
Abstract
Totiviridae, a viral family of double-stranded RNA (dsRNA) viruses, contain a single dsRNA genome 4.6–7.0 kb in length. Totiviridae were initially only known to infect fungi and other eukaryotes as well as plants, but an increase in totiviruses has been detected in insects, mosquitoes, and bats. Here, we describe the isolation and characterization of a strain belonging to the family Totiviridae isolated from Culex tritaeniorhynchus in Kenli, China, in 2016. We isolated a totivirus from field-collected mosquitoes in China by cell culture in Aedes albopictus C6/36 cells, identified the virus by morphological observation and complete genome sequencing, and characterized it by phylogenetic analysis. Transmission electron microscopy identified icosahedral, non-enveloped virus particles with a mean diameter of 35–40 nm. The genome was 7612 bp in length, including two open reading frames (ORFs). ORF1 (5058 nt) encodes the capsid protein, while ORF2 (2216 nt) encodes the viral RNA-dependent RNA polymerase (RdRp). Nucleotide and amino acid homology analysis of isolate showed higher levels of sequence identity with isolate CTV_NJ2 (China, 2010) with 94.87% nucleic acid identity and 97.32% amino acid identity. The isolate was designated C. tritaeniorhynchus totivirus KL (CTV-KL). This is the first identification of a totivirus in a C. tritaeniorhynchus in northern China. Analysis of the virus’s morphology, characteristic and genome organization will further enrich our understanding of the molecular and biological characteristics of dsRNA Totiviridae viruses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Based on the Baltimore classification, viruses are classified into seven groups: double-stranded DNA (dsDNA), single-stranded DNA (ssDNA), double-stranded RNA (dsRNA), positive single-stranded RNA (ssRNA+), negative single-stranded RNA (ssRNA−), single-stranded retro-transcribing RNA (ssRNA-RT), and double-stranded retro-transcribing DNA (dsDNA-RT) (Baltimore 1971). The latest release of the International Committee on Taxonomy of Viruses (ICTV) recognized 256 species of dsRNA viruses, which accounts for less than 4.6% of total established viral species. Due to their rarity, there is an inadequate appreciation of the diversity of dsRNA viruses (King et al. 2018).
Totiviridae, a viral family of dsRNA viruses, consist of a single dsRNA that is 4.6–7.0 kb in length with two open reading frames (ORFs) encoding a capsid protein (CP) and an RNA-dependent RNA polymerase (RdRp) (de Lima et al. 2019; Ghabrial 2008). Viral particles of Totiviridae are approximately 40 nm in diameter with icosahedral symmetry. The Totiviridae family consists of five genera: Totivirus, Victorivirus, Giardiavirus, Leishmaniavirus, and Trichomonasvirus (Ghabrial and Nibert 2009; Goodman et al. 2011). With the availability of more advanced detection techniques, viruses from a wider range of host organisms are being discovered at an increasing pace. Totiviridae was previously known to infect fungi and other eukaryotes as well as plants; however, recently a handful of related but yet unassigned totivirus-like viruses have been isolated from insects, mosquitoes, and bats (Huang et al. 2018; Yang et al. 2012; Zhai et al. 2010). The identification of these viruses will contribute to our understanding of the diversity and evolution of dsRNA viruses.
Mosquitoes are vectors of parasites and viruses that cause diseases of immense importance to public health. Mosquitoes are the major vector of Flaviviridae (ssRNA+) and Togaviridae (ssRNA+) viruses, with Reoviridae (dsRNA) and Birnaviridae (dsRNA) being reported recently (Kuwata et al. 2015; Shapiro et al. 2005; Strauss and Strauss 1994; van Cleef et al. 2014). In particular, mosquito-borne viruses are of increasing concern as arboviral diseases emerge and re-emerge in conjunction with global warming, which facilitates the geographic expansion of mosquito species (Kobayashi et al. 2002). Here, we report the isolation of a strain belonging to the family Totiviridae from mosquitoes collected in the Shandong Province of northern China during a 2016 arbovirus survey.
