
Abstract
In vitro genotoxicity testing that employs metabolically active human cells may be better suited for evaluating human in vivo genotoxicity than current bacterial or non-metabolically active mammalian cell systems. In the current study, 28 compounds, known to have different genotoxicity and carcinogenicity modes of action (MoAs), were evaluated over a wide range of concentrations for the ability to induce DNA damage in human HepG2 and HepaRG cells. DNA damage dose–responses in both cell lines were quantified using a combination of high-throughput high-content (HTHC) CometChip technology and benchmark dose (BMD) quantitative approaches. Assays of metabolic activity indicated that differentiated HepaRG cells had much higher levels of cytochromes P450 activity than did HepG2 cells. DNA damage was observed for four and two out of five indirect-acting genotoxic carcinogens in HepaRG and HepG2 cells, respectively. Four out of seven direct-acting carcinogens were positive in both cell lines, with two of the three negatives being genotoxic mainly through aneugenicity. The four chemicals positive in both cell lines generated HTHC Comet data in HepaRG and HepG2 cells with comparable BMD values. All the non-genotoxic compounds, including six non-genotoxic carcinogens, were negative in HepaRG cells; five genotoxic non-carcinogens also were negative. Our results indicate that the HTHC CometChip assay detects a greater proportion of genotoxic carcinogens requiring metabolic activation (i.e., indirect carcinogens) when conducted with HepaRG cells than with HepG2 cells. In addition, BMD genotoxicity potency estimate is useful for quantitatively evaluating CometChip assay data in a scientifically rigorous manner.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Genotoxicity testing is an important part of the safety assessment of all xenobiotics, generally being performed for predicting carcinogenic potential. A battery of short-term genetic toxicity tests, including the Ames test, mouse lymphoma assay (MLA), and in vitro micronucleus (MNvit) or chromosomal aberration (CA) assay, has been recommended by several international authoritative bodies, such as the Organization for Economic Co-operation and Development (OECD), International Council for Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), and International Cooperation on Harmonization of Technical Requirements for Registration of Veterinary Medicinal Products (VICH) for assisting regulatory agencies’ decision-making (ICH 2011; OECD 2015; VICH 2013). The performance of the three most commonly used assays (Ames, MLA, and MNvit or CA) has been evaluated in terms of their sensitivity, specificity, and positive and negative predictivity using data from 700 rodent carcinogens and non-carcinogens (Kirkland et al. 2005). The three-test battery of mammalian cell-based assays had high sensitivity but a surprisingly low specificity (< 45%) for testing chemicals for carcinogenic potential. In addition, 75–95% of non-carcinogens were positive in at least one assay of the test battery. These observations highlight the limitations of the current test battery and the need for developing testing systems with better sensitivity and specificity for predicting genotoxicity.
In vitro genotoxicity assays generally are performed in human or rodent cell lines that have restricted metabolic and DNA repair capacities, such as TK6 human lymphoblastoid cells, V79 Chinese hamster lung fibroblasts, or mouse lymphoma cells, which may be a source of at least some false-positive or false-negative responses (Kirkland et al. 2016; Le Hegarat et al. 2014). Typically, an exogenous metabolic activation system, such as the rat liver S9 fraction, is used to provide the metabolic activation that occurs in vivo. However, researchers have been increasingly aware of differences in drug metabolism between humans and commonly used nonclinical test species (i.e., mice, rats, and dogs) with improvements in methods for the identification and measurement of drug metabolites, because some metabolites are identified only in humans. (Josse et al. 2012; Le Hegarat et al. 2010; Robison and Jacobs 2009).
Many human carcinogens require metabolic activation to elicit adverse effects including genotoxicity (Luch 2005), and primary human hepatocytes (PHHs) are considered to be the golden standard for the in vitro evaluation of metabolism, drug–drug interactions, and hepatotoxicity (Zeilinger et al. 2016). However, PHHs have well-known drawbacks, such as inter-donor variability, limited availability, and the lack of proliferative capacity, that restrict their application in genotoxicity testing. HepG2 cells are a relatively easy-to-handle and widely used human hepatoma cell line for genotoxicity evaluations. However, HepG2 cells express only low levels of cytochrome P450 (CYP450s), resulting in limited metabolic capacities as compared to PHHs (Westerink and Schoonen 2007). HepaRG cells, a human non-tumorigenic hepatoma cell line, retain much of the metabolic activity of primary liver cells by expressing Phase I and Phase II enzymes, various transporters, and nuclear receptors at the levels comparable to those found in PHHs. Thus, HepaRG cells are considered as a promising surrogate to PHHs for use in studies on xenobiotic drug metabolism and liver toxicity (Antherieu et al. 2010; Guillouzo et al. 2007; Marrone et al. 2016; Tryndyak et al. 2018).
Inclusion of a metabolically competent human cell line as part of a genotoxicity test battery has the potential of advancing the drug development, as it would provide a test where unique human metabolites might potentially be generated and characterized for genetic toxicity. A limited number of studies have demonstrated that traditional genotoxicity assays, such as MNvit and Comet assays, can be conducted using HepaRG cells (Le Hegarat et al. 2010, 2014). However, it remained to be determined whether HepaRG cells could be adapted to a 96-well format for high-throughput genetic toxicity evaluations. In the current study, we used the Comet assay as a genotoxicity indicator test for detecting primary DNA damage. The Comet assay, also known as the single cell gel electrophoresis assay, is a sensitive method for detecting various types of DNA damage, including alkali-labile sites, abasic sites, and single and double stranded breaks at a single cell level (Ge et al. 2015). However, the Comet assay has some recognized shortcomings, for example, the labor intensive and relatively low throughput (Sykora et al. 2018; Wood et al. 2010). Recently, a novel 96-well CometChip platform was developed (Ge et al. 2015). The CometChip uses the micro-patterned agarose in the chip wells to facilitate high-throughput analysis, with a high level of reproducibility, and the potential for generating a large number of data points covering a wide range of chemical concentrations.
Quantitative approaches for assessing dose–response relationships and for deriving point-of-departure (PoD) metrics have been increasingly utilized in genetic toxicology studies (Gollapudi et al. 2013; Zeller et al. 2017). The benchmark dose (BMD) is the PoD metric recommended by the Working Group on Quantitative Approaches to Genetic Toxicology Risk Assessment (QWG) of the International Workshops on Genotoxicity Testing (IWGT). BMDs can be used for estimating reference exposure doses and for potency ranking using genetic toxicology data (MacGregor et al. 2015; Sand et al. 2017). Our previous studies demonstrated the feasibility of the BMD approach for discriminating the mutagenic responses and DNA damaging effects produced by chemicals and cigarette whole smoke solutions using data generated from the MLA and Comet assay (Guo et al. 2015, 2018b, 2018a). Furthermore, our studies have indicated the value of increasing the number of test article concentrations/doses for improving the BMD precision.
