, as well as a diminished adaptive antioxidant and detoxification response compared to WT mice. Together these data indicate that Nrf2 plays a key role in preventing or mitigating Cd-induced renal toxicity.
In the present study, Cd is expected to enter bloodstream soon after i.p. injection, then binds with hemoglobin, serum albumin, amino acids or reduced glutathione (GSH) in the blood (Jarup 2002). These Cd complexes are transported to the liver, where they generate various forms of the metal-binding proteins containing Cd, namely Cd–MTs, which tend to reduce the toxic effects of free Cd ion on biological macro-molecules (Satarug et al. 2017). As liver tissues turn over, Cd–MTs are released to the blood, and eventually reach the kidney where they are reabsorbed by the proximal convoluted tubule after filtration by the glomerulus. This reabsorption may lead to renal tubular injury since the Cd–MTs complexes are broken-down by the kidney cells and quickly release their Cd ions (Abouhamed 2007). Only about 0.005% of the total body burden of Cd is excreted in the urine (Jarup and Akesson 2009), so the overwhelming portion makes it back into the kidney. In the current study, we measured Cd levels in the blood, kidney, liver and urine. Cd in the blood, liver and kidney showed an increase with dose of exposure in both the Nrf2-WT and Nrf2-KO mice, although there were no differences between the genotypes in tissue content after Cd exposure. This suggests Nrf2 does not impact the process of Cd bio-deposition at least via blood into liver and kidney after acute exposure. It was also reported previously that Nrf2 or Keap1 deficiency in primary hepatocytes did not change Cd accumulation (Shinkai 2016). However, in the 2 mg Cd/kg group, urinary Cd concentrations were higher in the Nrf2-KO mice than Nrf2-WT mice, indicating the kidney re-absorption was disrupted, which is consistent with the enhanced renal tubular injury and proximal tubular cell apoptosis in these Cd-exposed Nrf2-KO mice. Of note, the amount of urinary Cd excretion was limited and cannot significantly affect the Cd accumulation in the tissues. In addition, the Cd exposure was performed by single injection per day for 7 days, leading to overwhelmed amount of Cd accumulated into the tissues.
The key findings in the current study are not entirely surprising as Nrf2 has been shown to have a protective role against systemic toxicity with several xenobiotic chemicals via regulation of antioxidant and detoxification enzymes (Yamamoto et al. 2018; Wu et al. 2012; Akiyama 2019). Interestingly, Cd is not a Fenton-reactive metal, and it does not induce redox reactions directly. However, once free Cd ion enters renal proximal tubular cells, ROS may be increased. The imbalance of ROS in response to Cd exposure is thought to be a major mechanism of Cd toxicity (Wang 2018). 4-HNE is a product of lipid peroxidation, an activator of Nrf2-ARE, and thus a biomarker of oxidative stress (Csala 2015). It has been shown that Cd exposure increases renal 4-HNE levels in vivo (Kuang 2017). In line with the findings above, our data indicated that the increased levels of 4-HNE adducts in the kidney caused by Cd exposure were much higher in Nrf2-KO mice than those in Nrf2-WT mice with the same treatment. This indicates that more oxidative injury occurred in the kidney of Cd-exposed Nrf2-KO mice. Consistent with the renal dysfunction and oxidative damage, the inflammatory indicators, renal Cd68 and Tnf mRNA levels, were also increased in 2 mg Cd/kg exposed Nrf2-KO mice. In a prior study, Chen et al. reported that Nrf2 activation conferred protection against Cd-induced apoptosis in rat proximal tubule-derived NRK-52E cells in vitro (Chen and Shaikh 2009). Our novel in vivo data presented here highlight the importance of Nrf2-mediated antioxidant response in protection against the renal tubular injury induced by subacute Cd exposure (Fig. 7). Taken together, accumulating evidence, including our own, indicate that the modulation of Nrf2 activity is a potential protective strategy against Cd intoxication in future (Ashrafizadeh 2019; Das et al. 2019).

