
Abstract
Summary
Odanacatib (ODN) was investigated as an osteoporosis treatment in 292 men. Compared with placebo, odanacatib improved bone mineral density and led to sustained bone resorption decreases while producing relatively little bone formation reduction that leveled off with time. However, increased risk of stroke in another study stopped further odanacatib development.
Introduction
ODN, a selective oral cathepsin K inhibitor, was in development for osteoporosis treatment. This phase 3, double-blind, randomized, placebo-controlled, 24-month study investigated ODN safety and efficacy in men with osteoporosis.
Methods
Men with idiopathic osteoporosis or osteoporosis due to hypogonadism and a lumbar spine or hip (total hip [TH], femoral neck [FN], or trochanter) bone mineral density (BMD) T-score of ≤ − 2.5 to ≥ − 4.0 without prior vertebral fracture or ≤ − 1.5 to ≥ − 4.0 with one prior vertebral fracture were randomized (1:1) to once-weekly ODN 50 mg or placebo. All received 5600 IU vitamin D3 weekly and calcium supplementation as needed (≥ 1200 mg daily). The primary efficacy outcome was changed from baseline in lumbar spine BMD versus placebo.
Results
Overall, 292 men, mean age 68.8 years, were randomly assigned to ODN or placebo. Versus placebo, ODN increased BMD from baseline at the lumbar spine, TH, FN, and trochanter by 5.6%, 2.0%, 1.7%, and 2.1%, respectively (all p < 0.01), and decreased uNTx/Cr (68%, p < 0.001), sCTx (77%, p < 0.001), sP1NP (16%, p = 0.001), and sBSAP (8%, p = 0.019). The between-group bone formation marker decrease peaked at 3 months, then returned toward baseline. The safety profile, including cardiovascular events, was similar between groups.
Conclusion
Though a promising osteoporosis therapy for men, ODN development was discontinued due to increased risk of stroke in the LOFT phase 3 trial.
Trial registration
Clinicaltrials.gov NCT01120600 (registered May 11, 2010).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Osteoporosis in men is associated with significant morbidity, mortality, and societal expense [1]. The lifetime osteoporosis-related fracture risk of a 50-year-old man is 13–22%, including a hip fracture risk of 3–11% [2]. Approximately one-third of all hip fractures occur in men [3] and are associated with higher mortality than in women, with 1-year mortality rates estimated at 32.5% and 21.9%, respectively [4].
Cathepsin K (CatK) is the primary protease involved in the osteoclastic degradation of organic bone matrix proteins [5]. Odanacatib (ODN), an oral selective inhibitor of CatK, does not reduce either osteoclast formation, general metabolic activity, or number; instead, it only reduces bone resorption mediated through inhibition of CatK released by the osteoclasts into resorption lacunae [6,7,8,9,10,11]. Clinical studies in postmenopausal women treated with ODN demonstrated relatively less reduction in bone formation than in bone resorption markers compared with bisphosphonates [12,13,14,15] and progressive increases in bone mineral density (BMD) up to 5 years [16]. Unlike bisphosphonates [17, 18], the effect of ODN on bone resorption was rapidly reversible, with BMD decreasing to baseline following discontinuation [19]. This 24-month, placebo-controlled, phase 3 study investigated the effect of ODN treatment of men with osteoporosis.
Materials and methods
Study design
This was an international, multicenter, double-blind, randomized, placebo-controlled, 24-month study (protocol MK-0822-053; ClinicalTrials.gov NCT01120600). Fifty-six centers in 13 countries (Chile, Colombia, Denmark, Estonia, France, Italy, Japan, Latvia, Mexico, the Netherlands, Russia, the UK, and the USA) participated on an out-patient basis. The study was conducted in accordance with the principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies. All participants provided written informed consent before the initiation of study procedures. The original study was divided into two parts: the primary analysis was planned at 24 months (part 1) and patients were to remain in the study for an additional 12 months (part 2). Part 2 was removed (protocol amendment 1 year into the study) to minimize the risk of bone loss to patients who would be on placebo during months 24 to 36.
Eligible men were randomized by a computer-generated allocation schedule using an interactive voice response system in a 1:1 ratio to receive ODN 50 mg once weekly (OW) or matching placebo OW, taken without regard to food. All participants received vitamin D3 (5600 IU OW, a dose selected to alleviate vitamin D deficiency; additional supplementation limited to < 400 IU daily as part of a multivitamin only) and daily calcium supplementation as needed to ensure a total daily calcium intake of 1200 mg. Participants were stratified by gonadal function (serum testosterone < 250 or ≥ 250 ng/dL), with a stratification goal of at least one-third of participants being hypogonadal. Investigator site personnel, study participants, and sponsor’s personnel were blinded to treatment allocation during the 24-month study period. Data collected during the study included BMD, biochemical markers of bone turnover, adverse events (AEs), laboratory safety evaluations, and spine radiographs (for morphometric fracture assessment). One statistician, not involved in the conduct of the study, reported unblinded safety results to an external data monitoring committee that performed periodic unblinded safety assessments of this and other ongoing ODN clinical trials. There were seven planned clinic visits (screening, randomization, and after months 3, 6, 12, 18, and 24) and a posttreatment follow-up telephone call (≥ 14 days after the last dose of blinded study therapy or study discontinuation, whichever occurred later). An optional transilial bone biopsy procedure was performed for qualitative histology and quantitative histomorphometry for consenting volunteers at month 24.