In the present study, we have evaluated the suitability of using metabolically competent human HepaRG cells and high-throughput high-content (HTHC) CometChip technology for detecting genotoxic potential. This evaluation was performed by testing a series of agents known to have different genotoxic and carcinogenic modes of action (MoAs), including genotoxic carcinogens, non-genotoxic carcinogens, genotoxic non-carcinogens, and cytotoxic non-genotoxic non-carcinogens (Table 1). The BMD approach was employed for determining PoDs using the dose–response data generated from the HTHC in vitro Comet assay. The resulting quantitative genotoxicity responses were compared with similar data generated using HepG2 cells.
Materials and methods
Materials
Twenty-eight test agents (Table 1) were selected from the compounds recommended by the European Centre for the Validation of Alternative Methods (ECVAM) for the assessment of new or improved genotoxicity tests (Kirkland et al. 2008, 2016). The compounds include 12 genotoxic carcinogens [benzo[a]pyrene (B[a]P), cyclophosphamide (CPA), 7,12-dimethylbenz[a]anthracene (DMBA), dimethylnitrosamine (DMNA), 2,4-diaminotoluene (2,4-DAT), cadmium chloride (CdCl2), cisplatin, colchicine, N-ethyl-N-nitrosourea (ENU), hydroquinone (HQ), methyl methanesulfonate (MMS), and 4-nitroquinoline 1-oxide (4-NQO)], 6 non-genotoxic carcinogens [amitrol, di-(2-ethylhexyl) phthalate (DEHP), diethanolamine, melamine, methyl carbamate, and progesterone], 5 genotoxic non-carcinogens [o-anthranilic acid, curcumin, ethionamide, 2-ethyl-1,3-hexanediol, and resorcinol], and 5 cytotoxic non-genotoxic non-carcinogens [ampicillin trihydrate, alosetron, D-mannitol, phenformin HCl, and sodium diclofenac]. All the 28 test compounds, along with other chemicals used for this study (phenacetin, bupropion, diclofenac, midazolam, and dextromethorphan) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The CometChip and other Comet supplies were obtained from Trevigen (Gaithersburg, MD, USA). Kits for performing the ATP, CellTiter-Blue cell viability assays, and cell proliferation (MTS) assay were obtained from Promega (Madison, WI, USA).
Cell culture
The HepaRG human hepatoma cell line was obtained from Biopredic International (Saint Grégoire, France) and cultured according to the supplier’s protocol. Briefly, the cells were seeded at a density of 1.3 × 104 cells/cm2 and cultured in William’s E medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 2 mM l-glutamine (Sigma-Aldrich) and growth additives (Lonza, Walkersville, MD, USA) for 14 days at 37 °C in a humidified atmosphere with 5% CO2. The cells were then differentiated by adding a differentiation supplement (Lonza) to the medium for an additional 14 days. The culture medium was changed every 2–3 days.
The HepG2 human hepatoma cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were cultured in William’s Medium E with l-glutamine (Sigma-Aldrich) supplemented with 2.2 g/l sodium bicarbonate (Sigma-Aldrich), 10% fetal bovine serum (FBS, Atlanta Biologicals, Flowery Branch, GA, USA), and 1% Antibiotic–Antimycotic supplement (Invitrogen, Carlsbad, CA, USA) at 37 °C in a humidified atmosphere with 5% CO2.
CYP activity measurement by LC–MS/MS
CYP450 activities in HepaRG and HepG2 cells were measured by the high-performance liquid chromatography–tandem mass spectrometry (LC–MS/MS) method as described by Dierks et al. with minor modifications (Dierks et al. 2001). Briefly, the cells were incubated for 2 h with 100 µl of medium without FBS but containing substrate cocktails of 100 µM phenacetin (CYP1A2), 100 µM bupropion (CYP2B6), 20 µM diclofenac (CYP2C9), 50 µM midazolam (CYP3A4), and 20 µM dextromethorphan (CYP2D6), at 37 °C in a humidified atmosphere with 5% CO2. At the end of the incubation, the medium was collected and 10 volumes of ice-cold acetonitrile were added to precipitate proteins. After centrifugation at 16,000g for 5 min, the supernatants were transferred to sample vials and 10 µl of the supernatants were injected onto a Shimadzu Prominence UFLC coupled with an AB SCIEX 3200 QTRAP mass spectrometer (SCIEX LLC, Framingham, MA). The analytes were eluted at a flow rate of 0.5 ml/min on a Waters Atlantis T3 C18 column (4.6 × 150 mm, 5 µm) at 40 °C using a gradient mobile phase containing water (Solvent A) and acetonitrile (Solvent B), both containing 0.1% formic acid. Elution started with 10% Solvent B for 0.2 min, followed by a linear gradient of 10–90% Solvent B for 9.8 min, returning to 10% Solvent B for 0.5 min; and the column was re-equilibrated for 4.5 min. The eluates were monitored by mass spectrometry using the positive electrospray mode (ESI+) and the multiple reaction monitoring (MRM) mode. The MRM transitions monitored for individual CYP metabolites were m/z 152.2–150.0 for 4-acetamidophenol (CYP1A2), m/z 256.2–130.2 for hydroxybupropion (CYP2B6), m/z 312.1–230.0 for 4-hydroxydiclofenac (CYP2C9), m/z 342.1–203.1 for 1-hydroxymidazolam (CYP3A4), and m/z 258.2–157.2 for dextrorphan (CYP2D6). For determining the protein content, the cells were lysed with RIPA buffer (Thermo Fisher Scientific) and the protein concentrations were measured using a Direct Detect® spectrometer (EMD Millipore, Burlington, MA, USA). Final CYP activities were expressed as pmol metabolite/min/mg protein.
Cell treatments
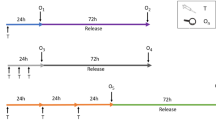
Figure 1 shows the experimental protocols used to treat the HepaRG cells (Fig. 1a) and HepG2 cells (Fig. 1b). Briefly, fully differentiated HepaRG cells (28 days after initial seeding) were replated at a density of 5 × 104 cells/well into a 96-well plate in William’s E differentiation medium and cultured for 3 days prior to the chemical treatment. Rapidly dividing HepG2 cells were seeded at a lower density of 2 × 104 cells/well into a 96-well plate and cultured for 24 h prior to the treatments. Dose range-finding tests were conducted to establish an appropriate concentration range for each chemical to avoid excessive cytotoxicity (approximately 80−100% cell viability). All chemicals were dissolved in DMSO (Sigma-Aldrich), except for CdCl2 and DMNA, which were dissolved in deionized water, and cisplatin, which was dissolved in 0.9% NaCl (Sigma-Aldrich). The 100 × chemical working solutions for each test chemical concentration were prepared fresh from stocks. The working solutions were diluted in cell culture medium to make the top primary concentrations, and serial dilution was performed to generate the lower treatment concentrations (Fig. 1c). The final concentration of DMSO in the medium never exceeded 1%. HepaRG and HepG2 cells were exposed to various concentrations of the test chemical in a total volume of 100 µl for 24 h at 37 °C in a humidified atmosphere with 5% CO2. The cytotoxicity and CometChip assays were performed following the treatment. The experiments were repeated independently at least three times for each chemical as described below.

