
Abstract
Osteosarcoma chemotherapy is often limited by chemoresistance, resulting in poor prognosis. Combined chemotherapy could, therefore, be used to prevent resistance to chemotherapeutics. Here, the effects of fisetin on osteosarcoma cells were investigated, as well as cytostatic potential in combination with the anti-cancer drug etoposide. For this, different osteosarcoma cell lines were treated with fisetin, with etoposide and with respective combinations. Fisetin was associated with decrease in colony formation in Saos-2 and in U2OS cells but not in MG-63 cells. Notwithstanding, upon evaluation of cellular growth by crystal violet assay, MG-63 and Saos-2 cells showed decreased cell proliferation at 40 and 20 µM fisetin, respectively. Depending on the relative concentrations, fisetin:etoposide combinations showed negative-to-positive interactions on the inhibition of cell proliferation. In addition, fisetin treatment up to 50 µM for 48 h resulted in G2-phase cell cycle arrest. Regardless of the combination, fisetin:etoposide increased % cells in G2-phase and decreased % cells in G1-phase. In addition, mixtures with more positive combined effects induced increased % cells in S-phase. Compared to etoposide treatment, these combinations resulted in decreased levels of cyclins B1 and E1, pointing to the role of these regulators in fisetin-induced cell cycle arrest. In conclusion, these results show that the combination of fisetin with etoposide has higher anti-proliferative effects in osteosarcoma associated with cell cycle arrest, allowing the use of lower doses of the chemotherapeutic agent, which has important implications for osteosarcoma treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteosarcoma is the most common primary bone malignancy and is characterized by histologic heterogeneity and high genetic instability (Luetke et al. 2014). Despite the drastic improvement in survival rates introduced by optimized chemotherapy programs, over the past decades, few advances have been made for patients with poor response to the conventional therapies (Anderson 2016). Typically, osteosarcoma chemotherapy involves agents such as doxorubicin, cisplatin, cyclophosphamide, methotrexate, and etoposide, which are commonly used alone or in combination. Nonetheless, chemotherapy can be toxic to healthy tissues or organs, and induce secondary malignancies. Moreover, chemoresistance is still a limitation to therapy (Luetke et al. 2014).
A diet rich in plant polyphenols has been documented to protect against several diseases, e.g., cardiovascular, neurodegenerative and gastrointestinal disorders, and cancer (Gomes et al. 2008; González-Gallego et al. 2010; Pandey and Rizvi 2009). Bone health maintenance is the consequence of a dynamic equilibrium between bone formation and degradation. Regarding this, polyphenols have been documented to show positive effects on the interaction between osteoblast and osteoclast activities, as reviewed (Horcajada and Offord 2012; Shen et al. 2012). In a collagen-induced arthritis model, the flavonoid fisetin (3,3′,4′,7-tetrahydroxyflavone) was found to decrease the incidence and severity of arthritis (Lee et al. 2009). Moreover, fisetin was reported to modulate bone mineral density, micro-architecture parameters, and bone markers, by repressing RANKL-induced osteoclast differentiation/activity and by stimulating osteoblast differentiation (Choi et al. 2012; Kim et al. 2014; Léotoing et al. 2013, 2014; Sakai et al. 2013). Regarding its anti-cancer properties, fisetin has been described as an anti-inflammatory agent inhibitor of NF-κB activation (Liao et al. 2009; Sung et al. 2007) and as an apoptosis inducer in multiple human cancer cell lines, e.g., (Khan et al. 2008; Murtaza et al. 2009; Suh et al. 2009; Ying et al. 2012). In addition, apoptosis induction by fisetin has been linked to p53-dependent pathways, e.g., (Chen et al. 2002; Li et al. 2011; Lim and Park 2009). Concerning osteosarcoma, fisetin was previously reported as inducer of apoptosis in the wild-type p53 U2OS cell line (Li et al. 2015).
In response to cell damage, p53 can induce cell cycle arrest and cell death commitment. Previously, fisetin treatment resulted in decreased proliferation and apoptosis in the wt p53 and wt Rb U2OS cell line (Li et al. 2015; Ying et al. 2012). This cell line exhibits epithelial morphology and is described as near hypertriploid, with most chromosomes showing abnormalities. The MG-63 and Saos-2 cell lines are both hypotriploid, and whereas the MG-63 cell line grows with fibroblastic shape, the Saos-2 cell line displays epithelial morphology. In addition, the MG-63 cell line showed TP53 sequence rearrangements and promoter hypermethylation, while Saos-2 was found a p53-null mutant (Di Fiore et al. 2014; Masuda et al. 1987).