Participants
Participants were ambulatory men aged 40–95 years with idiopathic osteoporosis or osteoporosis due to hypogonadism, who had lumbar spine anatomy suitable for dual-energy X-ray absorptiometry and were candidates for osteoporosis therapy. A BMD T-score (male reference range) of ≤ − 2.5 at either the lumbar spine or total hip, femoral neck, or trochanter and ≥ −4.0 at all sites, without prior vertebral fracture (based on morphometric evaluation of a baseline spine radiograph), or a BMD T-score ≤ − 1.5 at either the lumbar spine or total hip or any of the above hip subregions and ≥ − 4.0 at all sites, with one prior vertebral fracture, was required for study inclusion. Participants unable or unwilling to use available osteoporosis treatments could be included with a BMD T-score ≤ − 2.5 at the lumbar spine or the hip regions noted above without prior vertebral fracture or ≤ − 1.5 with prior vertebral fracture.
Exclusion criteria included the following: prior clinical fragility hip fracture; any clinical fragility fracture within the past 12 months; > 1 prior vertebral fracture; metabolic bone diseases other than osteoporosis; daily calcium intake of < 1200 mg (diet plus supplemental); inadequately controlled primary or secondary hyperparathyroidism; vitamin D deficiency (25-hydroxyvitamin D < 15 ng/mL; participants who were otherwise eligible could be retested after vitamin D repletion); malignancy within the past 5 years (except for adequately treated skin cancer); inadequately controlled thyroid disease; and severe renal insufficiency (creatinine clearance ≤ 29 mL/min). Oral bisphosphonates in the 6 months prior to randomization or for > 6 weeks within the prior 3 years, or any lifetime use of intravenous bisphosphonate or RANK ligand inhibitor (i.e., denosumab), were not permitted; additional prohibited medications are listed in the Supplemental Material.
Efficacy endpoints
The primary efficacy endpoint was the percent change from baseline in lumbar spine BMD with ODN compared with placebo at 24 months. Secondary efficacy endpoints included the following: percent change from baseline in the femoral neck, trochanter, and total hip BMD with ODN versus placebo at 24 months and geometric mean percent change from baseline in biochemical markers of bone resorption (serum C-telopeptides of type I collagen [sCTx], urinary N-telopeptides of type I collagen/creatinine ratio [uNTx/Cr]) and biochemical markers of bone formation (serum bone-specific alkaline phosphatase [sBSAP], serum N-terminal propeptide of type I collagen [sP1NP]) at 24 months. Within-group changes from baseline were evaluated for primary and secondary BMD endpoints. Exploratory endpoints included percent change from baseline in total body and distal forearm BMD at 24 months with ODN versus placebo and effect on qualitative histomorphometry and skeletal microarchitecture of transilial bone biopsy specimens using quantitative 2D histomorphometry and 3D μ-computerized tomography at 24 months.
BMD and vertebral fracture assessment
BMD was measured by dual-energy X-ray absorptiometry (GE Medical Systems Lunar, Madison, WI, USA, or Hologic, Bedford, MA, USA) at the lumbar spine and hip at screening and distal forearm and total body at randomization. BMD at the hip and spine was measured at months 3 and 6, and BMD at all skeletal sites was measured at months 12 and 24 or at early termination. Scans were analyzed at a BMD analysis and quality control center (Perceptive Informatics, Inc. [PARAXEL], Billerica, MA, USA). BMD eligibility criteria were determined by the central imaging vendor based on T-scores determined using the NHANES III 1998 reference values for Caucasian young adult men [20].
Radiographic vertebral fractures were evaluated by a central imaging vendor (Perceptive Informatics, Inc. [PARAXEL]) by semi-quantitative [21] and morphometric analysis of the lateral spine radiographs at screening, month 24, and any other time a fracture was suspected. New morphometric fractures were defined as progression of vertebral deformity in a T4 through L5 vertebral body and were assessed using a Genant semi-quantitative scale (score 0 to 3).
Serum and urine biochemical measures
Participants provided fasting morning blood samples and a second morning voided urine specimen for bone turnover marker measurements at randomization and at months 3, 6, 12, 18, and 24 or early termination. Markers were measured using the following assays: sCTx, Elecsys–CrossLaps/serum ECL kit (Roche Diagnostics, Mannheim, Germany); uNTx/Cr, Vitros ECL/ECIQ (Ortho Clinical Diagnostics, Rochester, NY, USA); sBSAP, Access Ostase (Beckman Coulter, Brea, CA, USA); sP1NP, Elecsys–serum ECL kit (Roche Diagnostics) and urine creatinine, Konelab 20 analyzer (Thermo Fisher Scientific, Waltham, MA, USA). Analyses were performed by PPD Central Lab (Highland Heights, KY, USA) in a non-batched fashion. Total serum testosterone (morning samples; run on Siemens Centaur XP using chemiluminescence [ADVIA Centaur testosterone; Siemens Healthcare Diagnostics Inc., Tarrytown, NY, USA]) and 25-hydroxyvitamin D (DiaSorin LIAISON 25 OH Vitamin D TOTAL chemiluminescent immunoassay [DiaSorin, Saluggia, VC, Italy]) were measured at the screening. Sex hormone binding globulin levels were not measured, and levels of free testosterone were not calculated.
Bone biopsy
Volunteers were given locally sourced tetracycline at month 23 for 3 days, with a 14-day interval prior to a second 3-day course. Transilial bone biopsy was performed 5 to 14 days later. The 2D histomorphometry on transilial bone biopsies from month 24 included the following measurements: bone volume, osteoid volume, trabecular number, trabecular thickness, osteoid surface, osteoid thickness, mineral apposition rate, and mineralizing surface [22].