The proposed mechanism of Nrf2 in protection against renal damage from subacute Cd exposure. The pathogenesis of renal tubule injury induced by Cd likely can be attributed to the accumulation of ROS, leading to local oxidative damage and inflammation stress, which activates Nrf2 pathway. The activated Nrf2 pathway aids in restoring homeostasis by reducing oxidative stress via induction of antioxidant systems, which then protects from renal tubule damage induced by Cd in Nrf2-WT mice. However, in the Nrf2-KO mice, the muted Nrf2-mediated adaptive antioxidant and detoxification response has sensitized these mice to Cd-induced renal tubule injury
MTs are small cysteine-rich metal-binding proteins that are ubiquitously expressed. They have high affinity for a variety of heavy metals and they are clearly related to Cd detoxification (Tachibana 2014). The mRNA expression of Mts is mainly regulated by metal response elements (MRE) and ARE in their promoter region (Gu 2017; Ohtsuji 2008). Recent studies have found that Mt1 is regulated by ARE promoting and participating in the formation of biological defense system, while Mt2 is only regulated by MRE (Fujie et al. 2016). It was also shown that Nrf2 mRNA expression is decreased in Mt-knockout myocardial cells. In a hypoxia model, MT regulated Nrf2 protein level and transcriptional activity through the PI3K–AKT–GSK signaling pathway (Zhou 2017). In addition, in Nrf2-deficient vascular endothelial cells, the induction of Mt1 by Cd exposure was inhibited (Shinkai 2016), suggesting that Mt1 is one of the downstream target gene of Nrf2 (Ohtsuji 2008). Although the detail regulatory mechanism between MTs and Nrf2 is still not clear, MTs and Nrf2 may cooperate to maintain the redox and detoxification capacity homeostasis. In the current study, Cd-induced Mt1 expression was time dependent. Acute (12 h) Cd exposure induced Mt1 expression three fold, compared with more than ten fold in subacute exposure (7 days). Consistent with previous study, Cd-induced Mt1 expression was completely blocked in the kidney of Nrf2-KO mice, while Mt2 was not affected. In addition to Mt1, the mRNA induction of the well-known downstream genes of Nrf2, such as Gclc, Nqo1 and Ho1, was also attenuated in Nrf2-KO kidney. This is an indication that both Nrf2-mediated antioxidant and detoxification response contribute to its protective role against Cd-induced kidney damage.
In summary, the current study highlights that Nrf2-mediated antioxidant response is involved in the protection against Cd-induced renal toxicity, indicating that the modulation of Nrf2 pathway could be a potential protective strategy with Cd intoxication. However, to exclude the systemic effect of global Nrf2 manipulation, a conditional Nrf2 knockout model is needed to confirm the role of Nrf2 specifically in renal proximal tubular cells or other key target cell types during Cd-induced renal toxicity.
Abbreviations
- AREs:
-
Antioxidant response elements
- BUN:
-
Blood urea nitrogen
- Cd:
-
Cadmium
- CdCl2 :
-
Cadmium chloride
- Cd–MTs:
-
Cd–metallothioneins
- Cd68:
-
Cluster of differentiation 68
- GSH:
-
Glutathione
- Gclc:
-
Glutamate cysteine ligase catalytic subunit
- Gclm:
-
Glutamate cysteine ligase modifier subunit
- 4-HNE:
-
4-Hydroxynonenal
- Ho1:
-
Heme oxygenase 1
- ICP-MS:
-
Inductively coupled plasma mass spectrometry
- IL-6:
-
Interleukin-6
- IHC:
-
Immunohistochemistry
- KEAP1:
-
Kelch-like ECH-associated protein-1
- MT:
-
Metallothionein
- Nrf2:
-
Nuclear factor erythroid-derived 2-like 2
- MRE:
-
Metal response elements
- Nrf2-KO:
-
Nrf2 Knockout
- Nrf2-WT:
-
Nrf2 Wild-type
- NaCl:
-
Sodium chloride
- NGAL:
-
Neutrophil gelatinase-associated lipocalin
- Nqo1:
-
Nicotinamide adenine dinucleotide (phosphate):quinine oxidoreductase 1
- PAS:
-
Periodic acid–Schiff
- ROS:
-
Reactive oxygen species
- SCR:
-
Serum creatinine
- TNF:
-
Tumor necrosis factor
- TUNEL:
-
TdT-mediated dUTP nick-end labeling
References
Abouhamed M et al (2007) Knockdown of endosomal/lysosomal divalent metal transporter 1 by RNA interference prevents cadmium-metallothionein-1 cytotoxicity in renal proximal tubule cells. Am J Physiol Renal Physiol 293(3):F705–F712
Akiyama M et al (2019) Environmental electrophile-mediated toxicity in mice lacking Nrf2, CSE, or both. Environ Health Perspect 127(6):67002
Ashrafizadeh M et al (2019) Back to nucleus: combating with cadmium toxicity using Nrf2 signaling pathway as a promising therapeutic target. Biol Trace Elem Res 197(1):52–62
Boudreau J et al (1988) Toxicity of inhaled cadmium chloride: early responses of the antioxidant and surfactant systems in rat lungs. J Toxicol Environ Health 23(2):241–256
Chen J, Shaikh ZA (2009) Activation of Nrf2 by cadmium and its role in protection against cadmium-induced apoptosis in rat kidney cells. Toxicol Appl Pharmacol 241(1):81–89
