
Abstract
Aims/hypothesis
Obesity caused by overeating plays a pivotal role in the development of type 2 diabetes. However, it remains poorly understood how individual meal size differences are determined before the development of obesity. Here, we investigated the underlying mechanisms in determining spontaneous food intake in newly established Oikawa–Nagao Diabetes-Prone (ON-DP) and Diabetes-Resistant (ON-DR) mice.
Methods
Food intake and metabolic phenotypes of ON-DP and ON-DR mice under high-fat-diet feeding were compared from 5 weeks to 10 weeks of age. Differences in leptin status at 5 weeks of age were assessed between the two mouse lines. Adipose tissue explant culture was also performed to evaluate leptin production capacity in vitro.
Results
ON-DP mice showed spontaneous overfeeding compared with ON-DR mice. Excessive body weight gain and fat accumulation in ON-DP mice were completely suppressed to the levels seen in ON-DR mice by pair-feeding with ON-DR mice. Deterioration of glucose tolerance in ON-DP mice was also ameliorated under the pair-feeding conditions. While no differences were seen in body weight and adipose tissue mass when comparing the two mouse lines at 5 weeks of age, the ON-DP mice had lower plasma leptin concentrations and adipose tissue leptin gene expression levels. In accordance with peripheral leptin status, ON-DP mice displayed lower anorexigenic leptin signalling in the hypothalamic arcuate nucleus when compared with ON-DR mice without apparent leptin resistance. Explant culture studies revealed that ON-DP mice had lower leptin production capacity in adipose tissue. ON-DP mice also displayed higher DNA methylation levels in the leptin gene promoter region of adipocytes when compared with ON-DR mice.
Conclusions/interpretation
The results suggest that heritable lower leptin production capacity plays a critical role in overfeeding-induced obesity and subsequent deterioration of glucose tolerance in ON-DP mice. Leptin production capacity in adipocytes, especially before the development of obesity, may have diagnostic potential for predicting individual risk of obesity caused by overeating and future onset of type 2 diabetes.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.

Introduction
The ever-increasing prevalence of obesity has become a global health problem [1, 2]. Although overeating is the major cause of obesity, not all people become obese, some remain lean, even in the modern ‘obesogenic’ environment of plentiful food supply [3]. This shows the existence of individual differences in determining meal size relative to energy expenditure [4, 5]. Considering the heritability of obesity [6], hereditary differences in spontaneous meal size may play a critical role in determining susceptibility to obesity and its metabolic complications including type 2 diabetes.
Recently, we established two mouse lines (closed colonies) with different susceptibilities to high-fat diet (HFD)-induced glucose intolerance, designated as Oikawa–Nagao Diabetes-Prone (ON-DP) and Oikawa–Nagao Diabetes-Resistant (ON-DR) (formerly referred to as SDG-P and SDG-R, respectively) [7,8,9]. ON-DP and ON-DR mice were derived through selective breeding (using C57BL/6, C3H and AKR mice as background strains) for superior and inferior glucose tolerance after HFD feeding, respectively. In the selective breeding, post-challenge blood glucose levels were used as the sole selection criterion (data for food intake or growth rate were not used for the selection). As a result, ON-DP mice displayed higher food intake and excessive body weight gain, when compared with ON-DR mice, from shortly after weaning [7]. These findings strongly suggest that hereditary traits in determining spontaneous food intake play a pivotal role in deterioration of glucose tolerance accompanied by development of obesity.
The aim of the present study was therefore to elucidate the underlying mechanisms in determining the differences in spontaneous food intake when comparing ON-DP and ON-DR mice. As a potential determining factor, we demonstrated here the importance of leptin production capacity before the development of obesity. Individual differences in leptin production capacity in adipocytes may play a critical role in the development of obesity and subsequent onset of type 2 diabetes.
Methods
Animals
All animal studies were approved by the Animal Care and Use Committee of Nippon Medical School (26-128). Male ON-DP (Onrg:ON-DP) and ON-DR (Onrg:ON-DR) mice, established by selective breeding using C57BL/6J, C3H/HeJ and AKR/N mice as background strains for different glucose tolerance [7,8,9], were obtained from the Institute for Animal Reproduction (Kasumigaura, Japan) after weaning. The mice were housed in standard plastic cages with paper chip bedding (three to five mice per cage) and maintained in a 14 h/10 h light–dark cycle temperature-controlled room (22 ± 1°C) with free access to water and a standard rodent chow (MF; Oriental Yeast, Japan). For the two mouse lines, body-weight-matched male mice of 5 weeks of age were used for each experiment. For tissue collection, mice were killed under anaesthesia 5 ± 1 h after the beginning of the light phase unless otherwise specified.
Pair-feeding
For the pair-feeding experiment, 5-week-old mice were individually housed and fed an HFD (30% wt/wt energy fat, 16.6 kJ/g; Quick Fat; CLEA Japan, Japan) for 5 weeks. Food intake was measured daily (at the end of the light phase) using an MF-1 feeding box (Shinfactory, Japan) and an amount of food corresponding to the mean quantity consumed by the ON-DR mice (n = 12) was given to a group of ON-DP mice (pair-fed ON-DP; n = 12). Another group of ON-DP mice was allowed free access to the diet (ad libitum-fed ON-DP; n = 12). Their body weights were measured weekly.
Glucose metabolism tests
Glucose tolerance was evaluated by OGTT. After overnight food deprivation, mice were given 20% wt/vol. glucose solution (Otuska Pharmaceutical, Japan) by oral gavage (40 or 60 mg glucose/mouse for mice aged 5 and 10 weeks old, respectively). Blood glucose levels were measured by tail bleeding with a glucose sensor (Glutest Neo Super; Sanwa Kagaku Kenkyusho, Japan) before and at 15, 30, 60 and 120 min after glucose administration. Insulin concentrations were also measured in the blood plasma collected before and at 15 and 30 min after the administration using an Ultra Sensitive Mouse Insulin ELISA kit (Morinaga Institute of Biological Science, Japan).