CometChip assay study design. HepaRG cells (a) were cultured in complete growth medium for 14 days and then differentiated in complete differentiation medium for another 14 days. Differentiated cells were seeded at a density of 5 × 104 cells/well into a 96-well plate and maintained for 3 days prior to the chemical treatments. HepG2 cells (b) were cultured for 3 days, and then seeded at a density of 2 × 104 cells/well into a 96-well plate and incubated overnight prior to the treatments. HepaRG and HepG2 cells were treated with various chemicals using a consistent serial dilution method (c). Following a 24-h treatment, the CometChip and cytotoxicity assays were conducted in both cell lines
Cell viability, cell proliferation, and cellular ATP levels
Following a 24-h treatment, the cells were detached by adding 30 µl accutase (Sigma-Aldrich) at room temperature, resuspended in 250 µl 1 × PBS per well, and gently mixed by pipetting to produce a single cell suspension. Cell viability and cell proliferation (MTS) assays were measured as described previously using a CellTiter-Blue cell viability assay kit (Promega) and a CellTiter 96 aqueous one solution cell proliferation assay kit (Promega), respectively (Guo et al. 2018b). Briefly, the CellTiter-Blue or MTS reagents were added into each well at a ratio of 1:10 and were incubated for 1 h at 37 °C in a humidified atmosphere with 5% CO2. Fluorescence at 530EX/590EM (CellTiter-Blue) or absorbance at 490 nm (MTS assay) was measured with a Synergy H4 Hybrid multi-mode microplate reader (BioTek, Winooski, VT, USA). The relative cytotoxicity was calculated by comparing the intensity levels of the treated cells to that of the vehicle controls.
Cellular ATP levels were measured using a CellTiter-Glo luminescent cell viability assay kit (Promega). Cells in a white 96-well flat-bottomed plate were mixed with Cell-Titer-Glo reaction reagent at a ratio of 1:1 and the luminescence was recorded immediately with a Synergy H4 Hybrid multi-mode microplate reader (BioTek). The relative cellular ATP content was calculated by comparing the luminescence intensities of the treated cells to that of the vehicle controls.
CometChip assay
The CometChip assay was performed under alkaline conditions according to the manufacturer’s instructions (Trevigen) to detect single and double DNA strand breaks. Following the 24-h treatment, the cells were dislodged by trypsinization and 100 µl of treated cells were transferred into each well of a 96-well CometChip, with each well containing approximately 400–500 microwells. The cells were gravity loaded into the 30-micron sized microwells for 40 min at 37 °C in a humidified atmosphere with 5% CO2. Following cell loading, the CometChip was gently rinsed with 1 × PBS and sealed with 1% low melting point agarose in PBS (Thermo Fisher Scientific). The CometChip next was treated with lysis solution (Trevigen) for 1 h at 4 °C, and then submerged into a chilled alkaline buffer (0.2 M NaOH, 1 mM EDTA, 0.1% TritonX-100, pH > 13) for 40 min in the dark to unwind the DNA. Electrophoresis was performed at 22 V for 50 min at 4 °C in the same solution. After neutralization with 0.4 M Tris–HCl buffer (pH 7.4, Sigma–Aldrich) and equilibration with 0.02 M Tris–HCl buffer (pH 7.4), the CometChip was stained overnight at 4 °C with 0.2 × SYBR® Gold (Invitrogen) diluted in 0.02 M Tris–HCl buffer (pH 7.4) and then de-stained in 0.02 M Tris–HCl buffer (pH 7.4) for 1 h. The comet images were acquired using an imaging analysis system consisting of a Leica DMI4000 B fluorescence microscope and Leica application suite software. The percentage of DNA in tails was scored in more than 100 cells for each concentration using Trevigen Comet Analysis Software.
Benchmark dose analysis
The Comet dose–response data were analyzed by benchmark dose (BMD) analysis using PROAST software running in R (version 65.5, developed at the Netherlands National Institute for Public Health and the Environment, RIVM), and following the manual provided by the European Food Safety Authority (EFSA) (Hardy et al. 2017). BMDs were estimated without using covariant analysis. The BMD is the dose resulting in a predetermined change in the response calculated from mathematically modeled dose–response relationships. BMD5, BMD10, BMD50, and BMD100 were calculated based on 5%, 10%, 50%, or 100% (twofold) increases in response above the vehicle control, respectively. BMDL and BMDU values, the lower and upper bounds of the 95% confidence interval (CI) of the BMD, were also calculated for 5%, 10%, 50%, and 100% changes in response above the vehicle control. PROAST Software fits data to dose–response curves using two nested models, the exponential model and the Hill model. The model that had a better precision (i.e., the smaller BMDU/BMDL ratio) was chosen for making BMD estimates.
Statistical analysis
Data are presented as the mean ± standard deviation (SD) of measurements from at least three independent experiments. Statistical significance (p < 0.05) was determined by one-way analysis of variance (ANOVA) followed by Dunnett’s test for comparisons between different concentrations to the vehicle control using SigmaPlot 13.0 (Systat Software, San Jose, CA, USA).
Results
CYP activities in differentiated HepaRG cells and HepG2 cells
The activities of five major CYPs (CYP1A2, CYP2C9, CYP2D6, CYP2B6, and CYP3A4) involved in drug metabolism were monitored in two batches of differentiated HepaRG cells and HepG2 cells. Overall, HepaRG cells maintained much higher levels of CYP activities over a period of 28-day period as compared to CYP levels in HepG2 cells (Fig. 2). Under the present experimental conditions, CYP3A4 and CYP2C9 activities ranged from 22 to 58 pmol/min/mg protein in HepaRG cells. The activities of CYP1A2 and CYP2B6 ranged from 2.1 to 6.4 pmol/min/mg protein, and CYP2D6 had low but detectable activity (< 3 pmol/min/mg protein) in HepaRG cells. HepaRG cells showed stable or slightly enhanced activities for the five CYPs over an extended 28-day period in culture. In contrast to their activities in HepaRG cells, the activities of all five CYPs in HepG2 cells were undetectable or less than 1 pmol/min/mg protein.

