
Abstract
The complement system is a set of over 50 proteins that constitutes an essential part of the innate immune system. Complement system activation involves an organized proteolytic cascade. Overactivation of complement system activation is the main pathogenic mechanism of several diseases and contributes to the manifestations of many other conditions. This review describes the normal complement system and the role for complement dysregulation in critical illnesses, notably sepsis and acute respiratory distress syndrome. Complement activation is involved in the immune system response to pathogens but, when excessive, can contribute to tissue damage, runaway inflammation, and capillary leakage syndrome. Complement overactivation may play a key role in severe forms of coronavirus disease 2019 (COVID-19). Two diseases whose manifestations are mainly caused by complement overactivation, namely, atypical hemolytic and uremic syndrome (aHUS) and myasthenia gravis, are discussed. A diagnostic algorithm for aHUS is provided. Early complement-inhibiting therapy has been proven effective. When renal transplantation is required, complement-inhibiting drugs can be used prophylactically to prevent aHUS recurrence. Similarly, acetylcholine-receptor autoantibody-positive generalized myasthenia gravis involves complement system overactivation and responds to complement inhibition. The two main complement inhibitors used in to date routine are eculizumab and ravulizumab. The main adverse event is Neisseria infection, which is rare and preventable, but can be fatal. The complement system is crucial to health but, when overactivated, can cause or contribute to disease. Effective complement inhibitors are now available, although additional data are required to determine optimal regimens. Further research is also needed to better understand the complement system, develop advanced diagnostic tools, and identify markers that allow the personalization of treatment strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
While the complement system is integral to pathogen elimination and maintenance of host homeostasis, its overactivation can cause tissue damage and runaway inflammation. Complement system dysregulation, as well as genetic factors, contribute to the symptoms of numerous critical illnesses including sepsis and ARDS. The introduction of complement inhibitors have improved the outcomes of several diseases but new tools are needed to provide early diagnosis of complement-driven diseases. |
Introduction
The complement system is a crucial component of the innate immune system that protects against infections and plays a key role in host homeostasis [1, 2]. When the complement system is activated, over 50 proteins engage in a proteolytic cascade, in concert with antibodies and other immune system components. The cascade can be rapidly triggered in the presence of antigen–antibody complexes or activating surfaces such as bacteria or apoptotic necrotic cells, via three main pathways (classical, mannan-binding lectin, and alternative). One major result is the identification, opsonization, and elimination of pathogens. The complement system is also involved in modulating inflammation and interacting with the adaptive immune system to fine-tune the immune response. Over- or underactivity of the complement system can cause a variety of abnormalities [3]. Tight regulation by surface-bound and soluble factors normally ensures effective pathogen clearance without excessive tissue damage or inflammation. If regulation is inadequate, the complement alternative pathway (cAP) can act as an amplification loop for all three pathways, potentially inducing runaway inflammation with damage to host tissues [3]. The endothelium is one of the primary targets of dysregulated complement activity [4, 5]. Imbalance in complement-regulating proteins is associated with complement-mediated (atypical) hemolytic and uremic syndrome (aHUS) [6], myasthenia gravis (MG) [7], C3 glomerulopathy [8], and paroxysmal nocturnal hemoglobinuria [9]. Moreover, complement overactivation triggered by various pathophysiological processes is being increasingly reported in many other diseases (Table 1) [10,11,12].
This review examines the role of complement dysregulation in common critical illness and in complement-mediated diseases that often require intensive care unit (ICU) admission. The normal structure, function, and regulation of the complement system are reviewed. Then specific attention is given to aHUS and MG, in which complement dysregulation plays a pivotal role.
The normal complement system
The role of the complement system in the immune response goes far beyond acting as a mere complement to antibody response. The numerous components of the complement system are produced primarily by the liver and are found in the blood and lymph, on cell surfaces, and in subcellular locations. This system is crucial to cell homeostasis and plays a key role in combating infections by inducing a systemic inflammatory response, strengthening and modulating immune responses, and directly killing certain pathogens. It also protects against harm from endogenous sources such as immune complexes and apoptotic cells [1, 2].
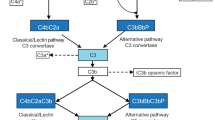
Figure 1 shows that the main functions of activated complement components are lysis (of cells, bacteria, or viruses), opsonization (phagocytosis), activation of the inflammatory response, and clearance of immune complexes. Figure 2 describes the three main pathways of complement system activation: the classical pathway is triggered by antibody–antigen complexes and the mannan-binding lectin pathway by carbohydrates in bacterial walls, whereas the cAP is activated non-proteolytically when the water in plasma hydrolyzes the thioester bond within C3, producing an active C3 convertase. After a sequence of serine protease reactions, the three pathways converge to generate C3 convertase and C5 convertase, which induce the release of anaphylatoxins (C3a and C5a) and opsonin (C3b/iC3b). The anaphylatoxins C3a and C5a bind to specific receptors, thereby inducing the production of inflammatory mediators, stimulating endothelial cells, and promoting cell migration and activation. Moreover, C5b binds with C6, C7, C8, and components of C9 to form C5b–9, also known as the membrane attack complex (MAC), at the pathogen surface, causing cell lysis (Fig. 2) [1, 2].

Main functions of activated complement components. Ag antigen, Ab antibody

Diagram of the complement system and its regulators. MBL mannan-binding lectin, MASPS mannan-binding lectin-associated serine proteases, MAC membrane attack complex
The classical and lectin pathways are initially triggered when pattern-recognition molecules (C1q and mannose-binding lectin) recognize pathogen-associated or damage-associated molecular patterns. In contrast, the cAP is always active at a low level (a process referred to as tickover) and involves spontaneous hydrolysis of C3, which releases small amounts of C3b. C3b binds covalently to activating surfaces, producing the C3 convertase C3b.Bb and initiating the amplification loop. Host-tissue damage is normally prevented via tight regulation of the cAP by the soluble factors H and I and by membrane-bound factors including membrane cofactor protein (CD46), complement receptor 1 (CD35), decay-accelerating factor (CD55), and CD59.