An ITT was performed to evaluate in vivo insulin sensitivity. After 6 h of food deprivation, human short-acting insulin (Humulin R; Eli Lilly Japan, Japan) was injected intraperitoneally into each mouse (0.5 U/kg of body weight). Blood glucose levels were measured before and at 20, 40, 60 and 80 min after the injection as described above.
Tissue lipid analysis
The lipid content of the liver and skeletal muscle (quadriceps femoris) was measured according to the method of Carr et al [10]. In brief, tissues were homogenised with PBS and the total lipids were extracted with chloroform–methanol. The total lipids were then resolubilised in water with Triton X-100 (X100PC; Sigma-Aldrich, MO, USA) and the triacylglycerol content was measured using Triglyceride E-Test colorimetric assay kit (Wako Pure Chemical, Japan).
Diurnal locomotor activity and food intake
Diurnal locomotor activity was measured using an ACTIMO-100 real-time monitoring system (Shinfactory). Locomotor activity of ON-DR and ON-DP mice (5 weeks old, individually housed in the monitoring cage; n = 7) was automatically measured by infrared beam breaks for 3 days. Food consumption of the mice (5 weeks old, individually housed in a standard cage; n = 9) was also measured at the end and beginning of the light phase for 3 days as described above.
Plasma leptin concentration and gene expression analysis
To evaluate in vivo leptin status, blood samples were collected from 5-week-old ON-DR and ON-DP mice (n = 9) by tail bleeding 6 ± 1 h after the beginning of the dark phase. Thereafter, the mice were killed by cervical dislocation and their brains were immediately collected and fixed for immunohistochemical analysis of phosphorylated signal transducer and activator of transcription 3 (STAT3). The epididymal white adipose tissue (eWAT) was collected for gene expression analysis and adipocyte size measurement.
Plasma leptin concentration was measured using Mouse/Rat Leptin Quantikine ELISA Kit (R&D Systems, MN, USA). Total RNA was isolated from eWAT using Isogen II Reagent (Nippon Gene, Japan) and the cDNA was synthesised using SuperScript VILO cDNA Synthesis Kit (Thermo Fisher Scientific, MA, USA). Quantitative PCR was performed using TaqMan Gene Expression Assays (Thermo Fisher Scientific) for leptin (Lep; Mm00434759_m1), CCAAT/enhancer binding protein α (Cebpa; Mm00514283_s1), peroxisome proliferator-activated receptor γ (Pparg; Mm00440940_m1), adiponectin (Adipoq; Mm00456425_m1) and fatty acid binding protein 4 (Fabp4; Mm01295675_g1). The expression levels were determined using the \( {2}^{-{\Delta \Delta C}_t} \) method and normalised to β-actin (Actb, Mm00607939_s1).
Adipocyte size measurement
Adipocyte size was measured according to the method of Parlee et al [11]. After eWAT was fixed with 4% wt/vol. paraformaldehyde in PBS and embedded in paraffin, three cross-sections (5 μm thick) were prepared at 100 μm intervals for each mouse. The sections were stained with H&E and the apparent cell size (cross-sectional area) was measured from the micrographs (~200 cells for each mice) using ImageJ software (National Institutes of Health, MD, USA).
Immunohistochemistry of p-STAT3
To evaluate leptin-induced anorexigenic signalling, quantitative analysis of p-STAT3-positive neurons in the hypothalamic arcuate nucleus (ARC) was performed immunohistochemically [12, 13]. After the brain was fixed with 4% wt/vol. paraformaldehyde in PBS and immersed with 25% wt/vol. sucrose in PBS, the coronal part including the hypothalamus was embedded in OCT compound (Sakura Finetek, Japan). The sections were cut into free-floating sections (30 μm thick) in a rostral-to-caudal manner, and were stored in an antifreeze solution (PBS–glycerol–ethylene glycol, 50:35:15 vol.) at −20°C until use [14]. For immunostaining, ten cross-sections covering the ARC (from bregma −1.4 mm at 120 μm intervals) were treated sequentially with 1% wt/vol. NaOH/1% wt/vol. H2O2 for 15 min, 0.3% wt/vol. glycine for 10 min and 0.03% wt/vol. SDS for 10 min. The sections were then blocked with 3% vol./vol. normal goat serum in PBS containing 0.25% vol./vol. Triton-X100 and 0.02% wt/vol. sodium azide (blocking buffer) for 1 h and incubated with rabbit anti-p-STAT3 (no. 9145, Cell Signaling Technology, MA, USA; 1:3000 in the blocking buffer) for 48 h at 4°C. The sections were further treated with biotinylated goat anti-rabbit IgG (BA-4000, Vector Laboratories, CA, USA; 1:1000 in the blocking buffer without sodium azide) for 1 h, followed by avidin–biotin–peroxidase complex reagents (Vectastain Elite; Vector Laboratories) in PBS containing 0.25% vol./vol. Triton X-100 for 45 min. Signals were developed with diaminobenzidine and H2O2 with cobalt chloride intensification [15]. The p-STAT3-positive nuclei in the ARC (stained dark blue) were counted from the micrographs. The stereotaxic coordinates of the brain regions were defined according to the mouse brain atlas [16].