CYP activity measured in HepG2 and differentiated HepaRG cells. Five major CYP450 activities were measured in differentiated HepaRG cells on Days 1, 2, 3, 5, 7, 14, 21, and 28 and in HepG2 cells on Day 1 after seeding into a 96-well plate, respectively. The cells were incubated for 2 h with William’s E medium without fetal bovine serum but containing enzyme substrate cocktails. Metabolites released into the medium were quantified by LC–MS/MS. CYP activities are expressed as pmol/min/mg protein and the results are presented as the mean ± SD of triplicate incubations
Cytotoxicity profiles of the 28 tested compounds in HepaRG cells and HepG2 cells
Cytotoxicity was evaluated following a 24-h exposure to each of the 28 tested compounds using assays measuring metabolically active cells (ATP assay), cell viability (CellTiter-Blue assay), and cell proliferation (MTS assay). When no cytotoxicity was observed with any of the three assays, the highest test concentration used for subsequent testing was 10 mM as recommended by the OECD guidance (OECD 2015).
Overall, the 28 compounds were more cytotoxic in HepaRG cells than HepG2 cells (Table 1). The five genotoxic carcinogens that require metabolic activation (2,4-DAT, B[a]P, CPA, DMNA, and DMBA) induced cytotoxicity of 12−40% in HepaRG cells, whereas three of them, CPA, DMNA, and DMBA, showed little or no cytotoxicity in HepG2 cells (Table 1; Fig. 3). The cytotoxicity of the remaining two compounds in this class, 2,4-DAT and B[a]P, was similar in HepaRG and HepG2 cells. The six direct-acting genotoxic carcinogens also induced similar cytotoxic effects in the two cell lines, except for 4-NQO and colchicine, which were more cytotoxic in HepaRG cells than in HepG2 cells (Table 1; Fig. 4). It is worth noting that although colchicine-induced cytotoxicity was not detected in HepG2 cells with the three assays, remarkable morphological changes indicative of cytotoxicity were observed in HepG2 cells treated with this test article (Supplementary Fig. 1).

Comparison of cytotoxicity and genotoxicity of indirect-acting genotoxic carcinogens in HepaRG and HepG2 cells. HepaRG (a) and HepG2 (b) cells were exposed to various concentrations of the indirect-acting genotoxic carcinogens (2.4-DAT, B[a]P, CPA, DMBA, and DMNA) for 24 h. DNA damage (% DNA tail intensity, black bar) was detected using the CometChip assay and the relative cell viability (% of control) was measured using the ATP, CellTilter-Blue, and MTS assays. The data are expressed as the mean ± SD (n ≥ 3). Significant differences between data from the treated cells and the corresponding control were determined by one-way ANOVA followed by Dunnett’s test (*P < 0.05, **P < 0.01, and ***P < 0.001). 2.4-DAT 2,4-diaminotoluene, B[a]P benzo[a]pyrene, CPA cyclophosphamide, DMBA 7,12-dimethylbenzanthracene, DMNA dimethylnitrosamine

Comparison of cytotoxicity and genotoxicity of direct-acting genotoxic carcinogens in HepaRG and HepG2 cells. HepaRG (a) and HepG2 (b) cells were exposed to various concentrations of the direct-acting genotoxic carcinogens (4-NQO, CdCl2, cisplatin, colchicine, ENU, HQ, and MMS) for 24 h. DNA damage (% DNA tail intensity, black bar) was detected using the CometChip assay and the relative cell viability (% of control) was measured using the ATP, CellTiter-Blue, and MTS assays. The data are expressed as the mean ± SD (n ≥ 3). Significant difference between data from the treated cells and the corresponding control were determined by one-way ANOVA followed by Dunnett’s test (*P < 0.05, **P < 0.01, and ***P < 0.001). 4-NQO 4-nitroquinoline 1-oxide, CdCl2 cadmium chloride, ENU N-ethyl-N-nitrosourea, HQ hydroquinone, MMS methyl methanesulfonate
All the 16 compounds in the non-genotoxic carcinogen, genotoxic non-carcinogen, and cytotoxic non-genotoxic non-carcinogen categories had little or no cytotoxicity in HepG2 cells at the highest tested concentration of 10 mM. In contrast to their weak cytotoxicity in HepG2 cells, all five genotoxic non-carcinogens and two out of six non-genotoxic carcinogens (melamine and progesterone) induced significant cytotoxicity in HepaRG cells, with values ranging from 17–55% in the three cytotoxicity assays; while two out of five cytotoxic non-genotoxic non-carcinogens (phenformin HCl, and sodium diclofenac) induced 20–58% cytotoxicity at various concentrations in HepaRG cells (Table 1 and Supplementary Fig. 2).
DNA damage profiles of the 28 tested compounds in HepaRG cells and HepG2 cells
The highest concentration used for each compound in the CometChip assay was either 10 mM for compounds displaying no cytotoxicity or the maximum concentration reducing viability no less than 70% in any of the three cytotoxicity assays that we conducted (ATP, CellTiter-Blue, and MTS assays). For the five carcinogens requiring metabolic activation (i.e., indirect-acting carcinogens), positive responses were observed for four test compounds in HepaRG cells and for two test compounds in HepG2 cells, thus revealing a sensitivity of 80% in HepaRG vs. 40% in HepG2 cells (Fig. 3; Table 2). Specifically, CPA, DMBA, and DMNA induced concentration-dependent increases in % tail DNA only in HepaRG cells (2.0-5.1-fold increase over the vehicle control), while the DNA damage produced by B[a]P was detected equally well in HepaRG and HepG2 cells (2.2- and 2.0-fold increase over control, respectively). In contrast, 2,4-DAT induced a weak but significant increase in DNA damage (1.8-fold at a concentration of 8 mM) only in HepG2 cells.
For the seven direct-acting genotoxic carcinogens or probable carcinogens tested, five had the same response calls in the two cell lines (Fig. 4; Table 2). Three compounds, 4-NQO, ENU, and MMS, induced significant DNA damage in both cell lines, i.e., 1.6-, 3.0-, and 16.2-fold increases in HepaRG and 3.4-, 2.3-, and 7.0-fold increases in HepG2 cells. No significant DNA damage was observed for colchicine or HQ in either cell line. Two compounds were positive in only one cell line, but the positive responses were very weak. CdCl2 induced a statistically significant increase (1.9-fold) in DNA damage only in HepG2 cells, while cisplatin was positive only in HepaRG cells (1.9-fold increase).
Sixteen compounds, including six non-genotoxic carcinogens, five genotoxic non-carcinogens, and five cytotoxic non-genotoxic non-carcinogens (Table 1) were negative for DNA breakage detected in the Comet assay in HepaRG cells (Supplementary Fig. 3).
