
Abstract
Aims/hypothesis
In obesity oxidative stress is thought to contribute to the development of insulin resistance, non-alcoholic fatty liver disease and the progression to non-alcoholic steatohepatitis. Our aim was to examine the precise contributions of hepatocyte-derived H2O2 to liver pathophysiology.
Methods
Glutathione peroxidase (GPX) 1 is an antioxidant enzyme that is abundant in the liver and converts H2O2 to water. We generated Gpx1 lox/lox mice to conditionally delete Gpx1 in hepatocytes (Alb-Cre;Gpx1 lox/lox) and characterised mice fed chow, high-fat or choline-deficient amino-acid-defined (CDAA) diets.
Results
Chow-fed Alb-Cre;Gpx1 lox/lox mice did not exhibit any alterations in body composition or energy expenditure, but had improved insulin sensitivity and reduced fasting blood glucose. This was accompanied by decreased gluconeogenic and increased glycolytic gene expression as well as increased hepatic glycogen. Hepatic insulin receptor Y1163/Y1163 phosphorylation and Akt Ser-473 phosphorylation were increased in fasted chow-fed Alb-Cre;Gpx1 lox/lox mice, associated with increased H2O2 production and insulin signalling in isolated hepatocytes. The enhanced insulin signalling was accompanied by the increased oxidation of hepatic protein tyrosine phosphatases previously implicated in the attenuation of insulin signalling. High-fat-fed Alb-Cre;Gpx1 lox/lox mice did not exhibit alterations in weight gain or hepatosteatosis, but exhibited decreased hepatic inflammation, decreased gluconeogenic gene expression and increased insulin signalling in the liver. Alb-Cre;Gpx1 lox/lox mice fed a CDAA diet that promotes non-alcoholic steatohepatitis exhibited decreased hepatic lymphocytic infiltrates, inflammation and liver fibrosis.
Conclusions/interpretation
Increased hepatocyte-derived H2O2 enhances hepatic insulin signalling, improves glucose control and protects mice from the development of non-alcoholic steatohepatitis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity is a major risk factor for the development of insulin resistance, a key factor in the aetiology of the metabolic syndrome and type 2 diabetes [1–3]. Obesity and insulin resistance promote the development of non-alcoholic fatty liver disease (NAFLD), which in a subset of individuals can progress to non-alcoholic steatohepatitis (NASH) [4]. NASH is characterised by fatty liver and overt inflammation that can lead to the death of steatotic hepatocytes, instigating reparative responses that result in fibrosis [4]. Oxidative stress accompanying the obese state is considered a key factor in the development of insulin resistance [5–7] and an important contributor to the development of NAFLD and NASH [4, 8–10]. Systemic and hepatic oxidative stress is evident in obesity and there is direct evidence for the involvement of reactive oxygen species (ROS) in the promotion of insulin resistance, NAFLD and NASH in obesity/type 2 diabetes in rodents [5–13].
Mitochondria are thought to be the primary contributors to oxidative stress in obesity. Superoxide (O2•–) is a natural byproduct of the single-electron transport chain [14, 15]. In obesity, the chronic uptake and oxidation of energy substrates is thought to generate reducing equivalents that exceed the rate of ATP utilisation, thus enhancing the generation of O2•– [14, 15]. In addition, anaplerotic/cataplerotic pathways in hepatocytes in NAFLD induce mitochondrial O2•– generation and inflammation [9], whereas in NASH, increased NADPH oxidase (NOX)-4 expression may also contribute to hepatic oxidative stress [8]. O2•– is converted to H2O2 by superoxide dismutase (SOD) and thereafter eliminated by antioxidant enzymes such as catalase, peroxiredoxins (PRDXs) and glutathione peroxidase (GPX) [14, 15]. Oxidative stress ensues when the production of O2•–/H2O2 exceeds the antioxidant capacity of a cell.