Leptin administration
Recombinant mouse leptin (R&D Systems) or vehicle was injected intraperitoneally into 5-week-old ON-DR and ON-DP mice (n = 10) on HFD twice a day (50 μg/mouse, at the end and beginning of the light phase) for 3 days. The mice were individually housed, their food intake was measured before each injection, and their body weight was measured daily. To confirm leptin signal transduction, hypothalamic ARC was dissected under a stereomicroscope at 40 min after a single injection of leptin or vehicle into mice that had been deprived of food overnight. STAT3 phosphorylation was assessed by western blotting using specific antibodies for phosphorylated and total STAT3 (no. 9145 and no. 9139, respectively, Cell Signaling Technology; 1:1000) with horseradish peroxidase-conjugated secondary antibodies for rabbit (no. 7074, Cell Signaling Technology; 1:2000) and mouse (NA931, GE healthcare, IL, USA; 1:10,000) IgG, respectively.
Glucose uptake in vivo
Glucose uptake rate was evaluated in vivo using 2-deoxyglucose (2-DG) as a tracer [17]. After overnight food deprivation, 5-week-old ON-DR and ON-DP mice (n = 10) were given 20% wt/vol. glucose solution (2 g/kg body weight) by oral gavage. Then, 15 min after the glucose challenge, a 2-DG solution (1% wt/vol. in saline [NaCl 154 mmol/l], 0.1 g/kg body weight) was injected intraperitoneally. Thirty minutes later, mice were killed by cervical dislocation and eWAT was quickly collected. The amount of 2-DG-6-phosphate (intracellular 2-DG metabolite) in eWAT was measured using 2-Deoxyglucose Uptake Measurement Kit (Cosmo Bio, Japan) [17].
Adipose tissue explant culture
Adipose tissue explant culture was performed according to the method of Thalmann et al [18]. After eWAT was collected from 5-week-old mice and cut into ~2 mm length pieces in M199 medium with 5% vol./vol. FCS and penicillin–streptomycin (culture medium), the tissue fragments were placed on a 100 μm nylon mesh and rinsed with the culture medium. After absorption of excess medium with sterile paper towels, the explants were weighed and incubated overnight in 24 well cell culture plates (~40 mg/well with 0.4 ml medium) for each experiment.
For in vitro glucose uptake assay (n = 8), the medium was changed to serum-free M199 (1% wt/vol. BSA) and the explants were further incubated for 6 h. The medium was then discarded and the explants were maintained in KRB (1% wt/vol. BSA) at 37°C for 20 min. Thereafter, insulin (I9278, Sigma-Aldrich; 10× solution) was added followed by further incubation for 15 min. Next, 2-DG (10× solution, 1 mmol/l final concentration) was added and incubation was continued for 5 min. The incubation was stopped by the addition of ice-cold 0.2 mmol/l phloretin (Sigma-Aldrich) in PBS and the explants were washed twice with the phloretin solution [17]. 2-DG uptake was measured as described above.
To evaluate leptin production capacity (n = 6), the explants were maintained in serum-free M199 medium (0.2% wt/vol. BSA, 5.5 mmol/l glucose) for 4 h. Then, the medium was changed to high-glucose (16.7 mmol/l) M119 medium with or without insulin. After incubation for 4 or 8 h, the medium was collected to measure leptin concentration. The explants were also collected after 4 h incubation for Lep gene expression analysis.
DNA methylation analysis
DNA methylation in the Lep promoter region was analysed by bisulphite sequencing. To isolate genomic DNA from mature adipocytes, eWAT (collected from 5-week-old mice; n = 4) was cut into small pieces and the tissue fragments were digested with collagenase (C7657, Sigma-Aldrich; 3 mg/ml) for 45 min at 37°C with occasional shaking. The cell suspension was then filtered through a 500 μm nylon mesh and allowed to stand for 10 min. The floating adipocytes were collected for DNA isolation using a Wizard Genomic DNA Purification Kit (Promega, WI, USA). Total RNA was also prepared from the isolated adipocytes for gene expression analysis. Bisulphite conversion was performed using EZ DNA Methylation-Gold Kit (Zymo Research, CA, USA) and 500 ng genomic DNA. For each bisulphite PCR, EpiTaq HS (Takara Bio, Japan) was used to catalyse the amplification. PCR was performed with the following thermocycling conditions: denaturation at 95°C for 3 min, then 40 cycles each consisting of incubation at 95°C for 30 s, 58°C for 30 s, and 72°C for 30 s, followed by final extension for 5 min at 72°C. The primer sequences used in the bisulphite PCR were as follows: forward primer-1, 5′-TTTGTTTTTGGGATTGGTTTAGA-3′; reverse primer-1, 5′-AACCAATAAACCAAAACTCTACCA-3′; forward primer-2, 5′-TTGGTAGAGTTTTGGTTTATTGG-3′; reverse primer-2, 5′-TCCACATAACCTCCTTCTTACCTC-3′. For sequencing, the PCR fragments were cloned into pGEM-T Easy vector (Promega). The vectors were sequenced by BigDye sequencing (Thermo Fisher Scientific). DNA methylation status in each sample was evaluated using QUMA software (http://quma.cdb.riken.jp/; date of access: 9 August 2018). No polymorphisms were found in the target sequence among the mice analysed.
Statistics
Values are expressed as mean ± SEM. All measurements were taken from distinct samples and the results were substantiated in at least two independent animal cohorts. Randomisation and blinding were not carried out. Mice that were blocked from free access to food due to the accidental clogging of a feeding box were excluded from the experiments. Statistical significance was determined by unpaired two-tailed t test or one-way ANOVA with Tukey’s post hoc test. A p value <0.05 was considered significant. All data were analysed using JMP software (ver. 9.0.2, SAS Institute, NC, USA).