Quantification of DNA damage potencies using benchmark dose (BMD) analysis
Quantitative analysis was performed on the compounds producing positive responses for DNA damage in the CometChip assays conducted with either HepaRG cells or HepG2 cells. BMD5, BMD10, BMD50, and BMD100 were calculated for each positive response, corresponding to 5%, 10%, 50%, or 100% increases in % tail DNA over the vehicle control, respectively (Table 3). For the eight positive responses in HepaRG cells, the exponential model was used for the 4-NQO, DMBA, MMS data, and the Hill model was used for the B[a]P, CdCl2, cisplatin, CPA, DMNA data. When compared by their BMD10s, the eight responses could be divided into three groups. Three compounds (4-NQO, B[a]P, and cisplatin) had the BMD10 values of < 0.6 µM; four compounds (CPA, DMBA, DMNA, and ENU) had values of between 2 and 4 µM; while MMS had the greatest BMD10 value, > 8 µM.
For the six positive responses in HepG2 cells, the exponential model was used for the MMS, ENU, 4-NQO and 2,4-DAT data, while the Hill model was used for data generated by treatments with B[a]P and CdCl2. Three compounds (4-NQO, B[a]P, and CdCl2) had BMD10 values of < 0.5 µM; two compounds (ENU and MMS) had moderate BMD10 values of between 2 and 5 µM; while 2,4-DAT had the greatest BMD10 value, 746 µM (Table 3).
The lowest effect concentration (LEC) for each positive response also was determined using one-way ANOVA, followed by pairwise comparison (Table 3). The LEC values for most of the compounds fell into a concentration range between the BMD50 and BMD100 values for both HepaRG and HepG2 cell lines. The exceptions were for DMNA and MMS, which had LEC values higher than their corresponding BMD100. For the four carcinogens that were positive in both cell lines, the calculated BMDs for B[a]P, ENU, and MMS in HepG2 cells were similar or slightly lower than those in HepaRG cells, and their 95% upper and lower BMD CIs for the responses in the two cell lines overlapped each other. The BMDs calculated for 4-NQO in the two cell lines were also similar, with one exception, the BMD100. The BMD100 for 4-NQO in HepaRG cells were two times higher than that in HepG2 cells, and the CIs for the two BMDs did not overlap (Table 3; Fig. 5).

Comparison of BMD values and their confidence intervals for chemical-induced DNA damage in HepaRG and HepG2 cells. The BMDs (BMD5, BMD10, BMD50, and BMD100) estimates producing a 5%, 10%, 50%, or 100% increase above the background responses were calculated by PROAST using the dose–response data from assays conducted with MMS, ENU, B[a]P, and 4-NQO. The bar represents the calculated lower and upper 95% confidence interval for each value (BMDU/BMDL), reflecting the uncertainty in the BMD estimates
Discussion
In the landmark report Toxicity Testing in the twenty-first Century: A Vision and a Strategy, the U.S. National Academy of Sciences envisioned using a series of human-cell-based, high- or medium-throughput pathway-based in vitro assays covering a broad dose range to yield quantitative dose–response data for performing toxicological evaluations (Krewski et al. 2010). Accordingly, the present study assessed the usefulness of DNA damage data generated from metabolically competent HepaRG cells and HepG2 cells using HTHC CometChip technology for quantitative genotoxicity evaluation. We first confirmed the stable high expression of CYP enzymes in HepaRG cells relative to HepG2 cells by measuring the activities of five primary CYPs over an extended 28-day culture period. Next, we evaluated the chemical-induced cytotoxicity in both cell lines, and minimized false-positive responses for subsequent CometChip assays by setting a cell viability cutoff value of ≥ 70%. Overall, a greater cytotoxic effect for approximately two-thirds of the tested chemicals was observed in HepaRG cells than HepG2 cells, regardless whether the chemical required metabolic activation or not (Table 1). Differentiated HepaRG cells contain two morphologically distinct populations: hepatocyte-like and biliary epithelial-like cells, and thus are a unique model that mimic the in vivo environment for drug metabolism (Cerec et al. 2007). Because HepaRG cells possess considerably high levels of CYP activity, we anticipated that they would be more sensitive than HepG2 cells to DNA damage induced by genotoxic carcinogens that require metabolic activation.
The expectation about metabolically competent HepaRG cells was confirmed. HepaRG cells were much more sensitive in assessing the DNA damage effects for carcinogens that require metabolic activation than were HepG2 cells (Table 2; Fig. 3). CPA, DMBA, and DMNA showed a significant induction of DNA damage following a 24-h treatment only in HepaRG cells. This observation is in agreement with results from previous studies in a traditional Comet assay where three compounds induced increased % DNA in tail with both HepaRG cells and HepG2 cells with S9, but not with HepG2 cells without S9 (Hong et al. 2018; Le Hegarat et al. 2010, 2014). CPA and DMNA require CYP2B6 and CYP2E1, respectively, for metabolic activation, which ultimately leads to DNA damage in the form of O6-and/or N7-alkylation of the guanine (Khoury et al. 2013; Kirkland et al. 2016; Le Hegarat et al. 2010). CYP1A1 and CYP1B1 are required for DMBA to produce the DMBA-3,4-diol-1,2-epoxide metabolite that generates DNA adducts (Quesnot et al. 2016). It was not surprising that CPA, DMBA, and DMNA were negative for both cytotoxicity and DNA damaging effects in HepG2 cells due to the low activities of CYP enzymes in these cells. B[a]P induced statistically significant DNA damage both in HepaRG and HepG2 cells in our study, with LECs of 20 µM and 32 µM, respectively (Table 3). These results are consistent with other studies in HepaRG cells showing that 24-h treatment with 50–250 µM B[a]P significantly increased DNA fragmentation (Le Hegarat et al. 2010), and increased comet tails % DNA and γH2AX foci formation in HepG2 cells in a dose- and time-dependent manner (Hong et al. 2018; Khoury et al. 2016; Uhl et al. 1999; Valentin-Severin et al. 2003). Activation by CYP1A1, CYP1B1 and/or CYP3A4 is required for the B[a]P to form B[a]P-7,8-diol-9,10-epoxide intermediate producing DNA adducts (Valentin-Severin et al. 2003). The observation that B[a]P produced similar responses in terms of cytotoxicity, % DNA in tail, as well as BMDs in HepaRG and HepG2 cells (Tables 1, 2, 3; Fig. 3) indicate that the lower CYP levels in HepG2 cells are not a limiting factor for the metabolism of B[a]P to DNA damaging species (Fig. 2).