Materials and methods
Mosquito collection
Kenli is a district in the city of Dongying, located in the northern Shandong province of eastern China near the mouth of the Yellow River. The arbovirus survey was conducted on September 7, 2016 in the residential regions of the village of Dongsui, a small river estuary town in the Kenli District (37°38′31.94″ N, 118° 51′ 41.69″ E; altitude, 2 m). Mosquitoes were collected from a pigsty at night using ultraviolet light traps (12 V; 300 mA; Wuhan Lucky Star Environmental Protection Tech Co., Ltd., Hubei, China). Traps were set from 7:00 p.m. to 7:00 a.m., which spanned the period from sunset to sunrise. The captured mosquitoes were frozen at − 20 °C for at least 40 min and identified according to morphological keys from Lu (1997a, b). Only female mosquitoes were selected for further study. Approximately 100 mosquitoes of the same species were pooled in cryogenic vials and stored in liquid nitrogen until further use (Li et al. 2010).
Virus isolation
Mosquitoes were removed from liquid nitrogen and immediately homogenized and centrifuged as previously reported (Sun et al. 2009). The supernatants were inoculated in a monolayer of Aedes albopictus mosquito C6/36, baby hamster kidney BHK-21, or African green monkey kidney cells in 24-well plates and incubated at 28 °C, 37 °C and 37 °C, respectively. After incubation for 24 h, the cells were observed for cytopathic effects (CPEs) every 8 h for 6–7 days. A specimen was regarded as a positive isolate if it caused CPEs in three successive cell passages. Infected cell supernatants were stored at − 80 °C until further analysis (Lu et al. 2009).
Virus electron microscopy
For negative staining, 1 ml of cell culture supernatant was ultracentrifuged at 100,000g for 10 min and the pellet was re-suspended in 50 μl infected cell supernatant. The virus was then absorbed onto Formvar and carbon-coated grids for 1 min and stained with 1% (w/v) phosphotungstic acid (pH 6.8) for 1 min. Grids were air-dried and observed under a Tecnai 12 transmission electron microscope (FEI, Eindhoven, Netherlands).
For ultrathin sections, cell pellets were fixed in a solution of 2% formaldehyde and 2.5% glutaraldehyde, post-fixed in 1% osmium tetroxide, dehydrated in an ethanol gradient, embedded in epoxy resin-812, and polymerized at 60 °C for 24 h. Ultrathin sections (80 nm) were obtained from the resin blocks, mounted on copper grids, and stained with uranyl acetate and lead citrate. Finally, the ultrathin sections were observed using transmission electron microscopy.
Extraction of nucleic acids and deep sequencing
Viral RNA was extracted from infected culture supernatants with positive CPEs using the QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA, USA). Complementary DNA (cDNA) synthesis was performed using Ready-to-Go RT-PCR Beads (Amersham Biosciences Co., Piscataway, NJ, USA). The library was prepared using an NEBNext Ultra RNA Library Prep Kit for Illumina (New England Biolabs, Hitchin, UK). The library was quantified on a Qubit 3.0 fluorometer (Thermo Fisher) and Agilent Bioanalyzer 2100 (Agilent). Paired-end sequencing [2 × 150 base pairs (bp)] was performed on an Illumina MiSeq v2 platform. Sequencing reads were filtered using previously described criteria to obtain valid data (Yang et al. 2011). For analysis, reads were filtered based on their length and mean quality values (Q30).
Sequence analysis
Contigs were prepared by de novo assembly and subjected to BLASTx alignment (E value < 10−5) against the non-redundant (NR) National Center for Biotechnology Information (NCBI) protein databases (https://www.ncbi.nlm.nih.gov). Mapping of sequence reads against the target genomes was done using the Bowtie 2 alignment algorithm in usegalaxy (https://usegalaxy.org/). RNA ligase-mediated (RLM)-RACE PCR amplification was used to confirm the 5′- and 3′-terminal sequences. ORFs were identified using GENETYX-MAC or EnzymeX version 3.3.3 (https://nucleobytes.com/enzymex/index.html). Sequence similarities were calculated using the BLASTx program available from NCBI. The complete nucleotide sequence of the virus reported in this study has been submitted to the GenBank database under accession number MN614415.