Results
Food restriction ameliorates adiposity and deterioration of glucose tolerance in ON-DP mice
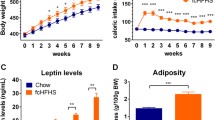
During the HFD feeding period (from 5 weeks to 10 weeks of age), ON-DP mice had a higher food intake and greater body weight gain than ON-DR mice under ad libitum-fed conditions (Fig. 1a, b). The excessive body weight gain of ON-DP mice was completely suppressed to the weight gain level seen in ON-DR mice when the amount of food available each day was restricted to the amount eaten by ON-DR mice (pair-fed ON-DP; Fig. 1a, b). After the feeding period, the deterioration in glucose tolerance and reduced insulin sensitivity shown in ad libitum-fed ON-DP mice were largely ameliorated by the pair-feeding (Fig. 1c–l). In addition, increased adipose tissue mass and lipid accumulation in the tissues of ON-DP mice were suppressed to the levels seen in ON-DR mice by the pair-feeding (Fig. 1m–o). Food intake, growth trajectory, and casual blood glucose levels from 5 to 10 weeks of age, including mice fed standard chow diet and female mice, are shown in electronic supplementary material (ESM) Fig. 1a–h. ON-DP mice of both sexes showed higher food intake and body weight gain than ON-DR mice, even under standard chow feeding, with the weight gain and higher blood glucose levels seen in the ON-DP mice being exaggerated by HFD feeding (ESM Fig. 1a–h).

During HFD feeding, excessive body weight gain, deterioration of glucose metabolism and accumulation of tissue lipid in ON-DP (DP) mice were suppressed by pair-feeding with ON-DR (DR) mice. (a, b) Daily food intake (a) and body weight (b) of ON-DR (white circles), ad libitum-fed ON-DP (black circles) and pair-fed ON-DP (grey circles) mice (n = 12 for each group). (c–e, h–j) Blood glucose and plasma insulin levels in OGTT in mice aged 5 weeks (c–e) and 10 weeks (h–j) (40 and 60 mg glucose/mouse, respectively). Plasma insulin levels are shown as the absolute concentration and the changes from the baseline. In (c–e), white circles, ON-DR (n = 11); black circles, ON-DP (n = 16). In (h–j), white squares, ON-DR (n = 11); black squares, ad libitum-fed ON-DP (n = 10); grey squares, pair-fed ON-DP (n = 10). (f, g, k, l) Blood glucose levels in ITT in mice aged 5 weeks (f, g) and 10 weeks old (k, l) (0.5 U insulin/kg body weight). Blood glucose levels are shown as the absolute concentration and the value relative to the baseline. In (f, g), white circles, ON-DR (n = 6); black circles, ON-DP (n = 6). In (k, l), white squares, ON-DR (n = 12); black squares, ad libitum-fed ON-DP (n = 12); grey squares, pair-fed ON-DP (n = 12). (m–o) eWAT weight (m) and triacylglycerol content in liver (n) and quadriceps femoris skeletal muscle (o) of ON-DR (white bars), ad libitum-fed ON-DP (black bars) and pair-fed ON-DP (grey bars) mice after the 5 week HFD feeding period (n = 12 for each group). (p, q) Locomotor activity (p) and food intake (q) during the dark and light phases in ON-DR (white bars) and ON-DP (black bars) mice at 5 weeks of age; n = 7 and n = 9 for each group in (p) and (q), respectively. Mean values without error bars have SEM within symbol size. In (a, c, e), *p < 0.05, **p < 0.01 and ***p < 0.001 vs ON-DR mice (unpaired two-tailed t test). In (b, h–l), *p < 0.05, **p < 0.01 and ***p < 0.001 for ON-DR vs ad libitum-fed ON-DP mice; †p < 0.05 and ††p < 0.01 for ON-DR vs pair-fed ON-DP mice; ‡p < 0.05, ‡‡p < 0.01 and ‡‡‡p < 0.001 for ad libitum-fed ON-DP vs pair-fed ON-DP mice (at each time point, ANOVA with Tukey’s post hoc test). In (m–o), ***p < 0.001 vs ON-DR mice; ‡‡‡p < 0.001 vs ad libitum-fed ON-DP mice (ANOVA with Tukey’s post hoc test). In (q), **p < 0.01 for ON-DR vs ON-DP mice (unpaired two-tailed t test)
No significant difference was observed in the locomotor activity between the two mouse lines at 5 weeks of age (Fig. 1p). Meanwhile, ON-DP mice showed higher food intake in the dark phase compared with ON-DR mice (Fig. 1q).
Lower leptin concentration and gene expression in ON-DP mice
At 5 weeks of age, with no differences in body weight and adipose tissue mass between the two mouse lines, plasma leptin concentrations were lower in ON-DP mice than in ON-DR mice (Fig. 2a–c). Lep gene expression level was also lower in the eWAT of ON-DP mice when compared with ON-DR mice, whereas no significant differences were observed in the expression levels of adipocyte differentiation marker genes (Cebpa and Pparg) or other genes primarily expressed in adipocytes (Adipoq and Fabp4) (Fig. 2d). The adipocyte size did not differ significantly between the two mouse lines (Fig. 2e, f). Plasma leptin and eWAT Lep gene expression levels at 10 weeks of age (after the 5 week HFD pair-feeding period) are shown in ESM Fig. 2a, b.