Complement is beneficial in attacking foreign invaders, but can also damage host cells. Four levels of control of complement system activation normally ensure proper function while preventing tissue damage. The first level involves early control at the stage of pathogen-associated or damage-associated molecular pattern recognition, through plasma C1 esterase inhibitor (C1-INH), which inhibits activated-C4 cleavage in the classical and lectin pathways. The second level prevents unwanted C3b/C4b deposition on host cells, via regulators such as factor I, factor H, C4BP, and complement receptor 1. The third level increases the dissociation of C3 and C5 convertases on host cells through factor H, C4BP, CD46, complement receptor 1, and CD55. Finally, at the fourth level, regulators such as CD59, clusterin, and vitronectin prevent MAC insertion on cell surfaces. Deficiencies in these protective controls or overactivation of the complement system may contribute to organ damage, infections, and other unwanted events.
The complement system in critical illnesses
The complement system plays a major role in the pathogenesis of critical illnesses [3]. A potent inflammatory response is the rule, notably in sepsis, and the complement system is therefore strongly activated [13, 14]. Notably, complement and coagulation proteolytic cascades are intimately linked at the crossroad between blood cells and microvascular endothelium [15]. The large amounts of the proinflammatory protein C5a released by sepsis induce tissue damage, increase the risk of infection [16], and enhance blood thrombogenicity, mainly through upregulation of tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) expression [15]. Hence, not surprisingly, both dysregulated complement and coagulation systems are critically involved in sepsis-related disseminated intravascular coagulation (DIC) [17]. C3a and C5a increase cytokine levels, leukocyte chemotaxis, and vascular endothelial permeability, thereby contributing to capillary leakage syndrome [13, 18]. The plasma serine protease C1-INH is the primary inhibitor of the classical complement pathway. Decreased C1-INH levels have been reported in patients with sepsis [19], notably those with refractory shock [20], and may promote complement overactivation. C1-INH administration has been associated with improved clinical outcomes of patients with sepsis [21, 22]. Similarly, the use of C3 blocker in a non-human primate model of sepsis decreased the coagulopathic response, preserved the endothelial anticoagulant properties, improved cardiac function, and mitigated kidney damage [23].
On the other hand, greater cAP activity has been associated with better survival of critically ill patients [24]. However, this finding may merely denote less complement consumption in survivors [25]. The red blood cell dysfunction and accelerated aging that occur during critical illness may be related to deposition on the cells of complement-activation products [26, 27].
Excessive complement activation may occur in acute respiratory distress syndrome (ARDS) [28]. However, findings from experimental models and clinical trials are conflicting [24, 29, 30]. Valuable insights have been produced by studies in patients with coronavirus disease 2019 (COVID-19) [31,32,33,34]. Prolonged complement overactivation may be a key pathophysiological factor in severe COVID-19 [35,36,37]. Clinical, transcriptomic, and genetic evidence suggest that complement overactivation may be associated with worse outcomes of severe COVID-19 [38]. Whether measurements of complement activation and assays of specific complement proteins are useful to guide intensive care deserves investigation. In sepsis, however, the extent of complement overactivation may be so marked that complement markers no longer predict the severity of inflammation or clinical outcomes [39].
In a nonrandomized study, adding a complement inhibitor to standard care was associated with better survival in patients with severe COVID-19 [35]. In a phase 2 randomized trial, the C3-blocker compstatin AMY-101 did not significantly improve the outcomes of severe COVID-19, perhaps due to the small sample size of only 31 patients [40]. However, in a randomized controlled trial in 202 patients receiving mechanical ventilation for severe COVID-19, adding the complement inhibitor ravulizumab produced no clinical benefits [41].
Bradykinin-related angioedema represents a classic, yet infrequent challenge in critical care settings [42], as opposed to histamine-mediated angioedema by its lack of response to traditional anti-allergic medications such as antihistamines, corticosteroids, and epinephrine [43]. This type of angioedema can be hereditary, as seen in hereditary angioedema or acquired, often linked to the use of angiotensin-converting enzyme inhibitors [44]. The underlying pathophysiology involves excessive production or impaired degradation of bradykinin, a potent vasodilator, which leads to increased vascular permeability and subsequent edema. Management of bradykinin-related angioedema necessitates targeted therapies [45], including C1-inhibitor concentrates, bradykinin receptor antagonists such as icatibant, and kallikrein inhibitors like ecallantide. In case of unavailability of these targeted treatments [46], attacks should be treated with solvent detergent-treated plasma, or alternatively with fresh frozen plasma. These specific treatments have been shown to be effective in alleviating symptoms and reducing the severity of attacks, underscoring the importance of early recognition and appropriate intervention in these critical scenarios. Understanding the distinct mechanisms and treatment options for bradykinin-related angioedema is essential for optimizing patient outcomes in the emergency and critical care settings.
Complement system inhibition
A radical breakthrough in recent years has been the development of drugs capable of inhibiting the complement system. The recombinant monoclonal antibody eculizumab inhibits C5 and was approved in the United States of America (USA) in 2007 and in Europe in 2011. C5 inhibition preserves opsonization by C3b and C4b, as well as immune signaling mediated by C3a, but prevents generation of the highly proinflammatory C5a and MAC. Eculizumab is generally well tolerated, with headache being one of the most common adverse effects. In a 2023 review of 10 years of post-marketing surveillance data from Japan, the safety profile was consistent with that seen in clinical studies, and no new safety signals were recorded [47]. However, complement inhibition impairs defenses against encapsulated bacteria, and Neisseria infection is a serious but rare complication. Over the 10 years from 2007 to 2016, 76 cases of meningococcal infection were reported in eculizumab-treated patients, including 8 with fatal outcomes [48]. Ravulizumab was engineered from eculizumab to extend the half-life, allowing for less frequent dosing [49, 50]. However, as pointed out by Benatar et al., research priorities are still needed to develop new patient-reported outcomes and biomarkers-guided clinical indications, and generate up-to-date health economic data [51]. Several drugs have been developed from the peptide compstatin AMY-101, which inhibits C3. Among them is pegcetacoplan, which is approved for paroxysmal nocturnal hemoglobinuria and geographic atrophy. Another peptide, zilucoplan, which inhibits C5, was approved in 2023 for the treatment of generalized myasthenia gravis (gMG) [52]. Interestingly, small proteins, including nomacopan and zilucoplan, which block C5 cleavage via the binding to a different C5 domain than eculizumab, elicit potent clinical efficacy in the carriers of C5 variant (R885H) unlike eculizumab/ravulizumab [53]. A good safety profile has been reported, with no cases of Neisseria infection when appropriate risk mitigation measures are taken [54]. However, caution regarding the risk of infection by encapsulated bacteria is warranted [54]. Many other complement inhibitors are being developed [55].