Phylogenetic analysis
Multiple alignments were performed with MAFFT version 7 (https://www.ebi.ac.uk/Tools/) using previously described parameters (Katoh and Toh 2008). The aligned matrix data were confirmed manually. Phylogenetic tree construction was based on a neighbor-joining (NJ) method. The statistical significance of the resulting tree was evaluated using a bootstrap test with 1,000 replicates. The phylogenetic tree (mid-point rooted) was visualized and refined with FigTree version 1.4.2 software (https://tree.bio.ed.ac.uk/software/).
Results
Virus isolation
On September 7, 2016, a total of 3000 mosquitoes consist of two species, Culex tritaeniorhynchus (2300, 76.7%) and Anopheles sinensis (700, 23.3%), were collected in Kenli District, Shandong Province, China.
The virus isolated from C. tritaeniorhynchus initially caused CPEs in A. albopictus C6/36 cells beginning at 72 h after inoculation and blind passage. The main characteristic of the CPEs was cell shedding, shrinking, and floating with eventual detachment from the growth surface (Fig. 1). No CPEs were observed in BHK-21 and Vero cells during three blind passages. Virus particles were enriched by ultracentrifugation from infected culture supernatants and stained negatively. The negative stained virus particles were non-enveloped and approximately 40 nm in diameter (Fig. 2a). Virus particles and virus inclusion bodies were observed in the cytoplasm on ultrathin sections (Fig. 2b). No such particles were observed in mock-infected controls (data not shown). For archiving purposes, the isolate was designated CTV_KL.

Cytopathic effects (CPEs) caused by CTV-KL in C6/36 cells. a Phase-contrast micrographs of control (mock-infected) C6/36 cells. b CTV-KL-infected cells 72 h post-infection. c CTV-KL-infected cells 120 h post-infection. Scale bars indicate 200 μm

Negative-contrast electron micrographs of CTV-KL particles. a Arrows indicate CTV-KL particles in infected cell culture supernatant. Scale bars indicate 100 nm. b Ultrathin sections show large, electron-dense viral inclusion body (arrow) within the cytoplasm composed of a large number of CTV-KL in infected C6/36 cells. Scale bars indicate 0.2 μm
Sequence analysis of the CTV_KL genome
To identify these viruses, the viral RNAs were subjected to next-generation sequencing and RACE analyses. The assembled genome of CTV_KL was 7612 bp with two ORFs (ORF1 and ORF2). The first ORF (ORF1) spans nucleotides 7–5061 (5055 nt in length) and encodes a 1685 aa protein, while the second ORF (ORF2) spans nucleotides 5319–7532 (2217 nt in length) and encodes 738 aa. The 5′ and 3′ untranslated regions of CTV_KL were 6 and 77 nt long, respectively. BLASTx analysis of the contigs obtained by de novo assembly showed that they matched most closely with the unassigned species of Totiviridae. While the CTV_KL ORFs only showed a 3–8% aa similarity to the classified Totiviridae viruses, it shared a 94.87% nt and 97.32% aa identity with the unclassified C. tritaeniorhynchus totivirus isolate CTV_NJ2 (KX456218) and 76.82% nt and 84.16% aa identity with totivirus-like virus ToV-TJ (JN391187) (Table 1).
Phylogenetic analysis of CTV_KL
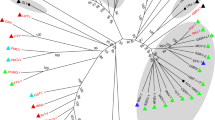
To study the evolutionary relationship of CTV_KL with other viruses in the family Totiviridae, RdRP amino acid sequences with ≥ 35% similarity with CTV_KL were downloaded from GenBank and used to construct a phylogenetic tree (Fig. 3). CTV_KL was classified into the clade of arthropod-infecting toti-like viruses. The phylogenetic analysis showed that CTV_KL was closely related to the unclassified Totiviridae C. tritaeniorhynchus totivirus, Omono River virus, and Penaeid shrimp infectious myonecrosis virus. However, CTV_KL formed a distinct cluster with Wuhan insect virus (KX882989), Wenzhou toti-like virus (KX882995), and Golden shiner totivirus (NC_030295).