In this study, the cytotoxic action and cell cycle effects of fisetin on osteosarcoma p53-deficient cell lines were investigated. For this, cells were exposed to fisetin, the chemotherapeutic agent etoposide, and fisetin:etoposide combinations. Cell cycle effects were analyzed and markers of cell cycle deregulation were identified. The study of differential regulation by fisetin:etoposide combinations allowed the identification of relevant biomarkers for the cytotoxic and cytostatic actions of fisetin on osteosarcoma.
Materials and methods
Cell culture media and reagents
Dulbecco’s modified Eagle’s medium, fetal bovine serum, l-glutamine, antibiotics, amphotericin B, and trypsin–EDTA (0.25% Trypsin, 1 mM EDTA) were purchased from Life Technologies (Carlsbad, CA, USA). Dimethyl sulfoxide (DMSO), sodium dodecyl sulfate (SDS), propidium iodide, and ribonuclease were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fisetin, etoposide, and crystal violet (CV) were purchased from Merck Millipore (Billerica, MA, USA).
Cell culture and exposure conditions
Human osteosarcoma cell lines MG-63, Saos-2, and U2OS were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). The cell lines were maintained and subcultured in complete medium, i.e., Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2.5 µg/ml amphotericin B. Cells were incubated in a humidified atmosphere of 5% CO2 at 37 °C. Subconfluent cells were trypsinized with trypsin–EDTA (0.25% trypsin, 1 mM EDTA) and subcultured at a split ratio of 1:6. For exposure experiments, concentrated stock solutions of fisetin (200 mM) or etoposide (60 mM) were prepared in DMSO. Final dilutions for exposure were prepared in complete medium. Final DMSO concentration was 0.1% for all conditions, including control. Each experiment was conducted with freshly thawed aliquots of these authenticated cell lines.
Clonogenic assay
MG-63, Saos-2, or U2OS cells were seeded in 6-well plates (100 cells/well) and allowed to adhere overnight at 37 °C. After adhesion, cells were washed with phosphate-buffered saline (PBS) and incubated in medium containing fisetin for 48 h. After exposure, cells were washed with PBS and incubated in complete Dulbecco’s modified Eagle’s medium for 7 days. During the 7-day recovery period, the medium was replaced with fresh medium every other day. After this period, the medium was carefully removed, and cells were fixed and stained for 30 min with crystal violet (CV) solution (0.2% CV and 10% ethanol). The culture plates were washed with distilled water and allowed to dry overnight. Colony image was acquired in a G:BOX CHEMI HR-16 BioImaging system (Syngene, Frederick, MD, USA) and clonogenic assay parameters colony number and area were determined with ImageJ (Abràmoff et al. 2004).
Cell growth and proliferation
Cell growth and proliferation was assessed by crystal violet (CV) assay. The CV assay allows quantification of cell growth based on the quantification of intracellularly retained CV. MG-63 and Saos-2 cells were seeded at a density of 5 × 104 cells/well in 24-well plates. After overnight adhesion, cells were exposed to fisetin, etoposide, or combinations for 48 h. Subsequently, cells were carefully washed with PBS and fixed/stained with CV solution (0.2% CV and 10% ethanol) for 30 min. After staining, cells were carefully washed with distilled water and, after drying, intracellular CV was dissolved in 900 μl of 1% SDS. The culture plates were shaken for 1 h and absorbance was measured at 570 nm using a Synergy HT Multi-mode Microplate Reader (BioTek Instruments, Winooski, VT, USA).
Combination index (CI) analysis
The CI analysis was performed according to the method of Chou-Talalay (Chou 2006) in MG-63, and Saos-2 cells. Briefly, data from CV assay dose–response curves for fisetin and etoposide were considered, together with CV assay data for the different mixtures fisetin:etoposide. The CI values for positive, independent, and negative interaction effects were determined using the CompuSyn software (Chou and Martin 2005). The CI is a quantitative measure of the degree of interaction between two or more drugs. For these studies, a CI = 1 denotes an independent effect of fisetin and etoposide, > 1 a negative interaction between the two drugs, and < 1 a positive interaction between the two drugs for the inhibition of cell growth and proliferation.
Cell cycle analysis
Cell cycle analysis was performed in MG-63 and Saos-2 cells as described in a previous work, with a few modifications (Ferreira de Oliveira et al. 2014). For this, cells on 6-well plates at the density of 3 × 105 cells/well were allowed to adhere at 37 °C overnight. After adhesion, cells were washed with PBS and treated with fisetin, etoposide, and combinations as indicated and additionally exposed for 48 h. Subsequently, cells were detached by trypsinization and centrifuged at 700 g for 5 min. The cells were resuspended in PBS, the cell suspension was recentrifuged as previously (700 g, 5 min) and the cell pellets were resuspended in 1 ml of 85% ethanol at 4 °C after which they were stored at − 20 °C until analysis. For cell cycle analysis, samples were centrifuged at 700 g for 5 min, and the cell pellets were resuspended in 0.8 ml of PBS. Cell suspensions were then filtered through a nylon mesh (35-µm pore), and ribonuclease (final 50 µg/ml) and propidium iodide (final 50 µg/ml) were added to the filtered suspension. For measurements of relative fluorescence intensity of PI-stained nuclei, a Coulter Epics XL Flow Cytometer (Beckman Coulter, Hialeah, FL, USA) equipped with an argon laser (15 mW, 488 nm) was used. Acquisitions were performed using SYSTEM II software v. 3.0 (Beckman Coulter, Hialeah, FL, USA). The number of events analyzed for each sample was ~ 5000. Histogram outputs were used for analysis and frequency results were determined with the software FlowJo (Tree Star, Ashland, OR, USA).