Atypical (complement-driven) hemolytic and uremic syndrome
HUS combines kidney failure, hemolytic anemia, and thrombocytopenia and can require ICU admission [56]. Both HUS and thrombotic thrombocytopenic purpura (TTP) belong to the thrombotic microangiopathy (TMA) family. TMAs are characterized by thrombi in the capillaries and arterioles due to endothelial injury and dysregulated coagulation. However, renal involvement is a hallmark of HUS [57].
The classification of TMAs now relies on pathogenic criteria rather than on clinical characteristics. TTP is due to a deficiency in ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13), whereas primary HUS, later renamed aHUS, is characterized by inherited or acquired dysregulation of cAP. Nevertheless, some forms of secondary TMA occurring concomitantly with other diseases or drug exposures have a more complex mechanism [57]. Thus, extrinsic factors may cause endothelial damage that interacts with complement dysregulation, blurring the line between aHUS and various forms of secondary HUS. The multifaceted nature of HUS thus continues to raise diagnostic and classification challenges.
In aHUS, complement dysregulation is a primary pathogenic driver, but can be exacerbated or triggered by external insults to the endothelial cells such as infections, medications, autoimmune diseases, and other factors that can independently cause endothelial damage or stress. Among patients with aHUS, up to 62% exhibit rare variants in complement genes or significant genomic rearrangements [58]. Over 500 rare variants have been identified, predominantly located in five key complement genes, namely, CFH, CFI, MCP, C3, and CFB [59,60,61,62].
Applying a standard diagnostic algorithm for aHUS (Fig. 3) is particularly valuable, as missing the diagnosis may delay appropriate complement-inhibiting therapy and result in underestimation of the relapse risk. In secondary HUS, whether complement activation is self-limited, inherent to the inaugural endothelial damage, or fueled by constitutive cAP dysregulation is relevant to the possible usefulness of complement-inhibition therapy [63].

Diagnostic flowchart for atypical hemolytic and uremic syndrome (aHUS) in adults. HUS hemolytic and uremic syndrome, aHUS atypical hemolytic and uremic syndrome, HIV human immunodeficiency virus, CT computed tomography, STEC-HUS HUS due to Shiga toxin-producing Escherichia coli, STX Shiga toxin
In adult patients with thrombotic microangiopathy and organ dysfunction requiring ICU admission, we have recommended starting therapeutic daily plasma exchange allowing a delivery of 60 ml/kg of plasma (1,5 blood volume) until TTP is excluded (detectable ADAMTS13 activity) and HUS due to Shiga toxin-producing Escherichia coli (STEC-HUS) and secondary HUS are ruled out [64, 65]. Then, eculizumab should be started without waiting for an additional complement biomarker, or any additional result. We do not support the use of C5 blockade only in case patients are refractory or dependent on plasma exchange because of the poor long-term results, particularly regarding renal outcomes [66]. Last, whether therapeutic plasma exchange can be considered as second-line therapy where C5 blockade proves ineffective has not been properly assessed.
Two clinical settings associated with HUS, namely, pregnancy and malignant hypertension, deserve note [56, 67]. Pregnancy-related HUS is a form of aHUS triggered by the systemic endothelial stress induced by pregnancy [68]. Rare complement variants have been detected in about half of patients with pregnancy-related HUS [69]. Eculizumab has been found to be safe and effective during pregnancy [70].
Malignant hypertension is a recognized cause of secondary TMA, but can also be a manifestation of (primary) aHUS, and the two must be distinguished. Hypertensive emergencies accounted for over half the presentations of aHUS in one study [37]. Interestingly, the prevalences of pathogenic gene variants and of gene variants of unknown significance did not differ across groups defined by hypertension severity. These variants were less common in patients of African ancestry who experienced hypertensive emergencies with TMA features [71]. Of 55 patients with aHUS, 36 (65%) had grade 2/3 hypertension, including 19 (35%) with malignant hypertension; and 19 (35%), including 13 with malignant hypertension, had pathogenic abnormalities in complement genes [72]. In the same study, only 6 of 110 (5%) patients with malignant hypertension but no aHUS had TMA. In the above-mentioned study of 55 patients with aHUS, eculizumab therapy was associated with better renal and hematological responses compared to plasmapheresis alone, with no differences according to hypertension severity or complement genetics.
It is known that untreated malignant hypertension causes widespread vascular damage affecting multiple organs, including the brain and kidneys, leading to acute renal failure, microangiopathic hemolytic anemia, disseminated intravascular coagulation, and hypertensive encephalopathy. In case of thrombotic microangiopathy and severe systolic or diastolic hyptertension, a fundoscopy is required to rule out malignant hypertension characterized by a bilateral grade 3 or grade 4 hypertensive retinopathy [72, 73].
A Cochrane systematic review [74] of articles on HUS treatment published up to 2020 identified five studies, all nonrandomized with no comparator group, including four on eculizumab [12, 75,76,77] and one on ravulizumab [78]. The 26-week complete remission rate was 60% with eculizumab and 54% with ravulizumab and, of the patients on dialysis at medication initiation, 70% were able to discontinue dialysis. Serious adverse events occurred in 42% of patients overall, including meningococcal infection in two eculizumab-treated patients. Complement inhibition has also been reported to improve the estimated glomerular filtration rate and health-related quality of life [79, 80]. Of note, the response to the meningococcal vaccine may be impaired in patients with aHUS, and vaccination of the family and other close contacts to decrease the risk of Neisseria infection may deserve consideration [58].