2,4-DAT, a hepatocarcinogenic aromatic amine, increased the percentage of tail DNA only at the highest concentration tested (8 mM) in HepG2 cells but not in HepaRG cells. Similarly, 2,4-DAT induced comet formation and γH2AX phosphorylation in HepG2 cells (Khoury et al. 2016; Severin et al. 2005), but has been negative response in HepaRG cells in previous studies (Le Hegarat et al. 2014). 2,4-DAT genotoxicity was linked to CYP1A2 bioactivation along with metabolism by Phase II enzymes, sulfotransferases (SULTs) or N-acetyltransferases (NATs), to form DNA-reactive metabolites (Cheung et al. 1996; Le Hegarat et al. 2014). The positive response for 2,4-DAT in HepG2 cells might be explained by higher SULT1A1 gene expression and higher SULT1A3/1A4 and NAT10 protein expression in HepG2 cells compared with HepaRG cells or PHH (Jennen et al. 2010; Sison-Young et al. 2015). In addition, 2,4-DAT was mutagenic in L5178Y cells only in the absence of exogenous bioactivation (rat liver S9 fraction) (Coppinger et al. 1984). Khoury and colleagues speculated that the positive response for 2,4-DAT in HepG2 cells may result from an oxidative stress-driven genotoxic MoA rather than DNA adduct formation (Khoury et al. 2016).
Among the seven direct-acting genotoxic compounds that were tested, 4-NQO, ENU, and MMS are highly mutagenic direct-acting clastogens and DNA alkylating agents (Kawaguchi et al. 2010; Khoury et al. 2016; Kirkland et al. 2016). The three compounds induced DNA damage in both HepaRG and HepG2 cells in the HTHC CometChip assay, a result consistent with the findings of other studies (Hong et al. 2018; Josse et al. 2012; Khoury et al. 2016; Le Hegarat et al. 2014; Valentin-Severin et al. 2003). However, differences in responses were observed between the two cell lines. For example, 4-NQO was more cytotoxic in HepaRG cells than HepG2 cells, but more genotoxic in HepG2 cells than HepaRG cells in the present study, and the % DNA in tail induced by MMS was twofold higher in HepaRG cells than in HepG2 cells. The mechanisms for the differences warrant further investigation. In fact, the amount of MMS-induced DNA damage was reduced in HepG2 cells after 24-h treatment as compared to a 4-h treatment (data not shown). The base excision repair pathway may account for the reduced DNA strand breaks by removing few oligonucleotides containing the MMS-induced damage during the 24-h treatment time (Hong et al. 2018). As different exposure periods can cause different responses to DNA damage and repair signaling for various chemicals, sampling time is a critical component to consider when performing the Comet assay (Sasaki et al. 2007).
The two known aneugens, colchicine and HQ, are positive in various genotoxicity tests, including in vitro and in vivo chromosome aberration and micronucleus tests, but have given negative responses in the in vitro and in vivo Comet assay (Hong et al. 2018; Josse et al. 2012; Kirkland et al. 2016). That was also the case in our study: colchicine and HQ were cytotoxic but did not produce DNA damage in either HepaRG or HepG2 cells in the CometChip assay (Fig. 3). Cisplatin, a cross-linking agent, has been reported to increase comet tails and H2AX phosphorylation (γH2AX) in HepG2 cells (Hong et al. 2018; Khoury et al. 2016). However, in our study, cisplatin-induced DNA damage was significantly increased only in HepaRG cells—not in HepG2 cells. CdCl2, an inorganic carcinogen, induced DNA damage in HepG2 cells, whereas no DNA damage was detected in HepaRG cells, a finding that was in agreement with previous studies (Le Hegarat et al. 2014; Skipper et al. 2016). Nucleotide excision repair and/or base excision repair and mismatch repair are important DNA repair pathways responsible for cisplatin- and CdCl2-induced DNA damage (Hartwig 2010; Zhang et al. 2010). As cells were harvested following a 24-h treatment in our study, any differences in the DNA repair capacities of HepaRG and HepG2 cells may be responsible for the different DNA damage responses in the two cell lines. In addition, oxidative stress plays a key role in CdCl2-induced DNA damage (Skipper et al. 2016). The high level of glutathione in HepaRG cells may attenuate the CdCl2-induced DNA damaging effects in these cells (Badisa et al. 2007; Xu et al. 2018).
To summarize these findings, the CometChip assay conducted with HepaRG cells had an 80% sensitivity for detecting genotoxic carcinogens requiring metabolic activation, and a 57% sensitivity for direct-acting genotoxic carcinogens. HepG2 cells had a sensitivity of 40% for carcinogens requiring metabolic activation and 57% for direct-acting genotoxic carcinogens. The overall sensitivities of HepaRG cells and HepG2 cells for detecting the 12 genotoxic carcinogens tested in the CometChip assay were 67% and 50%, respectively.
The five genotoxic non-carcinogens tested in our study (Table 1) are reported to be positive in one or more in vitro genotoxicity assays at high concentrations, but have negative or equivocal genotoxicity in vivo or have no available in vivo data (Kirkland et al. 2008). All five compounds, along with 11 non-genotoxic carcinogens or non-genotoxic non-carcinogens, had negative responses in the CometChip assay conducted with HepaRG cells, indicating a specificity of 100% (these compounds were not tested in the CometChip assay with HepG2 cells). In our study of 28 compounds, the HepaRG CometChip assay was positive only for compounds that were both carcinogens and genotoxicants.
Quantitative dose–response analysis was employed using the PROAST software to calculate the BMDs for the dose–responses generated from the HTHC CometChip assay. Taking advantage of the high throughput feature of the CometChip assay, we were able to test 20−22 closely spaced concentrations for each compound in an effort to improve the BMD precision by testing a large number of concentrations (Wills et al. 2016). By performing BMD analysis for the 14 dose–responses, and systemically removing one or more data points from the assays, we found that the number of concentrations giving the optimal BMD precision (as determined by the size of the BMD CIs) was in the range of 12–15, and that the shape of the dose–response curve affected the optimum number of doses. For compounds producing large responses over the background (i.e., > fivefold), increasing the number of low concentrations tested improves precision by decreasing the CIs for the BMD5 and BMD10. Significant responses that are less than threefold increases over the control usually produce large BMDU CIs, as was the case for ENU and B[a]P (Fig. 4). With the exception of the responses generated by DMNA and MMS, the LECs for all the positive responses fell between the BMD50 and BMD100. The LECs for DMNA and MMS were much higher than their corresponding BMD100. These results suggest that most of the positive responses began at concentrations producing 1.5–twofold or higher increases over the control, and that the BMD approach is often more sensitive than the LEC approach in determining the PoDs. In addition, BMD values generated from four carcinogens (4-NQO, B[a]P, ENU, and MMS) that were positive in both HepaRG and HepG2 cells had overlapping upper and lower BMD CIs, indicating that data generated from both cell types are equally useful for deriving PoDs, at least for these genotoxicants.