RNA extraction and qPCR
The oligonucleotide primers used were described previously (Coutinho et al. 2017). Primer3 platform (Rozen and Skaletsky 2000) was used for oligonucleotide primer design and primer specificity was confirmed by the UCSC In-Silico PCR tool (http://genome.ucsc.edu/cgi-bin/hgPcr?command=start). The TRIzol method was used to extract total RNA. For this, MG-63 and Saos-2 cells were lyzed in TRI reagent (Life Technologies, Carlsbad, CA, USA). Organic phase separation was achieved in Phase Lock Gel Heavy tubes (5 PRIME Inc., Boulder, CO, USA). The aqueous phase was mixed with 70% ethanol (1:1), and RNA was incubated with DNase I and purified using NZY Total RNA Isolation kit (NZYTech, Lisbon, Portugal). For cDNA synthesis, 2 μg total RNA was reverse-transcribed with 2.5 μM Oligo (dT)18, using the NZY First-Strand cDNA Synthesis Kit (NZYTech, Lisbon, Portugal). The cDNA samples were prediluted in ultrapure water (1:10). The final individual qPCR reactions contained iTaq Universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA), 150 nM each gene-specific primer, and 1:4 (v/v) prediluted cDNA (final 1:40). The qPCR program included denaturation for 1 min at 95 °C, and 60 cycles of amplification including denaturation for 5 s at 94 °C, annealing for 15 s at 58 °C, and extension for 15 s at 72 °C. After qPCR, a melting temperature program was performed. At least three qPCR technical replicates were performed per sample from each of three independent biological assays. Cycle thresholds and average PCR efficiencies were estimated from the baseline-corrected fluorescence data using Real-Time PCR Miner (Zhao and Fernald 2005). Gene expression of exposed cells was calculated relative to control cells and normalized with the GAPDH reference gene, following the Pfaffl method (Pfaffl 2001).
Statistical analysis
All data were derived from at least three independent experiments. The inhibitory concentrations (ICs) were determined from the growth assay with the R program for Windows version 3.1.1 (core team, VIE, Austria) from statistical packages drm and drc. Results are mean ± standard deviation (SD), except for gene expression data, where mean ± standard error of the mean (SE) are used. Results were analyzed by one-way ANOVA and compared with the Holm–Sidak statistical test. When normality could not be reached for at least one sample group, Kruskal–Wallis one-way ANOVA on ranks was performed, with post-hoc Tukey’s test analysis. For one-way ANOVA, the software package Sigmaplot for windows, version 11.0 (Systat Software Inc, San Jose, CA, USA) was used. For all comparisons, differences were considered statistically significant at p < 0.05.
Results
Inhibition of colony formation and cell recovery
Cell recovery was assessed by the number colonies relative to control and area occupied by colonies relative to total well area and relative to control (Fig. 1). After 48 h exposure to fisetin, U2OS and Saos-2 cell lines had decreased recovery in the exposures ranging from 10 to 20 µM in a dose-dependent manner (p < 0.05), as assessed by the two parameters. In contrast, MG-63 cell recovery was not significantly affected by fisetin for the same exposure period and concentrations. MG-63 and Saos-2 cells were then selected to test cytotoxic effects as well as cell cycle effects of fisetin used as single agent or combined with etoposide.

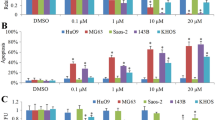
Colony formation ability after 48 h exposure to fisetin. After exposure, cells were allowed to recover in complete medium for 7 days after which colonies were stained with crystal violet and clonogenic assay parameters were analyzed with the ImageJ software. a Recovering colonies after exposure to fisetin; b number of colonies relative to control (%); c area occupied by colonies relative to total well area (%). The data are mean ± SD (n = 3). Statistical analysis was performed using the one-way ANOVA test (p < 0.05). Significant differences (asterisks) are shown relative to the control group
Inhibition of cell proliferation
MG-63 and Saos-2 cell growth decreased in a dose-dependent manner from 10 to 150 µM fisetin (Fig. 2). Compared to etoposide, fisetin was less cytotoxic. Considering only the tested concentrations, fisetin was significantly cytotoxic starting from 40 μM for MG-63 cell line and 20 μM for Saos-2 cell line (p < 0.05, Fig. 2b, c).