The optimal duration of complement-inhibition therapy in patients with aHUS remains under evaluation. Of 55 children and adults who discontinued eculizumab after a mean of 16.5 months, 13 (23%) relapsed, and risk factors for relapse were female sex, rare complement gene variants, and high plasma sC5b-9 level at discontinuation. Knowledge of these risk factors may help to guide individualized discontinuation decisions.
Complement-inhibiting therapy can also be used prophylactically. In a retrospective multicenter study of 126 kidney transplants in 116 adults with aHUS, the disease recurred after transplantation in 8% of patients with vs. 53% without prophylactic eculizumab therapy, which was independently associated with a lower risk of aHUS and with longer graft survival [6]. Prophylactic eculizumab was most effective in the sub-group at higher pre-transplantation risk for aHUS recurrence. The same study evaluated a population-based cohort of 397 adults with end-stage kidney disease and aHUS and found a sharp increase in the proportion who received kidney transplants between 2012 and 2016, from 46.2% to 72.3%, in close correlation with an increase in eculizumab use among transplant recipients [6]. Despite the clear evidence of efficacy, the extremely high cost of eculizumab is generating debate about the appropriateness of prophylactic eculizumab therapy. An individualized strategy reserving eculizumab prophylaxis for patients at moderate-to-high risk of aHUS recurrence after kidney transplantation may deserve evaluation.
Once aHUS has developed, early eculizumab initiation is crucial. In one study, among patients with aHUS recurrence after renal transplantation, the extent of renal function recovery decreased with increasing time from aHUS onset to eculizumab initiation [81]. The diagnosis of aHUS must therefore be made early. In aHUS, cAP activation occurs at the endothelial surface but not in the bloodstream, and C3 in plasma or serum therefore often remains normal. In a cohort of 214 patients, serum C3 was low at the first evaluation in only 77 (35.9%) of cases [61]. An enzyme-linked immunosorbent assay (ELISA) for soluble C5b-9 had 89% positive predictive value for active aHUS, but lacked sensitivity and remained positive in some patients who were in remission [82]. Research is needed to develop a reliable standardized test for detecting sub-clinical complement activation. Promising avenues are C3 or C5b-9 quantification in resting or ADP-activated cell lines incubated with patient serum or plasma [58] and assessment of C5b-9 deposition on cultured microvascular endothelium [83]. Rapid genome sequencing methods for detecting mutations in complement or ADAMTS-13 genes is developing at a brisk pace and can be expected to change clinical practice in the coming years [49, 84, 85].
Complement inhibition in myasthenia gravis
MG is a rare autoimmune disease of the neuromuscular junction that can result in chronic muscle function impairment [86]. Fluctuating severe weakness of voluntary muscles that worsens after exertion is typical [87,88,89]. Ocular muscle weakness with diplopia and ptosis is usually apparent at presentation and remains isolated in about 15% of patients, whereas generalized MG (gMG) develops in about 85% of patients [53, 54]. The clinical manifestations of gMG are highly variable over time in a given patient. Exacerbations may be triggered by exertion or other many other precipitating factors, and sometimes occur spontaneously. Bulbar weakness with difficulty chewing and swallowing, dysarthria, and dysphonia and respiratory muscle involvement with myasthenic crisis in the most severe form usually require consulting intensivist for potential ICU admission.
The autoantibodies that cause MG symptoms mainly (in about 85% of cases) target the nicotinic acetylcholine receptor complex (AChR, IgG1, and IgG3) at the postsynaptic membrane of the neuromuscular junction (Fig. 4) [40, 55]. Muscle-specific kinase (MuSK) autoantibodies are less common, and autoantibodies to low-density lipoprotein receptor-related protein have been reported in patients without AChR or MuSK autoantibodies [90].

Figure source: Pathophysiological mechanisms of myasthenia gravis at the neuromuscular junction, from [91]. Permission obtained
Anti-acetylcholine (ACh) receptor (AChR) antibodies activate the complement, causing production of the membrane attack complex (MAC), which damages the postsynaptic membrane at the neuromuscular junction. Anti-AChR antibodies can also cross-link AChRs, accelerating their internalization and degradation. Some antibodies can directly block the AChR-binding site. Anti-muscle-specific kinase (MuSK) antibodies do not activate the complement and typically prevent the interaction of MuSK and lipoprotein receptor-related protein 4 (LRP4), among other proteins, leading to reduced AChR clustering on the postsynaptic membrane. The pathogenicity of anti-LRP4 antibodies in myasthenia gravis (MG) remains to be established. Additional antibodies, such as anti-collagen Q (ColQ), anti-titin, anti-ryanodine receptor (RyR), anti-cortactin, and anti-voltage-gated potassium channel (Kv1.4) have been demonstrated in patients with MG, although their pathogenic significance remains unknown. Ach acetylcholine, AChR acetylcholine receptor, AChE acetylcholinesterase, MuSK muscle-specific kinase, LRP4 low-density lipoprotein receptor-related protein, MAC membrane attack complex, ColQ acetylcholinesterase collagenic tail peptide, RyR ryanodine receptor, rapsyn 43-kDa receptor-associated protein of the synapse, VGSC voltage-gated sodium channel, Kv1.4 1.4 voltage-gated potassium channel.
The AChR antibodies can activate the complement pathway generating excessive release of C5a causing inflammation, and of the MAC damaging the post-synaptic membrane [92]. Both eculizumab and ravulizumab have demonstrated clinically relevant improvement in patients with AChR-related gMG, with rapid and long-lasting disease stabilization enabling to substantially decrease exposure to corticosteroids and immunosuppressive drugs [51, 52, 56,57,58,59,60]. These treatments were also found to be well tolerated without evidence for increased incidence of meningococcal diseases pending appropriate risk mitigation strategy based on vaccination and/or antibiotic prophylaxis. Similar clinical benefit and safety profile was found with the peptide C5 inhibitor zilucoplan [52]. Recent clinical guidelines from Japan [93] and Germany [94] recommend, in active generalized MG, complement inhibitors for AChR-Ab-positive status. A more complete view on anti-complement drugs currently approved is provided in Fig. 5.