Lower plasma leptin concentration and eWAT Lep gene expression in ON-DP mice. (a–e) Body weight (a), eWAT weight (b), plasma leptin concentration (c), eWAT gene expression levels (d) and mean cross-sectional adipocyte size (e) in ON-DR (DR; white bars) and ON-DP (DP; black bars) mice at 5 weeks of age (n = 9 for each group). Blood and eWAT samples were collected 6 ± 1 h after the beginning of the dark phase. (f) Representative micrograph images for adipocyte size measurement; scale bar, 100 μm. **p < 0.01 and ***p < 0.001 for ON-DR vs ON-DP (unpaired two-tailed t test)
Lower leptin signalling in ON-DP mouse hypothalamus
In accordance with the lower plasma leptin concentration, the ON-DP mice showed fewer p-STAT3-positive neurons (i.e. lower anorexigenic signalling via leptin receptor) in the hypothalamic ARC when compared with ON-DR mice (Fig. 3a–c). On the other hand, peripheral leptin administration efficiently induced STAT3 phosphorylation in the ARC and consequently suppressed food intake and body weight gain in both mouse lines (Fig. 3d–f).

Lower leptin signalling in hypothalamic ARC leads to overfeeding in ON-DP mice. (a, b) Numbers of p-STAT3-positive neurons in ARC cross-sections taken from bregma −1.4 mm at 120 μm intervals (cross-section nos 1–10) (a) and the total counts (b) of ON-DR (DR; white bars) and ON-DP (DP; black bars) mice at 5 weeks of age (n = 9 for each group). Brain samples were collected 6 ± 1 h after the beginning of the dark phase. (c) Representative micrograph images used for counting p-STAT3-positive neurons (bregma −1.9 mm); scale bar, 100 μm; 3 V, third ventricle. (d) Western blot images of phosphorylated and total STAT3 in the ARC at 40 min after an i.p. injection of vehicle or leptin (50 μg/mouse). (e, f) Food intake (e) and body weight change (f) of ON-DR and ON-DP mice receiving i.p. injections of vehicle (white bars) or leptin (grey bars; 50 μg/mouse) twice a day for 3 days (n = 10 for each group). In (a, b): *p < 0.05 and **p < 0.01 for ON-DR vs ON-DP (unpaired two-tailed t test). In (e, f): ***p < 0.001 for vehicle- vs leptin-injected mice (unpaired two-tailed t test)
Glucose uptake rate and leptin production capacity in adipose tissue
When glucose uptake was assessed in vivo, the ON-DP mice showed a lower rate of glucose uptake (measured by 2-DG incorporation) in eWAT after oral glucose challenge compared with the ON-DR mice (Fig. 4a). In the eWAT explant culture study, explants from ON-DP mice also showed lower 2-DG uptake than those from ON-DR mice, even after exposure to the same concentration of insulin (Fig. 4b; albeit slightly above the significance threshold at 0.2 nmol/l insulin, p = 0.06). Explants from the ON-DP mice also showed lower levels of Lep gene expression and leptin secretion than explants from the ON-DR mice (Fig. 4c, d).

Potential determining factors for different leptin production capacity in ON-DR (DR) vs ON-DP (DP) mice. (a) In vivo 2-DG uptake into eWAT after oral glucose administration (2 g/kg body weight) in the two mouse lines (n = 10 for each group). (b) 2-DG uptake in eWAT explants from ON-DR (white bars) and ON-DP (black bars) mice, at different concentrations of insulin (n = 8 for each group). (c, d) Lep gene expression (c) and leptin secretion (d) in eWAT explants from ON-DR (white bars) and ON-DP (black bars) mice in the absence (−) or presence (+) of insulin (n = 6 for each group). (e, f) DNA methylation rate of CpG sites in the Lep gene promoter region in eWAT adipocytes from the two mouse lines (n = 4 for each group) (e) and its correlation with Lep gene expression levels (f) (Pearson correlation analysis: y = −0.03744x + 3.539, r2 = 0.8789, p < 0.001). White circles, ON-DR mice; black circles, ON-DP mice. In (a–e), *p < 0.05 and **p < 0.01 for ON-DR vs ON-DP (unpaired two-tailed t test). In (b), †p < 0.05, ††p < 0.01 and †††p < 0.001 vs no insulin (0 nmol/l) within in a mouse line (ANOVA with Tukey’s post hoc test). In (c, d), †p < 0.05, ††p < 0.01 and †††p < 0.001 vs no insulin (−) within a mouse line (at each time point, unpaired two-tailed t test)
DNA methylation analysis of isolated adipocytes revealed that the CpG methylation rate in the Lep promoter region was higher in ON-DP mice than in ON-DR mice (Fig. 4e). Furthermore, the methylation rate was inversely correlated with Lep gene expression levels in adipocytes (Fig. 4f). Overall DNA methylation profiles for each mouse are shown in ESM Fig. 3.
Discussion
The present results clearly demonstrated the role of leptin production capacity in determining spontaneous meal size and consequent susceptibility to obesity-induced diabetes in ON-DP and ON-DR mice. The development of obesity in ON-DP mice was completely suppressed when the feeding was restricted to the food consumption level of ON-DR mice (Fig. 1a, b). Consequently, the deterioration of glucose tolerance in ON-DP mice, accompanied by the emergence of insulin resistance, was largely ameliorated under pair-feeding conditions (Fig. 1c–l). Taken together with the finding of no significant difference in locomotor activity between the two mouse lines (Fig. 1p), the development of obesity and subsequent deterioration in glucose tolerance are most likely due to spontaneous overfeeding in ON-DP mice.