MG-63 and Saos-2 cell growth for single-agent exposures. Cells were exposed to fisetin or etoposide for 48 h and growth was evaluated by CV assay. a Cell morphology and confluence was assessed by inverted microscopy; scale bar: 100 μm; b MG-63 cell growth and proliferation; c Saos-2 cell growth and proliferation. The data are mean ± SD (n = 3). Statistical analysis was performed using the one-way ANOVA test (p < 0.05). Significant differences (asterisks) are shown relative to the control group. Fisetin concentrations: 0, 5, 10, 20, 40, 100, and 150 μM; etoposide concentrations: 0, 0.5, 1, 1.5, 2, 5, and 10 μM. The dose–response models presented are the Brain–Cousens log–logistic model for fisetin exposure and the Cedergreen–Ritz–Streibig model for etoposide exposure
From the dose–response curves shown in Fig. 2, inhibitory concentrations IC30 and IC50 were estimated (Table 1). Cells were then exposed, as indicated in Fig. 3, to the combinations of fisetin and etoposide near IC30 concentrations.

MG-63 and Saos-2 cell growth for double-agent combination exposures. Cells were exposed to fisetin and etoposide for 48 h, and growth was evaluated by CV assay. a Proliferation of MG-63 cells exposed to combination treatment; b proliferation of Saos-2 cells exposed to combination treatment. The data are mean ± SD (n = 3). Statistical analysis was performed using the one-way ANOVA test (p < 0.05) for MG-63 data and one-way ANOVA on ranks for Saos-2 data (p < 0.05). Significant differences between control and each experimental group are marked with asterisks. Significant differences between paired experimental groups are marked with triangles. CI combination index
A range of combination indices were observed, depending on the combination, between 1.51 (negative interaction fisetin:etoposide) and 0.59 (positive interaction fisetin:etoposide). For comparison, the effect of etoposide and fisetin at the largest concentrations is shown in Fig. 3, as isolated agents are presented in Table 1.
Cell cycle analysis
Exposure to the most anti-proliferative combinations investigated, i.e., 36 μM fisetin:0.5 μM etoposide for MG-63 cells and 45.5 μM fisetin:1 μM etoposide for Saos-2 cells, resulted in ~ 40% inhibition of cell growth/proliferation, as shown by CV assay (Fig. 3a, b). Exposure to fisetin or etoposide as single agents at ~ 15 and ~ 40% inhibitory concentrations was associated with decreased percentage of cells in G0/G1-phase and increased percentage of cells in G2/M-phase, for both cell lines (p < 0.05; Fig. 4a, b). For cells exposed to fisetin and etoposide as single agents, etoposide induced a larger difference in % G0/G1 and G2/M cells. When considering double-agent exposure, a decrease in % cells in G0/G1-phase and an increase in % cells in G2/M-phase were also observed in cells exposed to combinations of fisetin and etoposide, relative to control (p < 0.05; Fig. 4c, d). However, for both cell lines, despite the increase in % G2/M-phase cells compared to control, a significant decrease was observed in % G2/M-phase cells from the least to the most anti-proliferative combinations (p < 0.05; Fig. 4c, d). In addition to this, in Saos-2 cell line, despite the decrease in % S-phase cells compared to control, a significant increase was observed in % S-phase cells from the least to the most anti-proliferative combination (p < 0.05; Fig. 4d).

Cell cycle distribution of cells exposed to fisetin and etoposide. Cells were stained with propidium iodide and the cell cycle was analyzed by flow cytometry. Single-agent exposure in a MG-63 cells and b Saos-2 cells. Double-agent exposure in c MG-63 cells and d Saos-2 cells. The data are mean ± SD (n = 3). Statistical analysis was performed using the one-way ANOVA test (p < 0.05). Significant differences (asterisk) are shown relative to the control group
The gene expression of cell cycle regulators was analyzed after exposure to fisetin, etoposide, and combinations. For single-agent exposures, in the MG-63 cell line, etoposide increased cyclin E1 RNA levels, while in Saos-2 cells, the flavonoid fisetin decreased cyclin B1 RNA levels (p < 0.05, Fig. 5). For double-agent exposures (combinations), significant differences were found only in MG-63 cells, viz., decrease in cyclin B1, Cdk1, and Cdk2 RNA levels. Compared to the chemotherapeutic agent etoposide, the combinations fisetin:etoposide significantly decreased cyclin E1, Cdk1, and Cdk2 gene expression in MG-63 cells. In Saos-2 cells, the average cyclin E1 gene expression was lower when cells were exposed to fisetin:etoposide combination compared to etoposide as single agent, although not reaching statistical significance (p < 0.05, Fig. 5b).