Anti-complement drugs currently approved.  indicates small organic molecule.
indicates small organic molecule. indicates monoclonal antibodies .
indicates monoclonal antibodies . indicates small peptide coupled to polyethylene glycol (PEG).
indicates small peptide coupled to polyethylene glycol (PEG).  indicates peptide. Paroxysmal nocturnal hemoglobinuria (PNH); atypical hemolytic–uremic syndrome (aHUS); generalized myasthenia gravis (gMG) for patients who are anti-acetylcholine receptor antibody positive; neuromyelitis optica spectrum disorder (NMOSD) for patients who are anti-aquaporin-4 antibody positive. ANCA antineutrophil cytoplasmic antibodies. CHAPLE disease is also called CD55-deficient protein-losing enteropathy. GA geographic atrophy secondary to age-related macular degeneration (AMD). (HSCT-TMA) hematopoietic stem cell transplant-associated thrombotic microangiopathy. § Narsoplimab has received breakthrough therapy and orphan drug designations from the FDA
indicates peptide. Paroxysmal nocturnal hemoglobinuria (PNH); atypical hemolytic–uremic syndrome (aHUS); generalized myasthenia gravis (gMG) for patients who are anti-acetylcholine receptor antibody positive; neuromyelitis optica spectrum disorder (NMOSD) for patients who are anti-aquaporin-4 antibody positive. ANCA antineutrophil cytoplasmic antibodies. CHAPLE disease is also called CD55-deficient protein-losing enteropathy. GA geographic atrophy secondary to age-related macular degeneration (AMD). (HSCT-TMA) hematopoietic stem cell transplant-associated thrombotic microangiopathy. § Narsoplimab has received breakthrough therapy and orphan drug designations from the FDA
Conclusion
This review underscores the pivotal role of the complement system in both health and disease, emphasizing its importance in immune defense, inflammation modulation, and interaction with the adaptive immune system. While the complement system is integral to pathogen elimination and maintaining host homeostasis, complement system overactivation can cause tissue damage and runaway inflammation. Complement system dysregulation is a primary driver of several diseases, many of which require intensive care. Genetic factors can contribute to impair appropriate control of complement activation. Moreover, complement system dysregulation contributes to the symptoms of numerous critical illnesses including sepsis and ARDS. The introduction of complement inhibitors has radically improved the outcomes of several diseases including aHUS and gMG. Owing to the inhibition of the membrane attack complex by a certain complement inhibitor, meningococcal risk mitigation strategy with vaccination and or antibiotic prophylaxis is mandated. In addition, these medications are costly and more work is needed to determine the optimal treatment duration depending on the profile of each patient. Many other pharmacotherapy options are being evaluated. New tools capable of providing the early diagnosis of complement-driven diseases are needed.
Availability of data and materials
This study relies on previously published data.
References
Walport MJ (2001) Complement: first of two parts. N Engl J Med 344:1058–1066. https://doi.org/10.1056/NEJM200104053441406
Walport MJ (2001) Complement: second of two parts. N Engl J Med 344:1140–1144. https://doi.org/10.1056/NEJM200104123441506
Mannes M, Mastellos DC, Ekdahl KN et al (2022) Complement C3 activation in the ICU: disease and therapy as Bonnie and Clyde. Semin Immunol 60:101640. https://doi.org/10.1016/j.smim.2022.101640
Cofiell R, Kukreja A, Bedard K et al (2015) Eculizumab reduces complement activation, inflammation, endothelial damage, thrombosis, and renal injury markers in aHUS. Blood 125:3253–3262. https://doi.org/10.1182/blood-2014-09-600411
Blasco M, Guillén-Olmos E, Diaz-Ricart M, Palomo M (2022) Complement mediated endothelial damage in thrombotic microangiopathies. Front Med 9:811504. https://doi.org/10.3389/fmed.2022.811504
Zuber J, Frimat M, Caillard S et al (2019) Use of highly individualized complement blockade has revolutionized clinical outcomes after kidney transplantation and renal epidemiology of atypical hemolytic uremic syndrome. J Am Soc Nephrol JASN 30:2449–2463. https://doi.org/10.1681/ASN.2019040331
Iwasa K, Furukawa Y, Yoshikawa H et al (2023) CD59 expression in skeletal muscles and its role in myasthenia gravis. Neurol Neuroimmunol Neuroinflammation 10:e200057. https://doi.org/10.1212/NXI.0000000000200057
Weening JJ, Ronco P, Remuzzi G (2013) Advances in the pathology of glomerular diseases. Contrib Nephrol 181:12–21. https://doi.org/10.1159/000348639
Peffault de Latour R, Hosokawa K, Risitano AM (2022) Hemolytic paroxysmal nocturnal hemoglobinuria: 20 years of medical progress. Semin Hematol 59:38–46. https://doi.org/10.1053/j.seminhematol.2022.01.001
Hillmen P, Szer J, Weitz I et al (2021) Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med 384:1028–1037. https://doi.org/10.1056/NEJMoa2029073
Pittock SJ, Berthele A, Fujihara K et al (2019) Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med 381:614–625. https://doi.org/10.1056/NEJMoa1900866
Legendre CM, Licht C, Muus P et al (2013) Terminal complement inhibitor eculizumab in atypical haemolytic: uremic syndrome. N Engl J Med 368:2169–2181. https://doi.org/10.1056/NEJMoa1208981
Hack CE, Nuijens JH, Felt-Bersma RJ et al (1989) Elevated plasma levels of the anaphylatoxins C3a and C4a are associated with a fatal outcome in sepsis. Am J Med 86:20–26. https://doi.org/10.1016/0002-9343(89)90224-6
Younger JG, Bracho DO, Chung-Esaki HM et al (2010) Complement activation in emergency department patients with severe sepsis. Acad Emerg Med Off J Soc Acad Emerg Med 17:353–359. https://doi.org/10.1111/j.1553-2712.2010.00713.x