Transient, localised H2O2 generation can occur in response to physiological stimuli such as growth factors and hormones [14]. Both NOX and mitochondria have been implicated in the generation of O2•– and H2O2 in response to physiological stimuli [14, 16, 17]. An increase in H2O2 in response to stimuli such as insulin can facilitate signalling by oxidising and inactivating protein tyrosine phosphatases (PTPs) [18]. Several PTPs, including the tyrosine-specific PTP1B and the dual-specificity phosphatase and tensin homologue, can be oxidised in response to insulin to promote insulin receptor (IR) activation and downstream phosphatidylinositol 3-kinase (PI3K) signalling, respectively [16, 19–22].
GPX1 is a ubiquitous selenoenzyme that uses glutathione to catalyse the conversion of H2O2 into H2O [18]. Gpx1 –/– mice are healthy and fertile and do not show any obvious abnormalities [23, 24]. Indeed, when Gpx1 –/– mice are fed a high-fat diet that promotes moderate adiposity and insulin resistance, but not hyperglycaemia [25], GPX1 deficiency promotes insulin signalling in muscle and protects mice from the development of insulin resistance [22], indicating that increases in H2O2 in muscle may be beneficial. Moreover, even in the context of morbid obesity and hyperglycaemia, global GPX1 deficiency protects mice from the development of steatohepatitis and liver damage and improves glucose metabolism [25]. Given the extensive tissue crosstalk in the control of glucose and lipid homeostasis and the capacity of H2O2 to diffuse across membranes, it is difficult to definitively ascribe the effects of global GPX1 deficiency and heightened H2O2 on hepatic pathophysiology to any cell type or tissue and to exclude potential detrimental effects being masked by the global deletion of GPX1. Therefore we have ‘floxed’ the Gpx1 allele to allow us to explore the hepatocyte-specific contributions of GPX1 to the regulation of hepatic insulin sensitivity and the development of NAFLD and NASH.
Methods
Mice
Gpx1 lox/+ mice on a C57BL/6 J background were generated by the Monash Gene Targeting Facility as described in electronic supplementary material (ESM) Methods. Alb-Cre (C57BL/6 J) mice have been described previously [13]. Mice were maintained on a 12 h light–dark cycle in a temperature-controlled high-barrier facility with free access to food (6% wt/wt fat) and water. Where indicated, mice were fed an obesogenic diet (23.5% wt/wt fat, SF04-027; Specialty Feeds, Glen Forest, WA, Australia) or a choline-deficient amino-acid-defined (CDAA) diet (SF13-103; Specialty Feeds). Experiments were conducted on age-matched male mice and experimentors were blind to outcome assessment. Experiments were approved by the Monash University Animal Ethics Committee.
Metabolic analyses
Blood was collected for analysis of fed and fasted blood glucose and plasma insulin as described previously [22]. Insulin and pyruvate tolerance tests were performed in mice fasted for 4–6 h and hyperinsulinaemic–euglycaemic clamps were performed as described previously [22]. Other metabolic analyses and NASH scoring are described in ESM Methods.
Biochemical analyses
Tissue homogenates were analysed as described previously [22]. Immunoblotting antibodies are described in ESM Methods. Hepatic glycogen was extracted and debranched with amylo-α-l,4-α-1,6-glucosidase and glucose units were analysed using an enzymatic fluorometric method described previously [26].
For the analysis of PTP oxidation, frozen liver samples were homogenised under anaerobic conditions in the presence of N-ethylmaleimide to alkylate all reduced PTPs and then subsequently reduced and hyperoxidised to the sulfonic state as described previously [13]. Immunoblots were probed with a mouse monoclonal antibody (PTPox) raised against the signature motif of the prototypic PTP1B oxidised to the irreversible sulfonic state, which detects tyrosine-specific PTPs oxidised to the sulfonic state [27].
Total and oxidised glutathione levels in tissue supernatant fractions or blood were measured as described previously [25].