Gene expression of cell cycle regulators. Cells were exposed for 48 h to fisetin (single agent), etoposide (single agent), or combinations (double agent). Exposures in a MG-63 cells and b Saos-2 cells. The data are mean ± SD (n = 3). Statistical analysis was performed using the one-way ANOVA test (p < 0.05). Significant differences are shown relative to the control group (asterisks) or to other experimental groups (triangles)
Discussion
Targeted osteosarcoma therapy is still hindered by the genetic instability and complexity of the disease (Kansara et al. 2014; Yang and Zhang 2013). Combined therapy regimens could, therefore, prove beneficial, particularly for poor responders.
Herein, the main goal was to test cellular effects of fisetin in cell lines with deficient or absent wt p53 expression. In the wt p53 U2OS cells, Li et al. (2015) reported IC50 for fisetin near 70 micromolar for 48 h exposure, using MTS assay, while Ying et al. (2012) reported IC50 for fisetin near 130 micromolar (48 h exposure, MTT assay). In this study, a biphasic behavior was observed for the inhibition of cell growth by fisetin. Previously, at low concentrations, fisetin was found to enhance preosteoblast proliferation (Léotoing et al. 2014), and this effect could have played a role on the enhanced osteosarcoma proliferation up to 10 μmol observed in this study, resulting in a biphasic effect. Moreover, for some cell lines described in the literature, exposure to low fisetin concentrations did not result in significant inhibition of proliferation or viability (e.g., Kim et al. 2014; Touil et al. 2011a, b; Ying et al. 2012). Despite the increase in cell proliferation upon exposure to low-dose fisetin observed in this work, Saos-2 cells suffered inhibition of colony formation for 10 and 20 μmol concentrations. This observation shows that, even at subtoxic doses, fisetin decreased colony formation, suggesting a cytostatic effect in cells recovered from low-dose fisetin exposure. Depending on cell type and exposure times, in the previous studies, micromolar doses of fisetin showed various effects on the cell cycle, ranging from increase in % cells in G0/G1-phase and reduction in % cells in S-phase, to emergence of sub-G1 population with decreased % G0/G1 population, and increased % cells in S-phase and G2/M-phase, e.g., (Adan and Baran 2015; Chen et al. 2002; Haddad et al. 2006; Khan et al. 2008; Li et al. 2011; Lu et al. 2005; Pal et al. 2013; Ying et al. 2012).
Etoposide is frequently administered in patients with poor response to therapy. Despite its anti-cancer role as topoisomerase II inhibitor, a large body of evidence indicates that it could lead to secondary malignancies (Azarova et al. 2007). In this study, fisetin was shown less cytotoxic to osteosarcoma cells, compared to etoposide. Combination index values were calculated for different mixtures to determine positive or negative interactions between fisetin and etoposide. The two mixtures with highest fisetin concentration and lowest etoposide concentration showed more positive interaction effects for the inhibition of cell growth, pointing to fisetin usefulness in osteosarcoma combined therapy.
DNA damage triggers a set of cellular responses, including cell cycle arrest and commitment to cell death. It is well known that cell cycle arrest can increase the sensitivity of cancer cells to etoposide (Walker and Nitiss 2002). Comparing only cells exposed to fisetin:etoposide mixtures, an association was found with increased accumulation of cells in G1-phase and S-phase and decreased % of cells in G2-phase. CCNE1 (cyclin E1 gene) and CDK2 (cyclin-dependent kinase 2 gene) govern G1/S-phase transition and S-phase were previously found amplified and overexpressed in osteosarcoma (Lockwood et al. 2011; Vella et al. 2016). Moreover, silencing of CCNE1 or CDK2 was previously shown to suppress osteosarcoma proliferation (Vella et al. 2016; Wang et al. 2014). In general, when considering only fisetin:etoposide combinations, an increase was observed in % cells in S-phase for the mixtures with highest fisetin concentration for both cell lines. Combinations with lower CI values, i.e., more positive interactions between fisetin and etoposide, were associated with increased % of cells in S-phase, in both cell lines. In MG-63 cells, exposure to 38.6 μM fisetin alone (Fig. 4a) or to mixture fisetin:etoposide—36 μM:0.5 μM (Fig. 4c) originated very similar cell cycle profiles. Notwithstanding, when considering gene expression, there is no significant difference between control and cells exposed to fisetin for the genes analyzed in the MG-63 cell line. For this reason, it cannot be ruled out that other regulators of the cell cycle might be involved in accumulation at the S-phase, even though a decrease was observed in cyclin E1, Cdk1, and Cdk2 gene expression in cells exposed to fisetin:etoposide combination, and such decrease would be expected to impact cell cycle progression. Notwithstanding, in other cancer cell lines reported in the literature, fisetin was shown to decrease the protein levels of cyclin E, cyclin A, cyclin D1, Cdk1, Cdk2, and Cdk4, while increasing p21 levels (Khan et al. 2008; Li et al. 2011; Lu et al. 2005; Sung et al. 2007; Syed et al. 2011). The cell cycle effects of etoposide rely mostly on its action as topoisomerase II inhibitor, and previously, fisetin was described as a moderate topoisomerase II inhibitor in vitro (Bandele and Osheroff 2007; Strick et al. 2000). Therefore, it would be relevant in the future to investigate whether it shows similar effects on osteosarcoma cell lines. Concerning the possibility of induction of secondary malignancies by fisetin, at physiological concentrations, this flavonol induces the differentiation of pre-osteoblasts to mature osteoblasts, a feature opposite of that found in bone malignancy (Sung et al. 2007). More importantly, in a pharmacokinetics study in mice, fisetin was found to be accumulated at ~ 84 μg/(g tissue) in kidneys, corresponding to ~ 24 mmol/g, without negative effects being reported (Touil et al. 2011a). Moreover, in another work from this team, this fisetin dose was continuously administered in mice for 2 weeks and resulted in decreased Lewis lung tumor development, while showing no toxic effects (Touil et al. 2011b). These observations suggest that fisetin can inhibit tumorigenesis and would not induce secondary malignancies at the concentrations tested in this study.