Markiewski MM, Nilsson B, Ekdahl KN et al (2007) Complement and coagulation: strangers or partners in crime? Trends Immunol 28:184–192. https://doi.org/10.1016/j.it.2007.02.006
Wood AJT, Vassallo A, Summers C et al (2018) C5a anaphylatoxin and its role in critical illness-induced organ dysfunction. Eur J Clin Invest 48:e13028. https://doi.org/10.1111/eci.13028
Popescu NI, Lupu C, Lupu F (2022) Disseminated intravascular coagulation and its immune mechanisms. Blood 139:1973–1986. https://doi.org/10.1182/blood.2020007208
Huber-Lang M, Sarma VJ, Lu KT et al (2001) Role of C5a in multiorgan failure during sepsis. J Immunol Baltim Md 1950 166:1193–1199. https://doi.org/10.4049/jimmunol.166.2.1193
Charchaflieh J, Wei J, Labaze G et al (2012) The role of complement system in septic shock. Clin Dev Immunol 2012:407324. https://doi.org/10.1155/2012/407324
Hirose T, Ogura H, Takahashi H et al (2018) Serial change of C1 inhibitor in patients with sepsis: a prospective observational study. J Intensive Care 6:37. https://doi.org/10.1186/s40560-018-0309-5
Igonin AA, Protsenko DN, Galstyan GM et al (2012) C1-esterase inhibitor infusion increases survival rates for patients with sepsis*. Crit Care Med 40:770–777. https://doi.org/10.1097/CCM.0b013e318236edb8
Schlapbach LJ, Schibler A, Jensenius JC (2012) C1-esterase inhibitor treatment in sepsis-can we target the right patients? Crit Care Med 40:2735–2737. https://doi.org/10.1097/CCM.0b013e318258eb7a
Silasi-Mansat R, Zhu H, Popescu NI et al (2010) Complement inhibition decreases the procoagulant response and confers organ protection in a baboon model of Escherichia coli sepsis. Blood 116:1002–1010. https://doi.org/10.1182/blood-2010-02-269746
Bain W, Li H, van der Geest R et al (2020) Increased alternative complement pathway function and improved survival during critical illness. Am J Respir Crit Care Med 202:230–240. https://doi.org/10.1164/rccm.201910-2083OC
Bosmann M (2020) Complement activation during critical illness: current findings and an outlook in the era of COVID-19. Am J Respir Crit Care Med 202:163–165. https://doi.org/10.1164/rccm.202005-1926ED
Pietropaoli AP, Wexler O (2014) RBC dysfunction in critical illness: driven by complement? Crit Care Med 42:1323–1324. https://doi.org/10.1097/CCM.0000000000000275
Muroya T, Kannan L, Ghiran IC et al (2014) C4d deposits on the surface of RBCs in trauma patients and interferes with their function. Crit Care Med 42:e364-372. https://doi.org/10.1097/CCM.0000000000000231
Hammerschmidt DE, Weaver LJ, Hudson LD et al (1980) Association of complement activation and elevated plasma-C5a with adult respiratory distress syndrome: pathophysiological relevance and possible prognostic value. Lancet Lond Engl 1:947–949. https://doi.org/10.1016/s0140-6736(80)91403-8
Mulligan MS, Schmid E, Beck-Schimmer B et al (1996) Requirement and role of C5a in acute lung inflammatory injury in rats. J Clin Invest 98:503–512. https://doi.org/10.1172/JCI118818
Rittirsch D, Flierl MA, Day DE et al (2008) Acute lung injury induced by lipopolysaccharide is independent of complement activation. J Immunol Baltim Md 1950 180:7664–7672. https://doi.org/10.4049/jimmunol.180.11.7664
Peffault de Latour R, Bergeron A, Lengline E et al (2020) Complement C5 inhibition in patients with COVID-19 - a promising target? Haematologica 105:2847–2850. https://doi.org/10.3324/haematol.2020.260117
Lo MW, Kemper C, Woodruff TM (2020) COVID-19: complement, coagulation, and collateral damage. J Immunol Baltim Md 205:1488–1495. https://doi.org/10.4049/jimmunol.2000644
Helling H, Stephan B, Pindur G (2015) Coagulation and complement system in critically ill patients. Clin Hemorheol Microcirc 61:185–193. https://doi.org/10.3233/CH-151993
Ali YM, Ferrari M, Lynch NJ et al (2021) Lectin pathway mediates complement activation by SARS-CoV-2 proteins. Front Immunol 12:714511. https://doi.org/10.3389/fimmu.2021.714511
Annane D, Heming N, Grimaldi-Bensouda L et al (2020) Eculizumab as an emergency treatment for adult patients with severe COVID-19 in the intensive care unit: a proof-of-concept study. EClinicalMedicine 28:100590. https://doi.org/10.1016/j.eclinm.2020.100590
Defendi F, Leroy C, Epaulard O et al (2021) Complement alternative and mannose-binding lectin pathway activation is associated with COVID-19 mortality. Front Immunol 12:742446. https://doi.org/10.3389/fimmu.2021.742446
Holter JC, Pischke SE, de Boer E et al (2020) Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci U S A 117:25018–25025. https://doi.org/10.1073/pnas.2010540117
Ramlall V, Thangaraj PM, Meydan C et al (2020) Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat Med 26:1609–1615. https://doi.org/10.1038/s41591-020-1021-2
de Nooijer AH, Kotsaki A, Kranidioti E et al (2023) Complement activation in severely ill patients with sepsis: no relationship with inflammation and disease severity. Crit Care Lond Engl 27:63. https://doi.org/10.1186/s13054-023-04344-6
Skendros P, Germanidis G, Mastellos DC et al (2022) Complement C3 inhibition in severe COVID-19 using compstatin AMY-101. Sci Adv 8:eabo2341. https://doi.org/10.1126/sciadv.abo2341
Annane D, Pittock SJ, Kulkarni HS et al (2023) Intravenous ravulizumab in mechanically ventilated patients hospitalised with severe COVID-19: a phase 3, multicentre, open-label, randomised controlled trial. Lancet Respir Med 11:1051–1063. https://doi.org/10.1016/S2213-2600(23)00082-6
LoVerde D, Files DC, Krishnaswamy G (2017) Angioedema. Crit Care Med 45:725–735. https://doi.org/10.1097/CCM.0000000000002281