The 3D μ-computerized tomography performed on transilial bone biopsies from month 24 included the following measurements: percent bone volume fraction, bone surface density, trabecular number, trabecular thickness, and connectivity density. All bone biopsy evaluations were performed at a central laboratory (R. Recker, Creighton University, Omaha, NE, USA).
Safety measurements
Safety was assessed by review of AEs, vital signs, and laboratory safety evaluations. Clinical and laboratory evaluations (including serum chemistry, hematology, and urinalysis) were performed at screening and months 3, 6, 12, 18, and 24 or at early termination. Laboratory evaluations were performed centrally (PPD Laboratories, Highland Heights, KY, USA). AE assessments were performed throughout the study period, nominally through the final study visit. AEs reported after the last scheduled study visit but within 14 days after the last dose of study medication were also captured. Electrocardiograms were performed at randomization. All participants were assessed for vertebral fracture at screening and at month 24 with a lateral spine X-ray [21]. If an incident fracture was suspected based on clinical signs or symptoms, it was evaluated radiographically and, if present, reported as a fracture AE based on review by the study investigators. Lateral spine films were read by a radiologist at the central imaging vendor (Perceptive Informatics, Inc. [PARAXEL]). Participants with excessive BMD loss at the lumbar spine or total hip of ≥ 7% from baseline at any postbaseline time point were discontinued from both the study and study medication, and instructed to pursue treatment for their osteoporosis from their primary care physician. In countries where local regulatory guidelines are allowed, patients who were discontinued from the study were offered a 1-year supply of 70 mg OW alendronate, or 70 mg OW alendronate + 2800 IU vitamin D3, or 70 mg OW alendronate + 5600 IU vitamin D3 for optional prescription by their primary care physician. The following four specific AE categories of special interest were adjudicated by an expert committee, to confirm their occurrence and specific attributes: dental (to confirm or exclude osteonecrosis of the jaw); skin AEs with skin thickening and hardening suggestive of morphea or systemic sclerosis; delayed fracture unions; and serious respiratory events. Each committee comprised external and independent specialists (i.e., neither investigators nor study monitors) and was organized and run by an independent vendor (PAREXEL International, Waltham, MA, USA).
Adherence
Compliance with study treatment was monitored with participant-completed diary cards and tablet counts of returned medication. Compliance was reported as the proportion of the actual days with double-blind treatment intake to the expected number of days of treatment intake.
Statistical methods
Full statistical methods are provided in the Supplemental Materials. Briefly, a constrained longitudinal data analysis (cLDA) method [23] was used to obtain a point estimate and a 95% confidence interval (CI) of the between-treatment difference for BMD endpoints using the full analysis set (FAS) population, which included all participants who took at least one dose of study drug and had baseline measurements. Secondary and exploratory BMD endpoints were analyzed using a similar approach. Biochemical markers were analyzed in the per-protocol population, using a similar cLDA model as described for the BMD endpoints. Safety analyses were performed using the all-patients-as-treated (APaT) population, which included all participants who took at least one dose of the study drug.
Subgroup analyses
A prespecified subgroup analysis estimated (with a nominal 95% CI) whether or not the between-group treatment effect on lumbar spine BMD was consistent across the following subgroups: age (< 65 years, ≥ 65 years); geographical region (Europe, non-Europe); biochemical markers of bone turnover at baseline (tertiles); and height and weight at baseline (below/above median). Only summary statistics were calculated for subgroups of serum testosterone < 250 or ≥ 250 ng/dL.
Results
Patients
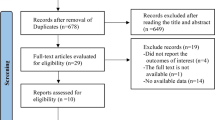
The study took place between January 6, 2010, and July 22, 2013. Of the 949 men screened for inclusion, 294 were enrolled and randomized (Fig. 1). Of the randomized participants, 292 took ≥ 1 dose of study medication (one patient in each group did not take any study medication) and were included in the APaT population; 269 were included in the FAS population for the primary efficacy parameter. Overall, 128 (87.1%) men in the ODN and 115 (78.2%) in the placebo group completed the study. Study discontinuation was numerically higher in the placebo group (n = 32, 21.8%) compared with the ODN group (n = 19, 12.9%). There were no clinically meaningful differences in reasons for discontinuation between groups, although the excessive bone loss was the reason for discontinuation in 4 men (2.7%) in the placebo group, compared with none in the ODN group (Fig. 1). Demographic and baseline characteristics were similar between the two treatment groups (Table 1). Mean age of the study population at baseline was 68.8 years, and total testosterone levels were < 250 ng/dL in 8 patients (5.8%) and 9 patients (6.2%) in the ODN and placebo groups, respectively.

Patient disposition
BMD
Treatment with ODN for 24 months significantly increased BMD from baseline at the lumbar spine and all three hip sites compared with placebo (Fig. 2, Supplemental Table 1). Least-squares (LS) mean differences in BMD versus placebo at 24 months were 5.6% at the lumbar spine (95% CI 4.5, 6.7; p < 0.001), 2.0% at the total hip (95% CI 1.3, 2.8; p < 0.001), 1.7% at the femoral neck (95% CI 0.5, 2.9; p = 0.008), and 2.1% at the trochanter (95% CI 0.9, 3.3; p < 0.001). At the lumbar spine, BMD increased continuously over time to month 24 in the ODN-treated group. Total body BMD also significantly increased from baseline with ODN treatment compared with placebo by 0.8% (95% CI 0.2, 1.5; p = 0.007 [data not shown]). Between-group increases were generally due to increases in the ODN treatment group, with relatively small decreases in the placebo group (Supplemental Table 1). No treatment difference was observed at 24 months between ODN and placebo for one-third radius BMD (0.01%; 95% CI − 0.87, 0.89; p = 0.976 [data not shown]).