In conclusion, fisetin combined with etoposide induced cell cycle arrest, which was found associated with decreased expression of cell cycle regulators commonly overexpressed in many osteosarcoma samples. For this reason, the use of fisetin combined with chemotherapeutic drugs warrants further studies.
References
Abràmoff MD, Magalhães PJ, Ram SJ (2004) Image processing with imageJ. Biophoton Int 11:36–41. https://doi.org/10.1117/1.3589100
Adan A, Baran Y (2015) Fisetin and hesperetin induced apoptosis and cell cycle arrest in chronic myeloid leukemia cells accompanied by modulation of cellular signaling. Tumor Biol. https://doi.org/10.1007/s13277-015-4118-3
Anderson ME (2016) Update on Survival in Osteosarcoma. Orthop Clin North Am 47:283–292. https://doi.org/10.1016/j.ocl.2015.08.022
Azarova AM, Lyu YL, Lin C et al (2007) Roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc Natl Acad Sci USA 104:11014–11019. https://doi.org/10.1073/pnas.0704002104
Bandele OJ, Osheroff N (2007) Bioflavonoids as poisons of human topoisomerase II alpha and II beta. Biochemistry 46:6097–6108. https://doi.org/10.1021/bi7000664
Chen YC, Shen SC, Lee WR et al (2002) Wogonin and fisetin induction of apoptosis through activation of caspase 3 cascade and alternative expression of p21 protein in hepatocellular carcinoma cells SK-HEP-1. Arch Toxicol 76:351–359. https://doi.org/10.1007/s00204-002-0346-6
Choi SW, Son YJ, Yun JM, Kim SH (2012) Fisetin inhibits osteoclast differentiation via downregulation of p38 and c-Fos-NFATc1 signaling pathways. Evid based Complement Alternat Med 2012:810563. https://doi.org/10.1155/2012/810563
Chou T (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–681. https://doi.org/10.1124/pr.58.3.10
Chou TC, Martin N (2005) CompuSyn for drug combinations: PC software and user’s guide: a computer program for quantitation of synergism and antagonism in drug combinations, and the determination of IC50 and ED50 and LD50 values. ComboSyn Inc, Paramus
Coutinho L, Oliveira H, Pacheco AR et al (2017) Hesperetin-etoposide combinations induce cytotoxicity in U2OS cells: Implications on therapeutic developments for osteosarcoma. DNA Repair 50:36–42. https://doi.org/10.1016/j.dnarep.2016.12.006
Di Fiore R, Marcatti M, Drago-Ferrante R et al (2014) Mutant p53 gain of function can be at the root of dedifferentiation of human osteosarcoma MG63 cells into 3AB-OS cancer stem cells. Bone 60:198–212. https://doi.org/10.1016/j.bone.2013.12.021
Ferreira de Oliveira JMP, Remédios C, Oliveira H et al (2014) Sulforaphane induces DNA damage and mitotic abnormalities in human osteosarcoma MG-63 cells: correlation with cell cycle Arrest and apoptosis. Nutr Cancer 5581:37–41. https://doi.org/10.1080/01635581.2014.864777
Gomes A, Fernandes E, Lima JLFC. et al (2008) Molecular mechanisms of anti-inflammatory activity mediated by flavonoids. Curr Med Chem 15:1586–1605
González-Gallego J, García-Mediavilla MV, Sánchez-Campos S, Tuñón MJ (2010) Fruit polyphenols, immunity and inflammation. Br J Nutr 104:S15–S27. https://doi.org/10.1017/S0007114510003910
Haddad AQ, Venkateswaran V, Viswanathan L et al (2006) Novel antiproliferative flavonoids induce cell cycle arrest in human prostate cancer cell lines. Prostate Cancer Prostatic Dis 9:68–76. https://doi.org/10.1038/sj.pcan.4500845
Horcajada MN, Offord E (2012) Naturally plant-derived compounds: role in bone anabolism. Curr Mol Pharmacol 5:205–218. https://doi.org/10.2174/1874467211205020205
Kansara M, Teng MW, Smyth MJ, Thomas DM (2014) Translational biology of osteosarcoma. Nat Rev Cancer 14:722–735. https://doi.org/10.1038/nrc3838
Khan N, Afaq F, Syed DN, Mukhtar H (2008) Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis 29:1049–1056. https://doi.org/10.1093/carcin/bgn078