Loules G, Parsopoulou F, Zamanakou M et al (2020) Deciphering the genetics of primary angioedema with normal levels of C1 inhibitor. J Clin Med 9:3402. https://doi.org/10.3390/jcm9113402
Zuraw BL, Christiansen SC (2011) Pathophysiology of hereditary angioedema. Am J Rhinol Allergy 25:373–378. https://doi.org/10.2500/ajra.2011.25.3661
Maurer M, Magerl M, Betschel S et al (2022) The international WAO/EAACI guideline for the management of hereditary angioedema: the 2021 revision and update. Allergy 77:1961–1990. https://doi.org/10.1111/all.15214
Longhurst HJ (2005) Emergency treatment of acute attacks in hereditary angioedema due to C1 inhibitor deficiency: what is the evidence? Int J Clin Pract 59:594–599. https://doi.org/10.1111/j.1742-1241.2005.00352.x
Nishimura J-I, Kawaguchi T, Ito S et al (2023) Real-world safety profile of eculizumab in patients with paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, or generalized myasthenia gravis: an integrated analysis of post-marketing surveillance in Japan. Int J Hematol 118:419–431. https://doi.org/10.1007/s12185-023-03630-x
Socié G, Caby-Tosi M-P, Marantz JL et al (2019) Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br J Haematol 185:297–310. https://doi.org/10.1111/bjh.15790
Vu T, Meisel A, Mantegazza R et al (2022) Terminal complement inhibitor ravulizumab in generalized myasthenia gravis. NEJM Evid 1:EVIDoa2100066. https://doi.org/10.1056/EVIDoa2100066
Meisel A, Annane D, Vu T et al (2023) Long-term efficacy and safety of ravulizumab in adults with anti-acetylcholine receptor antibody-positive generalized myasthenia gravis: results from the phase 3 CHAMPION MG open-label extension. J Neurol 270:3862–3875. https://doi.org/10.1007/s00415-023-11699-x
Benatar M, Cutter G, Kaminski HJ (2023) The best and worst of times in therapy development for myasthenia gravis. Muscle Nerve 67:12–16. https://doi.org/10.1002/mus.27742
Howard JF, Bresch S, Genge A et al (2023) Safety and efficacy of zilucoplan in patients with generalised myasthenia gravis (RAISE): a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Neurol 22:395–406. https://doi.org/10.1016/S1474-4422(23)00080-7
Goodship THJ, Pinto F, Weston-Davies WH et al (2017) Use of the complement inhibitor coversin to treat HSCT-associated TMA. Blood Adv 1:1254–1258. https://doi.org/10.1182/bloodadvances.2016002832
Shirley M (2024) Zilucoplan: first approval. Drugs 84:99–104. https://doi.org/10.1007/s40265-023-01977-3
Qu H, Ricklin D, Lambris JD (2009) Recent developments in low molecular weight complement inhibitors. Mol Immunol 47:185–195. https://doi.org/10.1016/j.molimm.2009.08.032
Fakhouri F, Zuber J, Frémeaux-Bacchi V, Loirat C (2017) Haemolytic uraemic syndrome. Lancet Lond Engl 390:681–696. https://doi.org/10.1016/S0140-6736(17)30062-4
Jokiranta TS (2017) HUS and atypical HUS. Blood 129:2847–2856. https://doi.org/10.1182/blood-2016-11-709865
Leon J, LeStang M-B, Sberro-Soussan R et al (2023) Complement-driven hemolytic uremic syndrome. Am J Hematol 98(Suppl 4):S44–S56. https://doi.org/10.1002/ajh.26854
Pickering MC, de Jorge EG, Martinez-Barricarte R et al (2007) Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med 204:1249–1256. https://doi.org/10.1084/jem.20070301
Bu F, Zhang Y, Wang K et al (2018) Genetic analysis of 400 patients refines understanding and implicates a new gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol JASN 29:2809–2819. https://doi.org/10.1681/ASN.2018070759
Fremeaux-Bacchi V, Fakhouri F, Garnier A et al (2013) Genetics and outcome of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol CJASN 8:554–562. https://doi.org/10.2215/CJN.04760512
Zipfel PF, Edey M, Heinen S et al (2007) Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet 3:e41. https://doi.org/10.1371/journal.pgen.0030041
Palma LM, Sridharan M, Sethi S (2021) Complement in secondary thrombotic microangiopathy. Kidney Int Rep. https://doi.org/10.1016/j.ekir.2020.10.009
Azoulay E, Knoebl P, Garnacho-Montero J et al (2017) Expert statements on the standard of care in critically ill adult patients with atypical hemolytic uremic syndrome. Chest 152:424–434. https://doi.org/10.1016/j.chest.2017.03.055
Azoulay E, Bauer PR, Mariotte E et al (2019) Expert statement on the ICU management of patients with thrombotic thrombocytopenic purpura. Intensive Care Med 45:1518–1539. https://doi.org/10.1007/s00134-019-05736-5
Noris M, Caprioli J, Bresin E et al (2010) Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol CJASN 5:1844–1859. https://doi.org/10.2215/CJN.02210310
Fakhouri FF, Frémeaux-Bacchi V (2021) Thrombotic microangiopathy in aHUS and beyond: clinical clues from complement genetics. Nat Rev Nephrol. https://doi.org/10.1038/s41581-021-00424-4
Fakhouri F, Scully M, Provôt F et al (2020) Management of thrombotic microangiopathy in pregnancy and postpartum: report from an international working group. Blood 136:2103–2117. https://doi.org/10.1182/blood.2020005221
Bruel A, Kavanagh D, Noris M et al (2017) Hemolytic uremic syndrome in pregnancy and postpartum. Clin J Am Soc Nephrol CJASN 12:1237–1247. https://doi.org/10.2215/CJN.00280117
Gaggl M, Aigner C, Csuka D et al (2018) Maternal and fetal outcomes of pregnancies in women with atypical hemolytic uremic syndrome. J Am Soc Nephrol JASN. https://doi.org/10.1681/ASN.2016090995
Timmermans SAMEG, Wérion A, Damoiseaux JGMC et al (2020) Diagnostic and risk factors for complement defects in hypertensive emergency and thrombotic microangiopathy. Hypertens Dallas Tex 1979(75):422–430. https://doi.org/10.1161/HYPERTENSIONAHA.119.13714