Neighbor-joining tree inferred from molecular phylogenetic analysis based on the amino acid sequences of the RDRP segments. The newly discovered CTV-KL strain is labeled with a solid circle. Numbers associated with branches indicate the percentage of 1000 bootstrap replicates that support the existence of these branches. Branches with < 60% bootstrap support have been collapsed
Discussion
We identified and characterized a new strain of Totiviridae virus designated CTV-KL, isolated from Culex mosquitoes in northern China. The CTV-KL particle was non-enveloped, isometric, and approximately 40 nm in diameter. The genome was composed of a single 7.6 kb dsRNA containing two ORFs. Genetic and phylogenetic analyses suggested that CTV-KL is most closely related to unassigned members of the family Totiviridae. In the NJ phylogenetic tree, CTV-KL fell in a high confidence branch with CTV_NJ2, ToV-TJ, and OMRV-AK4, and formed a distinct cluster with Golden shiner totivirus (NC_030295), Wuhan insect virus (KX882989), and Wenzhou toti-like virus (KX882995). C. tritaeniorhynchus totivirus (CTV_NJ2) was isolated in Nanjing, China, in 2016, ToV-TJ was isolated from bats in Tianjin, China in 2010, and three OMRV viruses were isolated in Vietnam and Japan from three different Culex species(Isawa et al. 2011). In addition, CTV_KL also fell into a cluster with unassigned Totiviridae infectious myonecrosis virus (IMNV) isolated from the skeletal muscle of Penaeid shrimp (Litopenaeus vannamei) in Brazil in 2003, Drosophila totivirus (DTV) found in S2-GMR Drosophila cells, and Armigeres subalbatus totivirus (AsTV) from Armigeres subalbatus (Poulos et al. 2006; Tang et al. 2005; Wu et al. 2010; Zhai et al. 2010).
Previously, viruses in Totiviridae were only associated with latent infections of their fungal or protozoan hosts until the isolation of IMNV from Penaeid shrimp (L. vannamei) in northeastern Brazil in 2005 and DTV from Drosophila melanogaster in the USA (Poulos et al. 2006; Wu et al. 2010; de Lima et al. 2019). With the rapidly evolving application of next-generation sequencing technology, more members of this family are being discovered in a wider range of host organisms than ever before (Kondo et al. 2016; Read et al. 2019). IMNV, AsTV, and DTV have recently been proposed as members of a novel Totiviridae genus, “Artivirus” (derived from “arthropod totivirus”) (Zhai et al. 2010). The widespread geographic distribution and broad host range in arthropods suggests that these totivirus-like viruses may be common worldwide (Fig. 2). The potential biological significance of these viruses and their vector hosts needs to be further investigated.
Mosquitoes are vectors of parasites and viruses that cause diseases of immense importance to public health. In particular, mosquito-borne viruses are of increasing interest and concern because of the recent emergence and re-emergence of arboviral diseases. In addition, ecological changes due to global warming may facilitate an expansion of the distribution of vector mosquitoes. The vector competence of mosquitoes, host competence for viral transmission, and viral pathogenicity are the three factors used for assessing the susceptibility of vertebrates to mosquito-borne viruses (Hardy et al. 1983). It has been discovered that OMRV establishes latent infection in host mosquitoes, IMNV causes progressive disease in crustacean hosts (Poulos et al. 2006), and DTV persistently infects cultured Drosophila cells together with four other RNA viruses (Wu et al. 2010). The CTV-KL genome sequence was 76.3% identical to OMRV-AK4 isolated from mosquitoes in Japan (Isawa et al. 2011), suggesting that CTV_KL may be a pathogen of mosquitoes. The virulence, host range, specificity to mosquitoes of CTV-KL will require further investigation. A detailed analysis of CTV-KL is needed for assessment of its biological properties and potential ecological impacts on host mosquitoes and other viruses.