Percent change from baseline in BMD at the a lumbar spine, b total hip, c femoral neck, and d trochanter (FAS population)
Biochemical markers of bone turnover
ODN treatment significantly decreased biochemical markers of bone resorption from baseline compared with placebo by month 3 (Fig. 3a, b). The geometric LS mean treatment differences were similar thereafter: at month 24, differences in LS mean were − 76.6% for sCTx (95% CI − 92.6, − 60.6; p < 0.001) and − 68.1% for uNTx/Cr (95% CI − 78.1, − 58.1; p < 0.001). While the between-group differences of indices in both bone resorption markers followed the same time course, the within-group changes were somewhat different. There was an increase in sCTx with placebo of 29.0% at month 3, 37.8% at month 6, and 50.7% at month 12, and it was stable thereafter (56.5% [95% CI 40.0, 75.0] at month 24). In contrast, the uNTx/Cr change was minimal over time (LS mean change 6.65% [95% CI − 2.68, 16.87] at month 24) in placebo-treated patients. In the ODN-treated group, there was a marked decrease in the geometric LS mean percent change from baseline in sCTx at month 3, increasing thereafter with only a small decrease from baseline at month 24. There was also a marked decrease in the geometric LS mean uNTx/Cr at month 3 in the ODN group that was maintained through month 24.

Geometric mean percent change from baseline in markers of bone resorption a sCTx and b uNTx/Cr, and markers of bone formation c sBSAP and d sP1NP (per-protocol population)
Biochemical markers of bone formation also significantly decreased following 24 months of ODN treatment compared with placebo (Fig. 3c, d). The geometric LS mean treatment differences at 24 months were as follows: − 7.9% for sBSAP (95% CI − 14.6, − 1.3; p = 0.019) and − 16.0% for sP1NP (95% CI − 25.7, − 6.3; p = 0.001). The maximum between-group difference in sBSAP was 16.0% and in sP1NP was 33.3% at month 3, and the differences between treatment groups decreased over time. In the ODN-treated group, the geometric LS mean percent change from baseline for sBSAP decreased by − 21.0% at month 6 and then increased from the nadir while remaining below baseline concentrations over time but with only a modest reduction from baseline at month 24. In the placebo group, sBSAP decreased slightly from baseline until month 6 (− 8.2%), then increased to baseline levels by month 24. For sP1NP, a prominent decrease in the geometric LS mean percent change was seen in the ODN group at month 3 (− 40.7%), with an increase from the month 3 nadir over the remainder of the double-blind treatment period, but remaining below baseline at month 24. In the placebo group, sP1NP decreased slightly by month 6 (− 9.0%) and then increased to baseline levels by month 12.
Bone biopsy histology and histomorphometry
Histologic evaluation of transilial bone biopsies from 26 patients (11 ODN, 15 placebo) revealed no qualitative abnormalities in either treatment group within the categories of abnormal osteoid, cortical trabecularization, marrow dyscrasia, marrow fibrosis, non-lamellar bone, osteomalacia, woven bone, or other pathologic findings (data not shown). Tetracycline labeling occurred in all biopsies; however, the proportion of biopsies with double-tetracycline labeling was lower in patients treated with ODN versus placebo (54.5% versus 100%). Histomorphometry showed no evidence of impaired bone formation or mineralization with ODN (Table 2), but due to the relatively small numbers of biopsies and the inability to evaluate all parameters in all biopsies, hypothesis testing for quantitative histomorphometry measures was not planned in the statistical analysis plan and therefore not performed.
Subgroup analyses
The treatment effect on the percent change from baseline in the lumbar spine (Supplemental Fig. S1) at month 24 was generally consistent across the subgroups defined by age (< 65, ≥ 65 years), region (Europe, non-Europe), and height and weight at baseline (below/above median). For the bone formation biomarkers sBSAP and sP1NP (analyzed by tertile), a larger BMD treatment difference was observed in patients in the tertile with the highest baseline concentrations. A similar pattern was observed for the bone resorption marker uNTx/Cr, with the largest BMD treatment difference in patients in the tertile subgroup with the highest, relative to the lowest, baseline concentration. In contrast, no clear pattern in the extent of BMD treatment difference was observed across subgroups by tertiles of baseline concentrations of the bone resorption marker sCTx. Analyses of the same subgroups performed for total hip, femoral neck, and trochanter BMD did not suggest any relationship between baseline biochemical markers and change in BMD at these femoral sites. Mean percent change in BMD at all four sites was ~ 2-fold greater in patients with testosterone < 250 ng/dL versus those with testosterone ≥ 250 ng/dL. However, the 95% CIs around the point estimates for each BMD region widely overlapped. Results of the subgroup analysis are based on relatively small numbers and should, therefore, be interpreted with caution.