As a possible cause of hyperphagic behaviour in ON-DP mice, we found lower plasma leptin concentration compared with ON-DR mice before the emergence of body weight difference (Fig. 2a, c). The ON-DP mice also expressed lower levels of Lep in eWAT (Fig. 2d), albeit with no apparent difference in adipocyte maturation between the mouse lines (Fig. 2b, e, f). In accordance with the lower plasma leptin concentration, ON-DP mice showed fewer p-STAT3-positive neurons in the hypothalamic ARC, a hallmark of leptin-induced anorexigenic signalling (Fig. 3a–c). On the other hand, apparent central leptin resistance was not observed in either of the mouse lines when exogenous leptin was administered peripherally (Fig. 3d–f). These findings suggest that the hyperphagic behaviour in ON-DP mice is primarily governed by lower circulating leptin levels. Therefore, we further investigated the underlying mechanisms involved in different leptin production capacity between the two mouse lines.
We previously reported that ON-DP mice show poor insulin secretion response to glucose, both in vivo and in isolated pancreatic islets, as the major cause of hereditary glucose intolerance [19]. Because Lep transcription is reported to be triggered by insulin-stimulated glucose uptake and subsequent intracellular glucose metabolism in adipocytes [20], we first hypothesised that the impaired insulin secretion determines lower circulating leptin levels in ON-DP mice, insofar as lower circulating insulin levels after feeding lead to lower glucose uptake rate in adipocytes, which in turn results in lower leptin secretion. As expected, we observed that glucose uptake into eWAT after glucose challenge was lower in ON-DP mice in vivo (Fig. 4a). However, we also found other possible causes that could lead to lower leptin secretion, independent of the impaired insulin secretion. As shown in Fig. 4b–d, eWAT explants from ON-DP mice showed lower glucose uptake, Lep gene expression and leptin secretion than those from ON-DR mice in vitro, even at the same concentration of insulin. The differences in glucose uptake under the same amount of insulin imply that there is heritable difference in adipose tissue insulin sensitivity between the two mouse lines. In concordance with the lower Lep gene expression levels, we found a higher level of DNA methylation in the Lep promoter region of ON-DP mouse adipocytes (Fig. 4e). The strong inverse correlation between the levels of methylation and gene expression implies that epigenetic modification plays a primary role in the quantitative regulation of Lep transcription (Fig. 4f). Although several previous reports have indicated the role of DNA methylation in Lep gene expression [21,22,23,24], the regulatory mechanisms of the methylation status per se have not yet been fully elucidated. Our findings strongly suggest that lower leptin production capacity in adipose tissue (due to lower glucose uptake rate and higher Lep promoter DNA methylation) determines hyperphagic behaviour in ON-DP mice, at least partially independent of lower pancreatic insulin secretion.
Ever since the discovery of leptin as the key molecule in appetite control, a number of prospective studies have addressed whether leptin levels predict future weight changes not only in adult but also in paediatric and adolescent populations. However, the results remain controversial [25,26,27]. One of the most plausible reasons for the inconsistent results is the difficulty in comparing the ‘relative level’ of leptin. In general, circulating leptin level is markedly increased with fat mass accompanied by decreased leptin sensitivity (leptin resistance) [28,29,30]. This dependency on body fat mass makes it difficult to appropriately assess the relative concentration of leptin (corresponding to the extent of leptin resistance for each individual) among populations with varying degree of obesity [25]. Within a population of relatively small background variation (normal-weight young adults entering university), Allard et al [31] recently demonstrated an inverse association between baseline leptin levels and subsequent 2 year body weight gain. In our preliminary study, there was also a strong correlation between adipose tissue mass and plasma leptin concentration in ON-DP and ON-DR mice (ESM Fig. 4a, b). We therefore used age- and body weight-matched mice for each experiment to minimise the influence of adiposity on leptin status. As a result, we demonstrated that the mice with relatively low leptin levels (ON-DP) were more prone to overfeeding than the control ON-DR mice. In contrast to the definitive role of total leptin deficiency in overfeeding (e.g. in ob/ob mice), information on the effect of its relative deficiency has been elusive in rodents as well as in humans. However, several reports have suggested a possible causative relationship between relative leptin levels and susceptibility to diet-induced obesity among different mouse strains [32, 33]. In addition, partial leptin deficiency due to the heterozygous mutation in the leptin-encoding gene has been reported to be associated with adiposity both in rodents and humans [34,35,36]. Collectively, these findings suggest that lower leptin levels before being overweight, especially in childhood or adolescence, may predict overeating and subsequent excessive weight gain.
Because the ON-DP and ON-DR mice were established by selective breeding for different susceptibilities to glucose intolerance, these mice likely mimic the polygenic and heritable aetiology of type 2 diabetes. Actually, the ON-DP mice display two major aetiological factors in the development of type 2 diabetes: impaired insulin secretion capacity and reduced insulin sensitivity (insulin resistance) [37, 38]. The latter is typically associated with the development of obesity. In the present study, insulin resistance was actually manifested in ON-DP mice accompanied by body weight gain and fat accumulation. As a cause of the development of obesity, we demonstrate here the effect of lower leptin production capacity on spontaneous hyperphagia. Despite the well-known role of leptin in feeding behaviour, little is known about the genetic or environmental factors that quantitatively regulate leptin production. As possible regulatory factors, we demonstrated the differences in glucose uptake rate and Lep gene promoter DNA methylation in adipose tissue when comparing ON-DP and ON-DR mice. However, it remains unclear how the methylation status had been regulated and inherited through the selective breeding. The elucidation of the underlying mechanisms for the epigenetic modulation (e.g. involvement of sequence variants in cis-regulatory elements or of trans-acting factors including non-coding RNAs) may lead to novel approaches for the prevention and treatment of obesity caused by overeating and its metabolic complications including type 2 diabetes. Recent reports demonstrated the potential role of a long non-coding RNA in Lep gene transcription in mice [39, 40]. A recent genome-wide association study also reported multiple loci robustly associated with circulating leptin levels in humans [41]. These findings warrant further research on the regulatory mechanisms of leptin gene expression (e.g. nutritional/metabolic regulation of epigenetics [42,43,44,45]).