Kim YH, Kim JL, Lee EJ et al (2014) Fisetin antagonizes cell fusion, cytoskeletal organization and bone resorption in RANKL-differentiated murine macrophages. J Nutr Biochem 25:295–303. https://doi.org/10.1016/j.jnutbio.2013.11.003
Lee JD, Huh JE, Jeon GS et al (2009) Flavonol-rich RVHxR from Rhus verniciflua Stokes and its major compound fisetin inhibits inflammation-related cytokines and angiogenic factor in rheumatoid arthritic fibroblast-like synovial cells and in vivo models. Int Immunopharmacol 9:268–276. https://doi.org/10.1016/j.intimp.2008.11.005
Léotoing L, Wauquier F, Guicheux J et al (2013) The polyphenol fisetin protects bone by repressing NF-kappaB and MKP-1-dependent signaling pathways in osteoclasts. PLoS One 8:1–14. https://doi.org/10.1371/journal.pone.0068388
Léotoing L, Davicco MJ, Lebecque P et al (2014) The flavonoid fisetin promotes osteoblasts differentiation through Runx2 transcriptional activity. Mol Nutr Food Res 58:1239–1248. https://doi.org/10.1002/mnfr.201300836
Li J, Cheng Y, Qu W et al (2011) Fisetin, a dietary flavonoid, induces cell cycle arrest and apoptosis through activation of p53 and inhibition of NF-kappa B pathways in bladder cancer cells. Basic Clin Pharmacol Toxicol 108:84–93. https://doi.org/10.1111/j.1742-7843.2010.00613.x
Li JM, Li WY, Huang MY, Zhang XQ (2015) Fisetin, a dietary flavonoid induces apoptosis via modulating the MAPK and PI3K/Akt signalling pathways in human osteosarcoma (U-2 OS) cells. Bangladesh J Pharmacol 10:820–829. https://doi.org/10.3329/bjp.v10i4.23039
Liao Y-C, Shih Y-W, Chao C-H et al (2009) Involvement of the ERK signaling pathway in fisetin reduces invasion and migration in the human lung cancer cell line A549. J Agric Food Chem 57:8933–8941. https://doi.org/10.1021/jf902630w
Lim DY, Park JHY (2009) Induction of p53 contributes to apoptosis of HCT-116 human colon cancer cells induced by the dietary compound fisetin. Am J Physiol Gastrointest Liver Physiol 296:G1060–G1068. https://doi.org/10.1152/ajpgi.90490.2008
Lockwood WW, Stack D, Morris T et al (2011) Cyclin E1 is amplified and overexpressed in osteosarcoma. J Mol Diagn 13:289–296. https://doi.org/10.1016/j.jmoldx.2010.11.020
Lu X, Jung J, Cho HJ et al (2005) Fisetin inhibits the activities of cyclin-dependent kinases leading to cell cycle arrest in HT-29 human colon cancer cells. J Nutr 135:2884–2890
Luetke A, Meyers PA, Lewis I, Juergens H (2014) Osteosarcoma treatment—Where do we stand? A state of the art review. Cancer Treat Rev 40:523–532. https://doi.org/10.1016/j.ctrv.2013.11.006
Masuda H, Miller C, Koeffler HP et al (1987) Rearrangement of the p53 gene in human osteogenic sarcomas. Proc Natl Acad Sci USA 84:7716–7719. https://doi.org/10.1073/pnas.84.21.7716
Murtaza I, Adhami VM, Hafeez B, Bin et al (2009) Fisetin, a natural flavonoid, targets chemoresistant human pancreatic cancer AsPC-1 cells through DR3-mediated inhibition of NF-κB. Int J Cancer 125:2465–2473. https://doi.org/10.1002/ijc.24628
Pal HC, Sharma S, Elmets CA et al (2013) Fisetin inhibits growth, induces G2/M arrest and apoptosis of human epidermoid carcinoma A431 cells: role of mitochondrial membrane potential disruption and consequent caspases activation. Exp Dermatol 22:470–475. https://doi.org/10.1111/exd.12181.Fisetin
Pandey KB, Rizvi SI (2009) Plant polyphenols as dietary antioxidants in human health and disease. Oxidative Med Cell Longev 2:270–278. https://doi.org/10.4161/oxim.2.5.9498
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:16–21. https://doi.org/10.1093/nar/29.9.e45
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386. https://doi.org/10.1385/1-59259-192-2:365