In conclusion, our results suggest that metabolically competent HepaRG cells and HepG2 cells can be adapted to the in vitro CometChip assay for quantifying DNA damage over a wide range of concentrations. HepaRG cells generally were superior to HepG2 cells in terms of detecting chemical-induced cytotoxicity and genotoxicity, especially for detecting the effects of indirect-acting genotoxic carcinogens. The HepaRG CometChip assay showed an overall sensitivity of 80% for genotoxic carcinogens requiring metabolic activation. Notably, the HepaRG CometChip assay had a specificity of 100% for identifying potential genotoxic carcinogens, as none of the 16 nongenotoxic compounds and genotoxic non-carcinogens tested positive in the assay. However, further studies are required to explore how the responses in HepaRG and HepG2 cells compare to those in PHHs and whether the sensitivity of the assay can be improved, especially by altering the treatment and/or sampling time. Our study also demonstrated improvement in the precision of BMD analysis by increasing the number of chemical concentrations that are tested. The integration of the HTHC HepaRG genotoxicity assay with quantitative analysis is a promising approach for evaluating human in vivo hazard by providing data in a scientifically rigorous manner.
References
Antherieu S, Chesne C, Li R et al (2010) Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab Dispos 38(3):516–525
Ates G, Mertens B, Heymans A et al (2018) A novel genotoxin-specific qPCR array based on the metabolically competent human HepaRG cell line as a rapid and reliable tool for improved in vitro hazard assessment. Arch Toxicol 92(4):1593–1608
Badisa VL, Latinwo LM, Odewumi CO et al (2007) Mechanism of DNA damage by cadmium and interplay of antioxidant enzymes and agents. Environ Toxicol 22(2):144–151
Bryce SM, Bernacki DT, Bemis JC, Dertinger SD (2016) Genotoxic mode of action predictions from a multiplexed flow cytometric assay and a machine learning approach. Environ Mol Mutagen 57(3):171–189
Cerec V, Glaise D, Garnier D et al (2007) Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology 45(4):957–967
Cheung YL, Snelling J, Mohammed NN, Gray TJ, Ioannides C (1996) Interaction with the aromatic hydrocarbon receptor, CYP1A induction, and mutagenicity of a series of diaminotoluenes: implications for their carcinogenicity. Toxicol Appl Pharmacol 139(1):203–211
Coppinger WJ, Brennan SA, Carver JH, Thompson ED (1984) Locus specificity of mutagenicity of 2,4-diaminotoluene in both L5178Y mouse lymphoma and AT3-2 Chinese hamster ovary cells. Mutat Res 135(2):115–123
Dierks EA, Stams KR, Lim HK, Cornelius G, Zhang H, Ball SE (2001) A method for the simultaneous evaluation of the activities of seven major human drug-metabolizing cytochrome P450s using an in vitro cocktail of probe substrates and fast gradient liquid chromatography tandem mass spectrometry. Drug Metab Dispos 29(1):23–29
Ge J, Chow DN, Fessler JL, Weingeist DM, Wood DK, Engelward BP (2015) Micropatterned comet assay enables high throughput and sensitive DNA damage quantification. Mutagenesis 30(1):11–19
Gollapudi BB, Johnson GE, Hernandez LG et al (2013) Quantitative approaches for assessing dose–response relationships in genetic toxicology studies. Environ Mol Mutagen 54(1):8–18
Guillouzo A, Corlu A, Aninat C, Glaise D, Morel F, Guguen-Guillouzo C (2007) The human hepatoma HepaRG cells: a highly differentiated model for studies of liver metabolism and toxicity of xenobiotics. Chem Biol Interact 168(1):66–73
Guo X, Heflich RH, Dial SL, Richter PA, Moore MM, Mei N (2015) Quantitative analysis of the relative mutagenicity of five chemical constituents of tobacco smoke in the mouse lymphoma assay. Mutagenesis 31(3):287–296
Guo X, Heflich RH, Dial SL, De M, Richter PA, Mei N (2018a) Quantitative differentiation of whole smoke solution-induced mutagenicity in the mouse lymphoma assay. Environ Mol Mutagen 59(2):103–113
Guo X, Seo JE, Bryce SM et al (2018b) Comparative genotoxicity of TEMPO and three of its derivatives in mouse lymphoma cells. Toxicol Sci 163(1):214–225
Hardy A, Benford D, Halldorsson T et al (2017) Update: use of the benchmark dose approach in risk assessment. EFSA Journal 15(1):4658
Hartwig A (2010) Mechanisms in cadmium-induced carcinogenicity: recent insights. Biometals 23(5):951–960
Hong YH, Jeon HL, Ko KY et al (2018) Assessment of the predictive capacity of the optimized in vitro comet assay using HepG2 cells. Mutat Res 827:59–67
ICH (2011) Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use S2(R1). ICH Expert Working Group. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S2_R1/Step4/S2R1_Step4.pdf. Accessed 12 Nov 2018
Jennen DG, Magkoufopoulou C, Ketelslegers HB, van Herwijnen MH, Kleinjans JC, van Delft JH (2010) Comparison of HepG2 and HepaRG by whole-genome gene expression analysis for the purpose of chemical hazard identification. Toxicol Sci 115(1):66–79
Josse R, Rogue A, Lorge E, Guillouzo A (2012) An adaptation of the human HepaRG cells to the in vitro micronucleus assay. Mutagenesis 27(3):295–304
Kawaguchi S, Nakamura T, Yamamoto A, Honda G, Sasaki YF (2010) Is the comet assay a sensitive procedure for detecting genotoxicity? J Nucleic Acids 2010:541050
Khoury L, Zalko D, Audebert M (2013) Validation of high-throughput genotoxicity assay screening using gammaH2AX in-cell western assay on HepG2 cells. Environ Mol Mutagen 54(9):737–746
Khoury L, Zalko D, Audebert M (2016) Evaluation of four human cell lines with distinct biotransformation properties for genotoxic screening. Mutagenesis 31(1):83–96
Kirkland D, Aardema M, Henderson L, Muller L (2005) Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens I. Sensitivity, specificity and relative predictivity. Mutat Res 584(1–2):1–256
Kirkland D, Kasper P, Muller L, Corvi R, Speit G (2008) Recommended lists of genotoxic and non-genotoxic chemicals for assessment of the performance of new or improved genotoxicity tests: a follow-up to an ECVAM workshop. Mutat Res 653(1–2):99–108
Kirkland D, Kasper P, Martus HJ et al (2016) Updated recommended lists of genotoxic and non-genotoxic chemicals for assessment of the performance of new or improved genotoxicity tests. M utat Res Genet Toxicol Environ Mutagen 795:7–30