H2O2 measurements
Hepatocytes were isolated using a two-step collagenase A perfusion method [13, 28]. Hepatocytes were serum-starved in low-glucose DMEM (ThermoFisher, Waltham, MA, USA) containing 0.1% (vol./vol.) FBS for 2 h and incubated in PBS containing 100 μmol/l Amplex Red reagent and 1 U/ml HRP (Amplex Red hydrogen peroxide assay kit; ThermoFisher) in the presence or absence of 100 nmol/l insulin and fluorescence measured and normalised to protein.
Quantitative PCR
Hepatic levels of Gpx1, Gpx3, Gpx4, Ho1 (also known as Hmox1), Txn1, Trxrd1 (Txnrd1), G6pc, Pck1, Gck, Pdk4, Lpk (Col2a1), Ppargc1α (Ppargc1a), Cpt1, Acadl, Srebp1c (Srebf1), Fasn, Saa1, Crp, Il1α (Il1a), Ifnγ (Ifng), Il6, Tnf, Mcp-1 (Ccl2), αSma (Acta2) and Tgfβ (Tgfb1) were assessed by quantitative real-time PCR (ΔΔCt) performed using the TaqMan Universal PCR Master Mix and TaqMan Gene Expression Assays (ThermoFisher Scientific) as described previously [13].
Statistical analyses
Analyses were performed using the two-tailed Student’s t test or ANOVA. A p value of < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001).
Results
Hepatocyte-specific GPX1-deficient mice
We generated a floxed allele of Gpx1 (Fig. 1a–c) by gene targeting in Bruce 4 ES cells. ‘Floxing’ the Gpx1 allele (Gpx1 lox/lox; C57BL/6) alone caused a reduction in GPX1 protein in epididymal fat and gastrocnemius muscle (Fig. 1d,e), which express low amounts of GPX1, but not in liver (Fig. 1f), where GPX1 expression is abundant. The reduction in GPX1 in fat or muscle did not result in systemic oxidative stress when GPX1-deficient mice were compared with C57BL/6 wild-type mice, as assessed by measuring reduced (GSH) and oxidised (GSSG) glutathione levels in blood and reflected by an unaltered GSH:GSSG ratio (a reduced ratio being indicative of oxidative stress; Fig. 2a). Moreover, the reduction in GPX1 in muscle of Gpx1 lox/lox (lox/lox) mice did not result in increased H2O2 when compared with C57BL/6 wild-type mice, as assessed using the H2O2 selective probe Amplex Red (Fig. 2e). Similarly, the reduction in GPX1 in fat or muscle did not result in oxidative stress as assessed by measuring GSH and GSSG levels (Fig. 2c,d). Therefore the remaining GPX1 protein in lox/lox mouse tissues such as adipose tissue or muscle is sufficient to prevent any increase in H2O2 and the development of oxidative stress. To conditionally delete Gpx1 in hepatocytes, we bred lox/lox mice with those expressing the Alb-Cre transgene, which recombines floxed alleles in hepatocytes [29]. GPX1 protein expression was specifically ablated in livers from 10-week-old Alb-Cre;Gpx1 lox/lox hepatocyte-specific Gpx1-knockout (HGKO) mice; this was not the case in other tissues including muscle and epididymal fat (Fig. 1d–f). Any remaining GPX1 in HGKO livers reflected that present in non-parenchymal cells, as no GPX1 protein was detected in hepatocytes from HGKO mice (Fig. 1f). Hepatic GPX1 deficiency did not result in compensatory increases in Gpx2, Gpx3 or Gpx4, or alterations in Txn1 (encoding thioredoxin), Trxrd1 (encoding thioredoxin reductase) or Ho1 (encoding haem oxygenase-1) (Fig. 1g). Moreover hepatic GPX1 deficiency did not result in compensatory increases in catalase or PRDX1 in liver and fat, or altered PRDX1 expression in muscle (Fig. 1d–f). However, hepatic GPX1 deficiency was accompanied by decreased catalase in gastrocnemius muscle (Fig. 1e), pointing towards crosstalk between hepatocyte and muscle antioxidant capacity.