In summary, our present results highlight the critical role of leptin production capacity from shortly after weaning as a determinant for the development of obesity, which in turn causes type 2 diabetes. In addition to lower insulin secretion capacity in pancreatic beta cells, lower leptin production capacity in adipocytes may be a heritable risk factor for the development of type 2 diabetes.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ARC:
-
Arcuate nucleus
- 2-DG:
-
2-Deoxyglucose
- eWAT:
-
Epididymal white adipose tissue
- HFD:
-
High-fat diet
- ON-DP:
-
Oikawa–Nagao Diabetes-Prone
- ON-DR:
-
Oikawa–Nagao Diabetes-Resistant
- STAT3:
-
Signal transducer and activator of transcription 3
References
Ng M, Fleming T, Robinson M et al (2014) Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 384:766–781. https://doi.org/10.1016/S0140-6736(14)60460-8
NCD Risk Factor Collaboration (NCD-RisC) (2016) Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet 387:1377–1396. https://doi.org/10.1016/S0140-6736(16)30054-X
Blundell JE, Macdiarmid JI (1997) Passive overconsumption. Fat intake and short-term energy balance. Ann N Y Acad Sci 827:392–407
Hetherington MM (2007) Individual differences in the drive to overeat. Nutr Bull 32:14–21. https://doi.org/10.1111/j.1467-3010.2007.00601.x
Llewellyn C, Wardle J (2015) Behavioral susceptibility to obesity: gene-environment interplay in the development of weight. Physiol Behav 152:494–501. https://doi.org/10.1016/j.physbeh.2015.07.006
Waalen J (2014) The genetics of human obesity. Transl Res 164:293–301. https://doi.org/10.1016/j.trsl.2014.05.010
Nagao M, Asai A, Kawahara M et al (2012) Selective breeding of mice for different susceptibilities to high fat diet-induced glucose intolerance: development of two novel mouse lines, selectively bred diet-induced glucose intolerance-prone and -resistant. J Diabetes Investig 3:245–251. https://doi.org/10.1111/j.2040-1124.2011.00175.x
Nagao M, Asai A, Sugihara H, Oikawa S (2015) Transgenerational changes of metabolic phenotypes in two selectively bred mouse colonies for different susceptibilities to diet-induced glucose intolerance. Endocr J 62:371–378. https://doi.org/10.1507/endocrj.EJ14-0241
Nagao M, Esguerra JLS, Wendt A et al (2020) Selectively bred diabetes models: GK rats, NSY mice, and ON mice. Methods Mol Biol 2128:25–54. https://doi.org/10.1007/978-1-0716-0385-7_3
Carr T, Andresen C, Rudel L (1993) Enzymatic determination of triglyceride, free cholesterol, and total cholesterol in tissue lipid extracts. Clin Biochem 26:39–42
Parlee SD, Lentz SI, Mori H, MacDougald OA (2014) Quantifying size and number of adipocytes in adipose tissue. Methods Enzymol 537:93–122. https://doi.org/10.1016/B978-0-12-411619-1.00006-9
Münzberg H, Flier JS, Bjørbæk C (2004) Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 145:4880–4889. https://doi.org/10.1210/en.2004-0726
Enriori PJ, Evans AE, Sinnayah P et al (2007) Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab 5:181–194. https://doi.org/10.1016/j.cmet.2007.02.004
Plum L, Ma X, Hampel B et al (2006) Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest 116:1886–1901. https://doi.org/10.1172/JCI27123
Hsu SM, Soban E (1982) Color modification of diaminobenzidine (DAB) precipitation by metallic ions and its application for double immunohistochemistry. J Histochem Cytochem 30:1079–1082
Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, 2nd edn. Academic Press, San Diego
Saito K, Lee S, Shiuchi T et al (2011) An enzymatic photometric assay for 2-deoxyglucose uptake in insulin-responsive tissues and 3T3-L1 adipocytes. Anal Biochem 412:9–17. https://doi.org/10.1016/j.ab.2011.01.022
Thalmann S, Juge-Aubry CE, Meier CA (2008) Explant cultures of white adipose tissue. Methods Mol Biol 456:195–199. https://doi.org/10.1007/978-1-59745-245-8_14
Nagao M, Asai A, Inaba W et al (2014) Characterization of pancreatic islets in two selectively bred mouse lines with different susceptibilities to high-fat diet-induced glucose intolerance. PLoS One 9:e84725. https://doi.org/10.1371/journal.pone.0084725
Mueller WM, Gregoire FM, Stanhope KL et al (1998) Evidence that glucose metabolism regulates leptin secretion from cultured rat adipocytes. Endocrinology 139:551–558
Melzner I, Scott V, Dorsch K et al (2002) Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J Biol Chem 277:45420–45427. https://doi.org/10.1074/jbc.M208511200
Marchi M, Lisi S, Curcio M et al (2011) Human leptin tissue distribution, but not weight loss-dependent change in expression, is associated with methylation of its promoter. Epigenetics 6:1198–1206. https://doi.org/10.4161/epi.6.10.16600
Schultz NS, Broholm C, Gillberg L et al (2014) Impaired leptin gene expression and release in cultured preadipocytes isolated from individuals born with low birth weight. Diabetes 63:111–121. https://doi.org/10.2337/db13-0621
Kuroda M, Tominaga A, Nakagawa K et al (2016) DNA methylation suppresses leptin gene in 3T3-L1 adipocytes. PLoS One 11:e0160532. https://doi.org/10.1371/journal.pone.0160532