Safety
Clinical AEs (treatment-emergent and posttreatment but during the study period) were reported for 230 (78.8%) of the APaT population, with the overall incidence balanced between treatment groups (Table 3). There was a low incidence of drug-related AEs and AEs leading to discontinuation in both groups. One patient in the ODN group discontinued due to myalgia that was considered by the study site investigator to be drug related. The incidence of serious AEs was similar between the two groups; these were not considered to be drug related. There was one death in the placebo group where the AE onset was reported during the double-blind period (alcohol-related hepatic failure); further three patients died due to AEs that were reported > 14 days after their last dose of study medication (two in the ODN group [sudden death of no specific cause identified and multiorgan failure following sepsis] and one in the placebo group [pancreatic carcinoma]). Two patients experienced laboratory AEs that led to discontinuation of study medication, both in the placebo group. Evaluations of laboratory safety tests (group mean changes and changes exceeding predefined limits), vital signs, and weight showed no clinically meaningful between-group differences. Serum calcium (mean) was approximately 0.1 to 0.2 mg/dL lower in the ODN group than in the placebo group, and serum phosphate was similar between groups. There were no AEs of clinical or laboratory (subclinical) hypocalcemia.
One patient in the ODN group had a skin event of clinical interest (erysipelas). No confirmed events of morphea/scleroderma occurred. A total of 13 patients (8 in the ODN group, 5 in the placebo group) had clinical fracture AEs. One patient in the ODN group experienced a confirmed non-serious AE of delayed fracture union. The fracture occurred following moderate energy trauma and could not be repositioned without surgery (which the patient refused). Among patients with evaluable spine X-ray pairs, two patients in the ODN (2/128, 1.6%) and four in the placebo group (4/123, 3.3%) had one new morphometric vertebral fracture during the 24-month treatment period. There were no cases of osteonecrosis of the jaw or atypical femoral fracture. One patient in the ODN group reported a serious respiratory infection (pneumonia and respiratory failure) confirmed by adjudication, but the investigator considered this unrelated to study treatment.
Discussion
This, the only study of ODN in men with osteoporosis, was designed to assess the effects of ODN on BMD at key skeletal sites. Treatment with ODN 50 mg weekly for 24 months increased BMD at the lumbar spine and all proximal femur sites. The increases were numerically comparable at the spine and perhaps less at the proximal femur sites than observed in a prior study of postmenopausal women after 24 months of treatment [12]. LOFT (Long-term Odanacatib Fracture Trial), which included ~ 16,000 women with postmenopausal osteoporosis, reported BMD increases versus placebo of 10.9% at the lumbar spine, 10.3% at the total hip, 10.1% at the femoral neck, and 14.6% at the trochanter, after 5 years of treatment [16].
Men who become hypogonadal due to disease or trauma, or as a consequence of androgen deprivation therapy, experience bone loss and increased fracture risk [24]. While it is standard to evaluate the effects of a drug in both men and women, there are no data to indicate that hypogonadal osteoporotic men would respond to ODN any differently from postmenopausal osteoporotic women. Given that many osteoporotic men are not hypogonadal, it was important to include them in this trial. Although that objective was achieved, this study did not reach the 1/3 target enrollment of hypogonadal men with osteoporosis, and only 17 of 292 osteoporotic men treated in this study were hypogonadal (baseline testosterone < 250 ng/dL). Many men with hypogonadism were excluded from enrollment due to prior testosterone treatment or a history of cancer ≤ 5 years prior.
The observed increase in lumbar spine BMD with placebo may be due to use of vitamin D and/or calcium by study participants or alternatively due to progression of spinal degenerative disease. Regardless, this phenomenon has been observed previously in other interventional studies of men with osteoporosis [25, 26]. However, BMD at the proximal femoral sites was stable in the placebo group.
ODN treatment resulted in rapid, substantial, and sustained decreases in bone resorption markers compared with placebo, similar to those observed with ODN in postmenopausal osteoporotic women [12]. There was a gradual return of sCTx to baseline with ODN, after an initial marked decrease, which is explained by increases in larger CTx species and tartrate-resistant acid phosphatase 5b, as shown by a post hoc batched biomarker study using data from LOFT [27]. In addition, there was an initial, modest decrease in markers of bone formation with ODN, which returned toward baseline levels (and levels in patients treated with placebo) at the end of the 24-month study, and which was also seen with ODN in postmenopausal osteoporotic women [12]. The observed transient reductions in sP1NP and sBSAP with ODN highlight the unique mechanism of action of ODN, whereby it is likely that bone resorption inhibition occurs while bone formation is preserved because the signaling between osteoclasts and osteoblasts is maintained.
The increase from baseline in sCTx in the placebo group over the first 6 months to 1 year of the study was unanticipated. After a decrease in sCTx in the ODN group at month 3, a similar “drift” upward was observed through month 24. The sCTx analysis method was reviewed as part of a central laboratory vendor data review; although several possible explanations were hypothesized, no problem with the assay, specimen collection, or handling was detected. While no assay-related problem was identified, this remains a possibility. What does appear clear is that treatment with ODN reduces sCTx versus placebo by 76.6% after 24 months. This is quantitatively similar to the reduction in uNTx/Cr versus placebo of 68.1% after 24 months. The between-group reduction in each resorption marker was reached at month 3 and sustained through month 24.
Iliac crest histologic examination revealed no qualitative abnormalities in any of the biopsies. Histomorphometric investigation did not indicate any impairment of bone mineralization: mineral apposition rate and mineralization lag time were similar in patients treated with ODN and placebo, and osteoid thickness, surface, area, and volume were generally lower in patients treated with placebo. However, our study did not assess resorption parameters, which is where it would be expected to see a difference between ODN- and placebo-treated men. These results are consistent with histologic and histomorphometry findings in postmenopausal women treated with ODN [28] and with the unique mechanism of action of ODN compared with other drugs that inhibit bone resorption.