Hepatocyte-specific Gpx1-deficient mice. (a) Gpx1 locus and targeting. (b) Southern blot analysis of C57BL/6 J wild-type (+/+) and Gpx1-targeted ES cells (figure provided by Monash Gene Targeting Facility, Monash University, Australia). (c) PCR analysis of +/+, Gpx1 lox/+ (lox/+) and Gpx1 lox/lox (lox/lox) C57BL/6 J mice. (d–f) Epididymal fat, gastrocnemius muscle, liver and isolated hepatocytes from +/+ (grey bars), lox/lox (black bars) and Alb-Cre;Gpx1 lox/lox (white bars) HGKO mice processed for immunoblotting. (g) Livers from lox/lox (black bars) and HGKO (white bars) processed for quantitative real-time PCR (n = 5 per genotype). Quantified results are means ± SE for the indicated number of mice

Liver GSH/GSSG and hepatocyte H2O2 levels. Ten-week-old male +/+ (grey bars, n = 4–7), lox/lox (black bars, n = 5–10) and HGKO (white bars, n = 5–10) mice were fed a chow diet. (a–d) Blood, liver, epididymal fat or gastrocnemius muscle total GSH (tGSH) and GSSG levels and normalised GSH/GSSG ratios. (e) H2O2 in muscle. Results are means ± SE. (f) Hepatocyte H2O2 production from lox/lox (black squares) and HGKO (white circles) mice. Representative results from three experiments are shown
H2O2 generation in HGKO mice
At 4–10 weeks of age no differences were evident in body weight or body/tissue composition between lox/lox and HGKO mice (ESM Fig. 1a–c). Food intake, oxygen consumption, ambulatory activity and energy expenditure were unaltered, as were respiratory exchange ratios, consistent with unaltered energy homeostasis and fuel utilisation (ESM Fig. 1d). To determine whether hepatocyte GPX1 deficiency results in oxidative stress we monitored alterations in GSH and GSSG in blood or liver homogenates from chow-fed lox/lox and HGKO mice. No alterations were evident in blood or liver as a consequence of GPX1 deficiency (Fig. 2a,b). Similarly, no changes in GSH or GSSG were evident in epididymal fat (Fig. 2c). Interestingly total GSH and GSSG were elevated in muscle of HGKO mice, but the GSH:GSSG ratio was unaltered (Fig. 2d), consistent with the increase in GSH being compensatory to counter any muscle oxidative stress that may otherwise occur as a result of decreased muscle catalase expression (Fig. 1e). In keeping with this, muscle H2O2 levels were not altered (Fig. 2e). To directly assess whether hepatic GPX1 deficiency results in increased H2O2, we isolated hepatocytes from chow-fed lox/lox and HGKO mice; we found that GPX1 deficiency resulted in increased hepatocyte H2O2 production (Fig. 2f). Therefore, GPX1 deficiency increases H2O2 in hepatocytes but the levels do not exceed the antioxidant capacity of the liver and do not promote overt hepatic or systemic oxidative stress in chow-fed mice.