Conclusions
We have identified and characterized a strain (CTV-KL) of family Totiviridae that may be biologically significant to mosquitoes and humans. This is the first identification of a totivirus in a C. tritaeniorhynchus in northern China. Several important questions related to its transmission, host maintenance, and the potential impact of infection on the host’s survival, evolution warrant further investigation.
Abbreviations
- ORFs:
-
Open reading frames
- RdRp:
-
RNA-dependent RNA polymerase
- CTV:
-
Culex tritaeniorhynchus totivirus
- CPEs:
-
Cytopathic effects
- dsDNA:
-
Double-stranded DNA
- ssDNA:
-
Single-stranded DNA
- dsRNA:
-
Double-stranded RNA
- ssRNA+:
-
Positive single-stranded RNA
- ssRNA−:
-
Negative single-stranded RNA
- ssRNA-RT:
-
Single-stranded retro-transcribing RNA
- dsDNA-RT:
-
Double-stranded retro-transcribing DNA
References
Baltimore D (1971) Expression of animal virus genomes. Bacteriol Rev 35:235–241
de Lima JGS, Teixeira DG, Freitas TT, Lima J, Lanza DCF (2019) Evolutionary origin of 2A-like sequences in Totiviridae genomes. Virus Res 259:1–9. https://doi.org/10.1016/j.virusres.2018.10.011
Ghabrial SA (2008) Totiviruses. In: Mahy BWJ, van Regenmortel MHV (eds) Encyclopedia of virology, vol 5, 3rd edn. Academic Press/Elsevier, San Diego, pp 163–174
Ghabrial SA, Nibert ML (2009) Victorivirus, a new genus of fungal viruses in the family Totiviridae. Adv Virol 154:373–379. https://doi.org/10.1007/s00705-008-0272-x
Goodman RP, Ghabrial SA, Fichorova RN, Nibert ML (2011) Trichomonasvirus: a new genus of protozoan viruses in the family Totiviridae. Adv Virol 156:171–179. https://doi.org/10.1007/s00705-010-0832-8
Hardy JL, Houk EJ, Kramer LD, Reeves WC (1983) Intrinsic factors affecting vector competence of mosquitoes for arboviruses. Annu Rev Entomol 28:229–262. https://doi.org/10.1146/annurev.en.28.010183.001305
Huang Y et al (2018) Discovery of two novel totiviruses from Culex tritaeniorhynchus classifiable in a distinct clade with arthropod-infecting viruses within the family Totiviridae. Adv Virol 163:2899–2902. https://doi.org/10.1007/s00705-018-3871-1
Isawa H et al (2011) Identification and molecular characterization of a new nonsegmented double-stranded RNA virus isolated from Culex mosquitoes in Japan. Virus Res 155:147–155. https://doi.org/10.1016/j.virusres.2010.09.013
Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Br Bioinform 9:286–298. https://doi.org/10.1093/bib/bbn013
King AMQ et al (2018) Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2018). Adv Virol 163:2601–2631. https://doi.org/10.1007/s00705-018-3847-1
Kobayashi S, Lauwereyns J, Koizumi M, Sakagami M, Hikosaka O (2002) Influence of reward expectation on visuospatial processing in macaque lateral prefrontal cortex. J Neurophysiol 87:1488–1498. https://doi.org/10.1152/jn.00472.2001
Kondo H et al (2016) Reprint of “Sequence and phylogenetic analyses of novel totivirus-like double-stranded RNAs from field-collected powdery mildew fungi”. Virus Res 219:39–50. https://doi.org/10.1016/j.virusres.2016.05.011
Kuwata R, Isawa H, Hoshino K, Sasaki T, Kobayashi M, Maeda K, Sawabe K (2015) Analysis of mosquito-borne flavivirus superinfection in Culex tritaeniorhynchus (Diptera: Culicidae) cells persistently infected with culex flavivirus (Flaviviridae). J Med Entomol 52:222–229. https://doi.org/10.1093/jme/tju059
Li WJ et al (2010) Mosquitoes and mosquito-borne arboviruses in the Qinghai–Tibet plateau-focused on the Qinghai area, China. Am J Trop Med Hyg 82:705–711. https://doi.org/10.4269/ajtmh.2010.09-0649