Csala M et al (2015) On the role of 4-hydroxynonenal in health and disease. Biochim Biophys Acta 1852(5):826–838
Das S et al (2019) Carnosic acid attenuates cadmium induced nephrotoxicity by inhibiting oxidative stress, promoting Nrf2/HO-1 signalling and impairing TGF-beta1/Smad/Collagen IV signalling. Molecules 24(22):4176
Edwards JR, Prozialeck WC (2009) Cadmium, diabetes and chronic kidney disease. Toxicol Appl Pharmacol 238(3):289–293
Faroon O, Ashizawa A, Wright S, Tucker P, Jenkins K, Ingerman L, Rudisill C (2012) Toxicological profile for cadmium. Agency for Toxic Substances and Disease Registry (US), Atlanta (GA). PMID: 24049863
Fu J et al (2015) Protective role of nuclear factor E2-related factor 2 against acute oxidative stress-induced pancreatic beta -cell damage. Oxid Med Cell Longev 2015:639191
Fujie T et al (2016) Transcriptional induction of metallothionein by tris(pentafluorophenyl)stibane in cultured bovine aortic endothelial cells. Int J Mol Sci 17(9):1381
Ghoochani M et al (2019) Association among sources exposure of cadmium in the adult non-smoking general population of Tehran. Biol Trace Elem Res 191(1):27–33
Gu J et al (2017) Metallothionein is downstream of Nrf2 and partially mediates sulforaphane prevention of diabetic cardiomyopathy. Diabetes 66(2):529–542
Itoh K et al (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236(2):313–322
Jarup L (2002) Cadmium overload and toxicity. Nephrol Dial Transplant 17(Suppl 2):35–39
Jarup L, Akesson A (2009) Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol 238(3):201–208
Kong W et al (2018) Nrf2 deficiency promotes the progression from acute tubular damage to chronic renal fibrosis following unilateral ureteral obstruction. Nephrol Dial Transplant 33(5):771–783
Kuang W et al (2017) Ligustrazine modulates renal cysteine biosynthesis in rats exposed to cadmium. Environ Toxicol Pharmacol 54:125–132
Menke A et al (2009) Cadmium levels in urine and mortality among U.S. adults. Environ Health Perspect 117(2):190–196
Ohtsuji M et al (2008) Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem 283(48):33554–33562
Satarug S, Vesey DA, Gobe GC (2017) Kidney cadmium toxicity, diabetes and high blood pressure: the perfect storm. Tohoku J Exp Med 241(1):65–87
Shi P, Jing H, Xi S (2019) Urinary metal/metalloid levels in relation to hypertension among occupationally exposed workers. Chemosphere 234:640–647
Shinkai Y et al (2016) Partial contribution of the Keap1-Nrf2 system to cadmium-mediated metallothionein expression in vascular endothelial cells. Toxicol Appl Pharmacol 295:37–46
Sun J et al (2018) NRF2 mitigates acute alcohol-induced hepatic and pancreatic injury in mice. Food Chem Toxicol 121:495–503
Suzuki T, Yamamoto M (2015) Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med 88(Pt B):93–100
Tachibana H et al (2014) Metallothionein deficiency exacerbates diabetic nephropathy in streptozotocin-induced diabetic mice. Am J Physiol Renal Physiol 306(1):F105–F115
Wang Y et al (2018) Roles of ROS, Nrf2, and autophagy in cadmium-carcinogenesis and its prevention by sulforaphane. Toxicol Appl Pharmacol 353:23–30
Wu KC, Liu JJ, Klaassen CD (2012) Nrf2 activation prevents cadmium-induced acute liver injury. Toxicol Appl Pharmacol 263(1):14–20
Yamamoto M, Kensler TW, Motohashi H (2018) The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 98(3):1169–1203
Zhang Q et al (2010) A systems biology perspective on Nrf2-mediated antioxidant response. Toxicol Appl Pharmacol 244(1):84–97
Zhang Q et al (2020) Ameliorative effects of resveratrol against cadmium-induced nephrotoxicity via modulating nuclear xenobiotic receptor response and PINK1/Parkin-mediated mitophagy. Food Funct 11(2):1856–1868
Zhou S et al (2017) Intermittent hypoxia-induced cardiomyopathy and its prevention by Nrf2 and metallothionein. Free Radic Biol Med 112:224–239
Funding
This work was supported by National Natural Science Foundation of China 82020108027 (J.P.), 81830099 (J.P.), 81573106 (J.P.), 81770698 (H.Z) and 81402635 (J.F.), Liaoning Key Research and Development Guidance Plan 2019JH8/10300012 (J.P.), Liaoning Province Natural Science Foundation (20180530011, J.F.), Educational Department of Liaoning Province Scientific Research Foundation (ZF2019035, J.F.), China Medical University Training Program for National Natural Science Fund for Excellent Young Scholars (YQ20170001, J.F.), and the Key Laboratory of Liaoning Province on Toxic and Biological Effects of Arsenic. We thank Dr. Michael P. Waalkes for editorial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no potential conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, C., Han, X., Wang, G. et al. Nrf2 deficiency aggravates the kidney injury induced by subacute cadmium exposure in mice. Arch Toxicol 95, 883–893 (2021). https://doi.org/10.1007/s00204-020-02964-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-020-02964-3