ODN was generally well tolerated in this study, with no unexpected safety findings. No AEs of morphea/scleroderma were reported. Morphea-like lesions in women treated with ODN were reported for the first time in LOFT (in 0.1% of patients treated with ODN [12/8043; plus one further case reported only after database lock] and 0.04% of patients [3/8028] given placebo) [16]. It is unlikely that any morphea-like AEs would have been observed in patients treated with ODN in the current study, based on sample size, if the risk in men was similar to the risk observed in postmenopausal women in LOFT. Further clinical development of ODN was stopped following an observed increase in the risk of stroke in LOFT [29]; however, it should be noted that neither the risk of myocardial infarction nor other major adverse cardiovascular events (MACE) were increased [16]. In the current study, cerebrovascular accident was reported for one patient (0.7%) in the ODN group and no patients in the placebo group. Myocardial infarction was reported for no patients in the ODN group and two patients (1.4%) in the placebo group.
There are differences in the pathophysiology of osteoporosis in men and women; consequently, it is important to assess new treatments in both sexes. While the potential causes of osteoporosis are similar in older men and women, hypogonadism is found in older women due to menopause. While hypogonadism causes similar bone loss in men, it is uncommon except in men with prostate cancer who are treated to induce hypogonadism. Age-related decreases in estrogen and testosterone are much more gradual in the great majority of men. A recent study has shown that ~ 60% of interventional trials of osteoporosis treatments enrolled only women, while only 4% of trials were specific to men [30]. Generally, only studies of glucocorticoid-induced osteoporosis enroll both men and women in the same trial. Nonetheless, most current treatments for postmenopausal osteoporosis (except for estrogens and selective estrogen receptor modulators) have been evaluated in men after they have been shown to reduce fracture risk in postmenopausal women. In men with osteoporosis receiving calcium and vitamin D supplementation, alendronate and risedronate demonstrated comparable BMD increases at 24 months to those reported here for ODN. Treatment with alendronate versus placebo resulted in BMD increases of 5.3% and 2.6% at the lumbar spine and femoral neck, respectively (p < 0.001 for both), as well as a significant reduction in vertebral (but not non-vertebral) fracture risk [26]. Compared with placebo, risedronate treatment resulted in BMD increases of 4.6% (p < 0.001) at the lumbar spine and approximately 1.2% at the femoral neck (p value not reported); a significant treatment difference was not seen for fracture risk reduction [31]. In men with primary or hypogonadism-associated osteoporosis, treatment with zoledronate increased lumbar spine BMD by 6.1% versus placebo (p < 0.001) at 24 months and reduced the risk of new morphometric vertebral fractures by 67% (relative risk 0.33; 95% CI 0.16, 0.70; p = 0.002) [32]. Treatment with the RANK ligand inhibitor denosumab has also been shown to increase BMD in men with low BMD [33, 34]. Parathyroid hormone (teriparatide, hPTH[1–34] analog) has also been investigated in men with low BMD, although the study was halted due to safety signals in preclinical models [35]. Increases in BMD in men treated with teriparatide were comparable with those seen in women with postmenopausal osteoporosis, although different treatment durations were used in the two studies, making decisive comparisons difficult [35, 36].
In addition to showing that ODN increases BMD in men as well as in postmenopausal women, the present study supports previous findings in postmenopausal women indicating that treatment with ODN results in relatively less reduction in markers of bone formation than bone resorption compared with bisphosphonates [12,13,14,15, 19]. This bone-formation–sparing antiresorptive effect of ODN differentiates it from other currently available treatments for osteoporosis.
A strength of the study was that the data were obtained in eugonadal men with idiopathic osteoporosis. Many men in the general population with osteoporosis are eugonadal and the etiology idiopathic. There have been no studies of ODN (or any other drug) in eugonadal premenopausal women with idiopathic osteoporosis that could be used for comparison. This study had a number of limitations, notably the inability to recruit the prespecified one-third of volunteers with hypogonadism, meaning that the results may have limited generalizability beyond eugonadal men with idiopathic osteoporosis. In addition, hypogonadism was only assessed by serum total testosterone and not serum free testosterone. Subgroup analyses indicated numerically greater BMD changes in the hypogonadal subgroup at both spine and proximal femoral sites. The population studied was predominantly white and from Europe or the USA. While the study assessed change in BMD with ODN at key skeletal sites, fracture prevention was not evaluated directly; a much larger cohort would be required to power the study to detect significant change in fracture risk.
In conclusion, in this study of men with osteoporosis, ODN resulted in significant increases in BMD versus placebo at key sites of fragility fracture (lumbar spine and proximal femur), reduced markers of bone resorption with comparatively small effects on bone formation, and was generally well tolerated.
Data availability
The data sharing policy of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, including restrictions, is available at http://engagezone.merck.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.