Krewski D, Acosta D Jr, Andersen M et al (2010) Toxicity testing in the 21st century: a vision and a strategy. J Toxicol Environ Health B Crit Rev 13(2–4):51–138
Le Hegarat L, Dumont J, Josse R et al (2010) Assessment of the genotoxic potential of indirect chemical mutagens in HepaRG cells by the comet and the cytokinesis-block micronucleus assays. Mutagenesis 25(6):555–560
Le Hegarat L, Mourot A, Huet S et al (2014) Performance of comet and micronucleus assays in metabolic competent HepaRG cells to predict in vivo genotoxicity. Toxicol Sci 138(2):300–309
Luch A (2005) Nature and nurture—lessons from chemical carcinogenesis. Nat Rev Cancer 5(2):113–125
MacGregor JT, Frotschl R, White PA et al (2015) IWGT report on quantitative approaches to genotoxicity risk assessment I. Methods and metrics for defining exposure–response relationships and points of departure (PoDs). Mutat Res Genet Toxicol Environ Mutagen 783:55–65
Marrone AK, Tryndyak V, Beland FA, Pogribny IP (2016) MicroRNA responses to the genotoxic carcinogens aflatoxin B1 and benzo[a]pyrene in human HepaRG cells. Toxicol Sci 149(2):496–502
OECD (2015) Guidance document on revisions to OECD genetic toxicology test guidelines. OECD Workgroup of National Coordinators for Test 42 Guidelines (WNT) https://www.oecd.org/chemicalsafety/testing/Genetic%20Toxicology%20Guidance%20Document%20Aug%2031%202015.pdf. Accessed 12 Nov 2018
Quesnot N, Rondel K, Audebert M et al (2016) Evaluation of genotoxicity using automated detection of gammaH2AX in metabolically competent HepaRG cells. Mutagenesis 31(1):43–50
Robison TW, Jacobs A (2009) Metabolites in safety testing. Bioanalysis 1(7):1193–1200
Sand S, Parham F, Portier CJ, Tice RR, Krewski D (2017) Comparison of points of departure for health risk assessment based on high-throughput screening data. Environ Health Perspect 125(4):623–633
Sasaki YF, Nakamura T, Kawaguchi S (2007) What is better experimental design for in vitro comet assay to detect chemical genotoxicity. AATEX 14:499–504
Severin I, Jondeau A, Dahbi L, Chagnon MC (2005) 2,4-Diaminotoluene (2,4-DAT)-induced DNA damage, DNA repair and micronucleus formation in the human hepatoma cell line HepG2. Toxicology 213(1–2):138–146
Sison-Young RL, Mitsa D, Jenkins RE et al (2015) Comparative proteomic characterization of 4 human liver-derived single cell culture models reveals significant variation in the capacity for drug disposition, bioactivation, and detoxication. Toxicol Sci 147(2):412–424
Skipper A, Sims JN, Yedjou CG, Tchounwou PB (2016) Cadmium chloride induces DNA damage and apoptosis of human liver carcinoma cells via oxidative stress. Int J Environ Res Public Health 13(1):88
Sykora P, Witt KL, Revanna P et al (2018) Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci Rep 8(1):2771
Tryndyak V, Kindrat I, Dreval K, Churchwell MI, Beland FA, Pogribny IP (2018) Effect of aflatoxin B1, benzo[a]pyrene, and methapyrilene on transcriptomic and epigenetic alterations in human liver HepaRG cells. Food Chem Toxicol 121:214–223
Uhl M, Helma C, Knasmuller S (1999) Single-cell gel electrophoresis assays with human-derived hepatoma (HepG2) cells. Mutat Res 441(2):215–224
Valentin-Severin I, Le Hegarat L, Lhuguenot JC, Le Bon AM, Chagnon MC (2003) Use of HepG2 cell line for direct or indirect mutagens screening: comparative investigation between comet and micronucleus assays. Mutat Res 536(1–2):79–90
VICH (2013) VICH topic GL23(R): Studies to evaluate the safety of residues of veterinary drugs in human food: Genotoxicity testing. https://www.ema.europa.eu/documents/scientific-guideline/international-cooperation-harmonisation-technical-requirements-registration-veterinary-medicinal_en-2.pdf. Accessed 12 Nov 2018
Westerink WM, Schoonen WG (2007) Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol In Vitro 21(8):1581–1591
Wills JW, Long AS, Johnson GE et al (2016) Empirical analysis of BMD metrics in genetic toxicology part II: in vivo potency comparisons to promote reductions in the use of experimental animals for genetic toxicity assessment. Mutagenesis 31(3):265–275
Wood DK, Weingeist DM, Bhatia SN, Engelward BP (2010) Single cell trapping and DNA damage analysis using microwell arrays. Proc Natl Acad Sci USA 107(22):10008–10013
Xu J, Oda S, Yokoi T (2018) Cell-based assay using glutathione-depleted HepaRG and HepG2 human liver cells for predicting drug-induced liver injury. Toxicol In Vitro 48:286–301
Zeilinger K, Freyer N, Damm G, Seehofer D, Knospel F (2016) Cell sources for in vitro human liver cell culture models. Exp Biol Med (Maywood) 241(15):1684–1698
Zeller A, Duran-Pacheco G, Guerard M (2017) An appraisal of critical effect sizes for the benchmark dose approach to assess dose–response relationships in genetic toxicology. Arch Toxicol 91(12):3799–3807
Zhang R, Niu Y, Zhou Y (2010) Increase the cisplatin cytotoxicity and cisplatin-induced DNA damage in HepG2 cells by XRCC1 abrogation related mechanisms. Toxicol Lett 192(2):108–114
Acknowledgements
J.E.S. and K.D. were supported by appointments to the Postgraduate Research Program at the National Center for Toxicological Research (NCTR) administered by the Oak Ridge Institute for Science Education through an interagency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration (FDA). We greatly appreciate Dr. Lei Guo (DBT/NCTR) for generously providing HepG2 cells and thank Drs. Robert H. Heflich, Dayton Petibone, and Elvis-Yane Cuevas-Martinez for their critical review of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There was no conflict of interest declared.
Disclaimer
The information in this paper is not a formal dissemination of information by the U.S. FDA and does not represent the agency position or policy.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Seo, JE., Tryndyak, V., Wu, Q. et al. Quantitative comparison of in vitro genotoxicity between metabolically competent HepaRG cells and HepG2 cells using the high-throughput high-content CometChip assay. Arch Toxicol 93, 1433–1448 (2019). https://doi.org/10.1007/s00204-019-02406-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-019-02406-9