Lu BL (1997a) Fauna Sinica, Insecta vol 8, Diptera: Culicidae 1. Science Press, Beijing
Lu BL (1997b) Fauna Sinica, Insecta vol 9, Diptera: Culicidae II. Science Press, Beijing
Lu Z et al (2009) Tahyna virus and human infection, China. Emerg Infect Dis 15:306–309
Poulos BT, Tang KF, Pantoja CR, Bonami JR, Lightner DV (2006) Purification and characterization of infectious myonecrosis virus of penaeid shrimp. J Gen Virol 87:987–996. https://doi.org/10.1099/vir.0.81127-0
Read DA et al (2019) Diversity and distribution of maize-associated totivirus strains from Tanzania. Virus Genes. https://doi.org/10.1007/s11262-019-01650-6
Shapiro A, Green T, Rao S, White S, Carner G, Mertens PP, Becnel JJ (2005) Morphological and molecular characterization of a Cypovirus (Reoviridae) from the mosquito Uranotaenia sapphirina (Diptera: Culicidae). J Virol 79:9430–9438. https://doi.org/10.1128/JVI.79.15.9430-9438.2005
Strauss JH, Strauss EG (1994) The alphaviruses: gene expression, replication, and evolution. Microbiol Rev 58:491–562
Sun X et al (2009) Distribution of arboviruses and mosquitoes in northwestern Yunnan Province, China. Vector Borne Zoonotic Dis 9:623–630. https://doi.org/10.1089/vbz.2008.0145
Tang KF, Pantoja CR, Poulos BT, Redman RM, Lightnere DV (2005) In situ hybridization demonstrates that Litopenaeus vannamei, L. stylirostris and Penaeus monodon are susceptible to experimental infection with infectious myonecrosis virus (IMNV). Dis Aquat Organ 63:261–265. https://doi.org/10.3354/dao063261
van Cleef KW et al (2014) Mosquito and Drosophila entomobirnaviruses suppress dsRNA- and siRNA-induced RNAi. Nucleic Acids Res 42:8732–8744. https://doi.org/10.1093/nar/gku528
Wu Q, Luo Y, Lu R, Lau N, Lai EC, Li WX, Ding SW (2010) Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc Natl Acad Sci USA 107:1606–1611. https://doi.org/10.1073/pnas.0911353107
Yang J et al (2011) Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J Clin Microbiol 49:3463–3469. https://doi.org/10.1128/JCM.00273-11
Yang X, Zhang Y, Ge X, Yuan J, Shi Z (2012) A novel totivirus-like virus isolated from bat guano. Adv Virol 157:1093–1099. https://doi.org/10.1007/s00705-012-1278-y
Zhai Y et al (2010) Isolation and full-length sequence analysis of Armigeres subalbatustotivirus, the first totivirus isolate from mosquitoes representing a proposed novel genus (Artivirus) of the family Totiviridae. J Gen Virol 91:2836–2845. https://doi.org/10.1099/vir.0.024794-0
Acknowledgements
We thank the staff of the Kenli County Centers for Disease Control and Prevention for assistance with the mosquito collection.
Funding
This work was supported by National Science and Technology Major Project (2017ZX10104001); National Natural Science Foundation of China (31900156); National key research and development project (2016YFC1200905); the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
FL and HW were major contributors in writing the manuscript. WZ and AX collected the mosquitoes sample. SF, FL, WZ, QW, YH, and WL performed virus isolation. FL sequenced the genome of CTV-KL. JS performed the virus electron microscopy. FL, JD, and ZW did the Bioinformatics analysis. HW, LZ, and GL conceived the study and drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of supporting data
Not applicable.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Li, F., Du, J., Wu, Z. et al. Identification and genetic analysis of a totivirus isolated from the Culex tritaeniorhynchus in northern China. Arch Microbiol 202, 807–813 (2020). https://doi.org/10.1007/s00203-019-01788-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-019-01788-9