Cavero T, Arjona E, Soto K et al (2019) Severe and malignant hypertension are common in primary atypical hemolytic uremic syndrome. Kidney Int 96:995–1004. https://doi.org/10.1016/j.kint.2019.05.014
Poli F, Yusuf IH (2021) Retinopathy in malignant hypertension. N Engl J Med 385:1994. https://doi.org/10.1056/NEJMicm2109500
Pugh D, O’Sullivan ED, Duthie FA et al (2021) Interventions for atypical haemolytic uraemic syndrome. Cochrane Database Syst Rev 3:CD012862. https://doi.org/10.1002/14651858.CD012862.pub2
Fakhouri F, Hourmant M, Campistol JM et al (2016) Terminal complement inhibitor eculizumab in adult patients with atypical hemolytic uremic syndrome: a single-arm, open-label trial. Am J Kidney Dis Off J Natl Kidney Found 68:84–93. https://doi.org/10.1053/j.ajkd.2015.12.034
Greenbaum LA, Fila M, Ardissino G et al (2016) Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int 89:701–711. https://doi.org/10.1016/j.kint.2015.11.026
Licht C, Greenbaum LA, Muus P et al (2015) Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int 87:1061–1073. https://doi.org/10.1038/ki.2014.423
Rondeau E, Scully M, Ariceta G et al (2020) The long-acting C5 inhibitor, ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int 97:1287–1296. https://doi.org/10.1016/j.kint.2020.01.035
Mukherjee AA, Kandhare AD, Bodhankar SL (2018) Evaluation of health-related quality of life in hemolytic uraemic syndrome patients treated with eculizumab: a systematic evaluation on basis of EMPRO. Ren Fail 40:107–118. https://doi.org/10.1080/0886022X.2018.1427110
Azoulay E, Souppart V, Kentish-Barnes N et al (2023) Post-traumatic stress disorder and quality of life alterations in survivors of immune-mediated thrombotic thrombocytopenic purpura and atypical hemolytic and uremic syndrome. J Crit Care 76:154283. https://doi.org/10.1016/j.jcrc.2023.154283
Zuber J, Le Quintrec M, Krid S et al (2012) Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am J Transplant Off J Am Soc Transplant Am Soc Transpl Surg 12:3337–3354. https://doi.org/10.1111/j.1600-6143.2012.04252.x
Bu F, Meyer NC, Zhang Y et al (2015) Soluble c5b–9 as a biomarker for complement activation in atypical hemolytic uremic syndrome. Am J Kidney Dis Off J Natl Kidney Found 65:968–969. https://doi.org/10.1053/j.ajkd.2015.02.326
Galbusera M, Noris M, Gastoldi S et al (2019) An ex vivo test of complement activation on endothelium for individualized eculizumab therapy in hemolytic uremic syndrome. Am J Kidney Dis Off J Natl Kidney Found 74:56–72. https://doi.org/10.1053/j.ajkd.2018.11.012
Doreille A, Rafat C, Rondeau E, Mesnard L (2023) How I treat thrombotic microangiopathy in the era of rapid genomics. Blood 141:147–155. https://doi.org/10.1182/blood.2022015583
Owen MJ, Niemi A-K, Dimmock DP et al (2021) Rapid sequencing-based diagnosis of thiamine metabolism dysfunction syndrome. N Engl J Med 384:2159–2161. https://doi.org/10.1056/NEJMc2100365
Gilhus NE (2016) Myasthenia gravis. N Engl J Med 375:2570–2581. https://doi.org/10.1056/NEJMra1602678
Mantegazza R, Bonanno S, Camera G, Antozzi C (2011) Current and emerging therapies for the treatment of myasthenia gravis. Neuropsychiatr Dis Treat 7:151–160. https://doi.org/10.2147/NDT.S8915
Melzer N, Ruck T, Fuhr P et al (2016) Clinical features, pathogenesis, and treatment of myasthenia gravis: a supplement to the Guidelines of the German Neurological Society. J Neurol 263:1473–1494. https://doi.org/10.1007/s00415-016-8045-z
Meriggioli MN, Sanders DB (2009) Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol 8:475–490. https://doi.org/10.1016/S1474-4422(09)70063-8
Pevzner A, Schoser B, Peters K et al (2012) Anti-LRP4 autoantibodies in AChR- and MuSK-antibody-negative myasthenia gravis. J Neurol 259:427–435. https://doi.org/10.1007/s00415-011-6194-7
Gilhus NE, Tzartos S, Evoli A et al (2019) Myasthenia gravis. Nat Rev Dis Primers 5(1):30. https://doi.org/10.1038/s41572-019-0079-y
Sanderson NSR (2022) Complement and myasthenia gravis. Mol Immunol 151:11–18. https://doi.org/10.1016/j.molimm.2022proinf.08.018
Murai H (2024) The Japanese clinical guidelines 2022 for myasthenia gravis and lambert-eaton myasthenic syndrome: an overview. Brain Nerve Shinkei Kenkyu No Shinpo 76:7–12. https://doi.org/10.11477/mf.1416202551
Wiendl H, Abicht A, Chan A et al (2023) Guideline for the management of myasthenic syndromes. Ther Adv Neurol Disord 16:17562864231213240. https://doi.org/10.1177/17562864231213240
Funding
None.
Author information
Authors and Affiliations
Contributions
EA contributed to literature review, manuscript drafting, and designing tables and figures. JZ contributed to literature review and manuscript drafting. AAB contributed to literature review and manuscript drafting. YL contributed to literature review and data interpretation. YT contributed to literature review and data interpretation. SL contributed to literature review and data interpretation. ME contributed to literature review and data interpretation. DA contributed to literature review and manuscript drafting. All authors have approved the submitted version.
Corresponding author
Ethics declarations
Conflicts of interest
EA has received fees for lectures from Alexion, Sanofi, Gilead, and Pfizer. DA has received fees for lectures or to sit on advisory board from Alexion. JZ has received lecture fees, travel grants, and board member honorarium from Alexion Pharmaceuticals. AAB, YL, YT, SL, and ME report no conflict of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Azoulay, E., Zuber, J., Bousfiha, A.A. et al. Complement system activation: bridging physiology, pathophysiology, and therapy. Intensive Care Med (2024). https://doi.org/10.1007/s00134-024-07611-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00134-024-07611-4