Sakai E, Shimada-Sugawara M, Yamaguchi Y et al (2013) Fisetin inhibits osteoclastogenesis through prevention of RANKL-induced ROS production by Nrf2-mediated up-regulation of phase II antioxidant enzymes. J Pharmacol Sci 121:288–298. https://doi.org/10.1254/jphs.12243FP
Shen CL, von Bergen V, Chyu MC et al (2012) Fruits and dietary phytochemicals in bone protection. Nutr Res 32:897–910. https://doi.org/10.1016/j.nutres.2012.09.018
Strick R, Strissel PL, Borgers S et al (2000) Dietary bioflavonoids induce cleavage in the MLL gene and may contribute to infant leukemia. Proc Natl Acad Sci USA 97:4790–4795. https://doi.org/10.1073/pnas.070061297
Suh Y, Afaq F, Johnson JJ, Mukhtar H (2009) A plant flavonoid fisetin induces apoptosis in colon cancer cells by inhibition of COX2 and Wnt/EGFR/NF-κB-signaling pathways. Carcinogenesis 30:300–307. https://doi.org/10.1093/carcin/bgn269
Sung B, Pandey MK, Aggarwal BB (2007) Fisetin, an inhibitor of cyclin-dependent kinase 6, down—regulates nuclear factor-κB-regulated cell proliferation, antiapoptotic and metastatic gene products through the suppression of TAK-1 and receptor-interacting protein—regulated IκBα kinase activ. Mol Pharmacol 71:1703–1714. https://doi.org/10.1124/mol.107.034512
Syed DN, Afaq F, Maddodi N et al (2011) Inhibition of human melanoma cell growth by the dietary flavonoid fisetin is associated with disruption of Wnt/β-catenin signaling and decreased Mitf levels. J Invest Dermatol 131:1291–1299. https://doi.org/10.1038/jid.2011.6
Touil YS, Auzeil N, Saighi H et al (2011a) Fisetin disposition and metabolism in mice: Identification of geraldol as an active metabolite. 82:1731–1739. https://doi.org/10.1016/j.bcp.2011.07.097
Touil YS, Seguin J, Scherman D, Chabot GG (2011b) Improved antiangiogenic and antitumour activity of the combination of the natural flavonoid fisetin and cyclophosphamide in Lewis lung carcinoma-bearing mice. Cancer Chemother Pharmacol 68:445–455. https://doi.org/10.1007/s00280-010-1505-8
Vella S, Tavanti E, Hattinger CM et al (2016) Targeting CDKs with roscovitine increases sensitivity to DNA damaging drugs of human osteosarcoma cells. PLoS One 11:e0166233. https://doi.org/10.1371/journal.pone.0166233
Walker JV, Nitiss JL (2002) DNA topoisomerase II as a target for cancer chemotherapy. Cancer Invest 20:570–589. https://doi.org/10.1081/CNV-120002156
Wang J, Xu G, Shen F, Kang Y (2014) miR-132 targeting cyclin E1 suppresses cell proliferation in osteosarcoma cells. Tumor Biol 35:4859–4865. https://doi.org/10.1007/s13277-014-1637-2
Yang J, Zhang W (2013) New molecular insights into osteosarcoma targeted therapy. Curr Opin Oncol 25:398–406. https://doi.org/10.1097/CCO.0b013e3283622c1b
Ying TH, Yang SF, Tsai SJ et al (2012) Fisetin induces apoptosis in human cervical cancer HeLa cells through ERK1/2-mediated activation of caspase-8-/caspase-3-dependent pathway. Arch Toxicol 86:263–273. https://doi.org/10.1007/s00204-011-0754-6
Zhao S, Fernald RD (2005) Comprehensive algorithm for quantitative real-time polymerase chain reaction. J Comput Biol 12:1047–1064. https://doi.org/10.1089/cmb.2005.12.1047.Comprehensive
Acknowledgements
This work was supported by Grants from the Portuguese Foundation for Science and Technology—Fundação para a Ciência e a Tecnologia (SFRH/BPD/74868/2010 to J.M.P.F.O, SFRH/BPD/111736/2015 to H.O.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical statement
The manuscript does not contain clinical studies or patient data.
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ferreira de Oliveira, J.M.P., Pacheco, A.R., Coutinho, L. et al. Combination of etoposide and fisetin results in anti-cancer efficiency against osteosarcoma cell models. Arch Toxicol 92, 1205–1214 (2018). https://doi.org/10.1007/s00204-017-2146-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-017-2146-z