Jenkins AB, Campbell LV (2003) Does relative leptinemia predict weight gain in humans? Obes Res 11:373–374. https://doi.org/10.1038/oby.2003.49
Hivert M-F, Langlois M-F, Carpentier AC (2007) The entero-insular axis and adipose tissue-related factors in the prediction of weight gain in humans. Int J Obes 31:731–742. https://doi.org/10.1038/sj.ijo.0803500
Ahima RS (2008) Revisiting leptin’s role in obesity and weight loss. J Clin Invest 118:2380–2383. https://doi.org/10.1172/JCI36284
Arch JR, Stock MJ, Trayhurn P (1998) Leptin resistance in obese humans: does it exist and what does it mean? Int J Obes 22:1159–1163. https://doi.org/10.1038/sj.ijo.0800779
Enriori PJ, Evans AE, Sinnayah P, Cowley MA (2006) Leptin resistance and obesity. Obesity (Silver Spring) 14(Suppl 5):254S–258S. https://doi.org/10.1038/oby.2006.319
Myers MG, Leibel RL, Seeley RJ, Schwartz MW (2010) Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab 21:643–651. https://doi.org/10.1016/j.tem.2010.08.002
Allard C, Doyon M, Brown C et al (2013) Lower leptin levels are associated with higher risk of weight gain over 2 years in healthy young adults. Appl Physiol Nutr Metab 38:280–285. https://doi.org/10.1139/apnm-2012-0225
Surwit RS, Petro AE, Parekh P, Collins S (1997) Low plasma leptin in response to dietary fat in diabetes- and obesity-prone mice. Diabetes 46:1516–1520
Watson PM, Commins SP, Beiler RJ et al (2000) Differential regulation of leptin expression and function in A/J vs. C57BL/6J mice during diet-induced obesity. Am J Physiol Endocrinol Metab 279:E356–E365. https://doi.org/10.1152/ajpendo.2000.279.2.E356
Chung WK, Belfi K, Chua M et al (1998) Heterozygosity for Lepob or Leprdb affects body composition and leptin homeostasis in adult mice. Am J Phys 274:R985–R990. https://doi.org/10.1152/ajpregu.1998.274.4.R985
Farooqi IS, Keogh JM, Kamath S et al (2001) Partial leptin deficiency and human adiposity. Nature 414:34–35. https://doi.org/10.1038/35102112
Begriche K, Lettéron P, Abbey-Toby A et al (2008) Partial leptin deficiency favors diet-induced obesity and related metabolic disorders in mice. Am J Physiol Endocrinol Metab 294:E939–E951. https://doi.org/10.1152/ajpendo.00379.2007
Gerich JE (1998) The genetic basis of type 2 diabetes mellitus: impaired insulin secretion versus impaired insulin sensitivity. Endocr Rev 19:491–503. https://doi.org/10.1210/edrv.19.4.0338
Kahn SE (2003) The relative contributions of insulin resistance and beta cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia 46:3–19. https://doi.org/10.1007/s00125-002-1009-0
Lo KA, Huang S, Walet ACE et al (2018) Adipocyte long-noncoding RNA transcriptome analysis of obese mice identified Lnc-Leptin, which regulates leptin. Diabetes 67:1045–1056. https://doi.org/10.2337/db17-0526
Dallner OS, Marinis JM, Lu Y-H et al (2019) Dysregulation of a long noncoding RNA reduces leptin leading to a leptin-responsive form of obesity. Nat Med 25:507–516. https://doi.org/10.1038/s41591-019-0370-1
Kilpeläinen TO, Carli JFM, Skowronski AA et al (2016) Genome-wide meta-analysis uncovers novel loci influencing circulating leptin levels. Nat Commun 7:10494. https://doi.org/10.1038/ncomms10494
Lu C, Thompson CB (2012) Metabolic regulation of epigenetics. Cell Metab 16:9–17. https://doi.org/10.1016/j.cmet.2012.06.001
Sharma U, Rando OJ (2017) Metabolic inputs into the epigenome. Cell Metab 25:544–558. https://doi.org/10.1016/j.cmet.2017.02.003
Ling C, Rönn T (2019) Epigenetics in human obesity and type 2 diabetes. Cell Metab 29:1028–1044. https://doi.org/10.1016/j.cmet.2019.03.009
Sun L, Lin JD (2019) Function and mechanism of long noncoding RNAs in adipocyte biology. Diabetes 68:887–896. https://doi.org/10.2337/dbi18-0009
Acknowledgements
The authors thank M. Kawahara (Department of Endocrinology, Diabetes and Metabolism, Nippon Medical School, Tokyo, Japan) for excellent technical assistance.
Authors’ relationships and activities
The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Funding
This study was supported by the Japan Society for the Promotion of Science (25860300, 15K08434, 17K08780), the Nippon Medical School Alumni Association and the Lotte Shigemitsu Prize.
Author information
Authors and Affiliations
Contributions
AA and MN conceived and designed the study. AA, MN and KH acquired and analysed the data. TM, HS and SO contributed to the analysis and interpretation of the data. AA drafted the manuscript. MN, KH, TM, HS and SO contributed to critical revision of the manuscript for important intellectual content. All authors approved the final version of the manuscript to be published. AA is the guarantor of this work.
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM Figures
(PDF 399 kb)
Rights and permissions
About this article

Cite this article
Asai, A., Nagao, M., Hayakawa, K. et al. Leptin production capacity determines food intake and susceptibility to obesity-induced diabetes in Oikawa–Nagao Diabetes-Prone and Diabetes-Resistant mice. Diabetologia 63, 1836–1846 (2020). https://doi.org/10.1007/s00125-020-05191-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-020-05191-8