References
Willson T, Nelson SD, Newbold J, Nelson RE, LaFleur J (2015) The clinical epidemiology of male osteoporosis: a review of the recent literature. Clin Epidemiol 7:65–76
Johnell O, Kanis J (2005) Epidemiology of osteoporotic fractures. Osteoporos Int 16(Suppl 2):S3–S7
Johnell O, Kanis JA (2006) An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int 17:1726–1733
Brauer CA, Coca-Perraillon M, Cutler DM, Rosen AB (2009) Incidence and mortality of hip fractures in the United States. JAMA 302:1573–1579
Costa AG, Cusano NE, Silva BC, Cremers S, Bilezikian JP (2011) Cathepsin K: its skeletal actions and role as a therapeutic target in osteoporosis. Nat Rev Rheumatol 7:447–456
Reid IR, Miller PD, Brown JP, Kendler DL, Fahrleitner-Pammer A, Valter I, Maasalu K, Bolognese MA, Woodson G, Bone H, Ding B, Wagman RB, San Martin J, Ominsky MS, Dempster DW, Denosumab Phase 3 Bone Histology Study Group (2010) Effects of denosumab on bone histomorphometry: the FREEDOM and STAND studies. J Bone Miner Res 25:2256–2265
Masarachia PJ, Pennypacker BL, Pickarski M, Scott KR, Wesolowski GA, Smith SY, Samadfam R, Goetzmann JE, Scott BB, Kimmel DB, Duong LT (2012) Odanacatib reduces bone turnover and increases bone mass in the lumbar spine of skeletally mature ovariectomized rhesus monkeys. J Bone Miner Res 27:509–523
D’Amelio P, Grimaldi A, Cristofaro MA, Ravazzoli M, Molinatti PA, Pescarmona GP, Isaia GC (2010) Alendronate reduces osteoclast precursors in osteoporosis. Osteoporos Int 21:1741–1750
D’Amelio P, Grimaldi A, Di Bella S, Tamone C, Brianza SZ, Ravazzoli MG, Bernabei P, Cristofaro MA, Pescarmona GP, Isaia G (2008) Risedronate reduces osteoclast precursors and cytokine production in postmenopausal osteoporotic women. J Bone Miner Res 23:373–379
Migliaccio S, Brama M, Spera G (2007) The differential effects of bisphosphonates, SERMS (selective estrogen receptor modulators), and parathyroid hormone on bone remodeling in osteoporosis. Clin Interv Aging 2:55–64
Leung P, Pickarski M, Zhuo Y, Masarachia PJ, Duong LT (2011) The effects of the cathepsin K inhibitor odanacatib on osteoclastic bone resorption and vesicular trafficking. Bone 49:623–635
Bone HG, McClung MR, Roux C, Recker RR, Eisman JA, Verbruggen N, Hustad CM, DaSilva C, Santora AC, Ince BA (2010) Odanacatib, a cathepsin-K inhibitor for osteoporosis: a two-year study in postmenopausal women with low bone density. J Bone Miner Res 25:937–947
Rizzoli R, Benhamou CL, Halse J, Miller PD, Reid IR, Rodríguez Portales JA, DaSilva C, Kroon R, Verbruggen N, Leung AT, Gurner D (2016) Continuous treatment with odanacatib for up to 8 years in postmenopausal women with low bone mineral density: a phase 2 study. Osteoporos Int 27:2099–2107
Bonnick S, Saag KG, Kiel DP, McClung M, Hochberg M, Burnett SAM, Sebba A, Kagan R, Chen E, Thompson DE, de Papp AE (2006) Comparison of weekly treatment of postmenopausal osteoporosis with alendronate versus risedronate over two years. J Clin Endocrinol Metab 91:2631–2637
Reid DM, Hosking D, Kendler D, Brandi ML, Wark JD, Marques-Neto JF, Weryha G, Verbruggen N, Hustad CM, Mahlis EM, Melton ME (2008) A comparison of the effect of alendronate and risedronate on bone mineral density in postmenopausal women with osteoporosis: 24-month results from FACTS-International. Int J Clin Pract 62:575–584
McClung MR, O’Donoghue ML, Papapoulos SE, Bone H, Langdahl B, Saag KG, Reid IR, Kiel DP, Cavallari I, Bonaca MP, Wiviott SD, de Villiers T, Ling X, Lippuner K, Nakamura T, Reginster J-Y, Rodriguez-Portales JA, Roux C, Zanchetta J, Zerbini CAF, Park J-G, Im K, Cange A, Grip LT, Heyden N, DaSilva C, Cohn D, Massaad R, Scott BB, Verbruggen N, Gurner D, Miller DL, Blair ML, Polis AB, Stoch SA, Santora A, Lombardi A, Leung AT, Kaufman KD, Sabatine MS, Investigators LOFT (2019) Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol 7:899–911
Whitaker M, Guo J, Kehoe T, Benson G (2012) Bisphosphonates for osteoporosis--where do we go from here? N Engl J Med 366:2048–2051
Black DM, Bauer DC, Schwartz AV, Cummings SR, Rosen CJ (2012) Continuing bisphosphonate treatment for osteoporosis--for whom and for how long? N Engl J Med 366:2051–2053
Eisman JA, Bone HG, Hosking DJ, McClung MR, Reid IR, Rizzoli R, Resch H, Verbruggen N, Hustad CM, DaSilva C, Petrovic R, Santora AC, Ince BA, Lombardi A (2011) Odanacatib in the treatment of postmenopausal women with low bone mineral density: three-year continued therapy and resolution of effect. J Bone Miner Res 26:242–251
Looker AC, Wahner HW, Dunn WL, Calvo MS, Harris TB, Heyse SP, Johnston CC Jr, Lindsay R (1998) Updated data on proximal femur bone mineral levels of US adults. Osteoporos Int 8:468–489
Genant HK, Wu CY, van Kuijk C, Nevitt MC (1993) Vertebral fracture assessment using a semiquantitative technique. J Bone Miner Res 8:1137–1148
Dempster D, Compston JE, Drezner MK, Glorieux F, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM (2013) Standardized, nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 28:2–17
Zeger SL, Liang KY (1986) Longitudinal data analysis for discrete and continuous outcomes. Biometrics 42:121–130
Golds G, Houdek D, Arnason T (2017) Male hypogonadism and osteoporosis: the effects, clinical consequences, and treatment of testosterone deficiency in bone health. Int J Endocrinol 2017:4602129
Nakamura T, Matsumoto T, Sugimoto T, Hosoi T, Miki T, Gorai I, Yoshikawa H, Tanaka Y, Tanaka S, Sone T, Nakano T, Ito M, Matsui S, Yoneda T, Takami H, Watanabe K, Osakabe T, Shiraki M, Fukunaga M (2014) Clinical Trials Express: fracture risk reduction with denosumab in Japanese postmenopausal women and men with osteoporosis: denosumab fracture intervention randomized placebo controlled trial (DIRECT). J Clin Endocrinol Metab 99:2599–2607
Orwoll E, Ettinger M, Weiss S, Miller P, Kendler D, Graham J, Adami S, Weber K, Lorenc R, Pietschmann P, Vandormael K, Lombardi A (2000) Alendronate for the treatment of osteoporosis in men. N Engl J Med 343:604–610
Duong LT, Pickarsky M, Clark S, Giezek H, Cohn D, Massaad R, Stoch SA (2016) Differential effects of odanacatib therapy on markers of bone resorption and formation in postmenopausal women with osteoporosis: a subgroup study of the 5-year data from the extension of the phase 3 Long-term Odanacatib Fracture Trial (LOFT). American Society for Bone and Mineral Research (ASBMR), 2016 September 16–19, Atlanta, Georgia, USA. Abstract SA0299
Recker R, Dempster D, Langdahl B, Giezek H, Clark S, Ellis G, de Villiers T, Valter I, Zerbini CAF, Cohn D, Santora A, Duong LT (2020) Effects of odanacatib on bone structure and quality in postmenopausal women with osteoporosis: 5-year data from the Phase 3 Long-Term Odanacatib Fracture Trial (LOFT) and its extension. J Bone Miner Res 35(7):1289–1299.
Mullard A (2016) Merck & Co. drops osteoporosis drug odanacatib. Nat Rev Drug Discov 15:669
Barnard K, Lakey WC, Batch BC, Chiswell K, Tasneem A, Green JB (2016) Recent clinical trials in osteoporosis: a firm foundation or falling short? PLoS One 11:e0156068
Boonen S, Orwoll ES, Wenderoth D, Stoner KJ, Eusebio R, Delmas PD (2009) Once-weekly risedronate in men with osteoporosis: results of a 2-year, placebo-controlled, double-blind, multicenter study. J Bone Miner Res 24:719–725
Boonen S, Reginster JY, Kaufman JM, Lippuner K, Zanchetta J, Langdahl B, Rizzoli R, Lipschitz S, Dimai HP, Witvrouw R, Eriksen E, Brixen K, Russo L, Claessens F, Papanastasiou P, Antunez O, Su G, Bucci-Rechtweg C, Hruska J, Incera E, Vanderschueren D, Orwoll E (2012) Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med 367:1714–1723
Langdahl BL, Teglbjaerg CS, Ho PR et al (2015) A 24-month study evaluating the efficacy and safety of denosumab for the treatment of men with low bone mineral density: results from the ADAMO trial. J Clin Endocrinol Metab 100:1335–1342
Orwoll E, Teglbjaerg CS, Langdahl BL et al (2012) A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab 97:3161–3169
Orwoll ES, Scheele WH, Paul S, Adami S, Syversen U, Diez-Perez A, Kaufman JM, Clancy AD, Gaich GA (2003) The effect of teriparatide [human parathyroid hormone (1-34)] therapy on bone density in men with osteoporosis. J Bone Miner Res 18:9–17
Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mellström D, Oefjord ES, Marcinowska-Suchowierska E, Salmi J, Mulder H, Halse J, Sawicki AZ, Mitlak BH (2001) Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 344:1434–1441
Acknowledgments
Medical writing, under the direction of the authors, was provided by Annette Smith, PhD, of CMC AFFINITY, McCann Health Medical Communications, in accordance with Good Publication Practice (GPP3) guidelines. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. We also acknowledge Steven Doleckyj for prior clinical trial, data and operations management.
Funding
Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
BBS, HG, and ACS are, or were at the time of study conduct, employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and may own stock and/or stock options in the company. NB has received research support from Radius and is a consultant for Amgen. RC has been a consultant and/or has given talks for AbbVie, Amgen, Bristol-Myers Squibb, Chugai, Janssen-Cilag, Lilly, MSD, Pfizer, Radius, Sandoz, and UCB. BLL has received research grants (institution) from Amgen and Novo Nordisk and has been involved in advisory boards and lecturing for Amgen, Eli Lilly, MSD, and UCB. EO has received research support from Lilly and Mereo and provides consulting for Bayer and Amgen. ACS is currently a consultant and Chief Medical Officer of Entera Bio Ltd.
Ethics approval
The study was conducted in accordance with the principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies.
Consent to participate
All participants provided written informed consent before any study procedure was initiated.
Consent for publication
Not applicable.
Code availability
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(PDF 310 kb).
Rights and permissions
About this article

Cite this article
Binkley, N., Orwoll, E., Chapurlat, R. et al. Randomized, controlled trial to assess the safety and efficacy of odanacatib in the treatment of men with osteoporosis. Osteoporos Int 32, 173–184 (2021). https://doi.org/10.1007/s00198-020-05701-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-020-05701-9