Glucose homeostasis in HGKO mice
In the postprandial state insulin acts in the liver via the PI3K/Akt2 pathway with the following effects: (1) repressed transcription of gluconeogenic genes, including Pck1 (encoding phosphoenolpyruvate carboxykinase [PEPCK]) and G6pc (encoding glucose 6-phosphatase [G6P]) and (2) increased transcription of Gck (encoding glucokinase [GCK]) and other genes that promote glycogen storage or glycolysis, to coordinately repress hepatic glucose production (HGP) and prevent postprandial hyperglycaemia [1–3]. We found that the expression of the gluconeogenic genes G6pc and Pck1 was reduced in the livers of fasted mice (Fig. 3a), accompanied by a 7.2-fold increase in Gck expression (Fig. 3a); GCK catalyses the conversion of glucose to G6P and serves as the first step in glycolysis or glycogen synthesis. In keeping with this, glycogen levels were increased in the livers of HGKO mice (Fig. 3b). In addition, the expression of Pdk4 (encoding pyruvate dehydrogenase kinase-4, which is repressed by insulin and inhibits glucose oxidation) was decreased by 50% (Fig. 3a). No differences were evident in Srebp1c and Fasn lipogenic gene expression (Fig. 3c) or in hepatic steatosis as assessed by histology (Fig. 3d). No changes were evident for Lpk (encoding pyruvate kinase, which catalyses the conversion of phosphoenolpyruvate to pyruvate) or for Ppargc1a (encoding proxisome proliferator-activated receptor γ-1α) and other mitochondrial genes involved in fatty acid oxidation (Fig. 3a). These results are consistent with GPX1 deficiency enhancing the repression of HGP and promoting glucose storage and utilisation without effects on lipogenesis.

Glucose homeostasis in chow-fed mice. (a–d) Livers from lox/lox (n = 5–9) and HGKO (n = 6–8) mice processed for quantitative PCR (a, c), glycogen measurement (b) or histology (d) (scale bar, 50 μm). Black bars, lox/lox mice; white bars, HGKO mice. (e) Blood glucose and plasma insulin levels from +/+ (grey bars, n = 10), lox/lox (black bars, n = 12) and HGKO (white bars, n = 11) mice. (f) Pyruvate tolerance test (2 mg/g) and insulin tolerance test (0.5 mU/g) from lox/lox (black squares; n = 9 and 12, respectively) and HGKO (white circles; n = 9 and 13, respectively) mice. AUCs were determined and arbitrary units (AU) are shown. (g) Hyperinsulinaemic–euglycaemic clamps in 10-week-old male fasted (4 h) lox/lox (black bars, n = 6) and HGKO (white bars, n = 5) mice. Glucose infusion (GIR) and disappearance (RD) rates and basal and clamped endogenous glucose production (EGP) were determined. Results are means ± SE. † p = 0.06
We next examined glucose homeostasis in chow-fed C57BL/6 (+/+), lox/lox and HGKO mice. Although blood glucose levels of fed mice and mice fasted for 6 h remained the same, 12 h fasted blood glucose levels were significantly reduced in HGKO mice compared with +/+ or lox/lox mice (Fig. 3e). The reduced blood glucose levels in mice fasted for 12 h is consistent with HGP being reduced in HGKO mice. No differences were observed in plasma insulin levels of fed mice (Fig. 3e), consistent with unaltered pancreatic insulin secretion, but 12 h fasted plasma insulin levels were reduced, consistent with improved insulin sensitivity; no differences were observed between +/+ and lox/lox mice. To assess the effect on HGP we performed pyruvate tolerance tests; pyruvate increases blood glucose by promoting gluconeogenesis. Administration of pyruvate increased blood glucose in +/+ and lox/lox mice, but the increase was attenuated in HGKO mice (Fig. 3f; ESM Fig. 2a). Next we assessed insulin sensitivity in insulin tolerance tests. We found that insulin responses were moderately improved in HGKO mice (Fig. 3f); no differences were observed between +/+ and lox/lox mice (ESM Fig. 2b). These results point towards hepatocyte-specific GPX1 deficiency repressing HGP and enhancing insulin sensitivity in chow-fed mice.
To further characterise glucose homeostasis we subjected chow-fed lox/lox and HGKO mice to hyperinuslinaemic–euglycaemic clamps (Fig. 3g). We found that the glucose infusion rate necessary to maintain euglycaemia was significantly increased in HGKO mice (Fig. 3g), consistent with enhanced whole-body insulin sensitivity. Moreover glucose disappearance, which reflects hepatic and peripheral glucose metabolism, tended to be higher in HGKO mice than in lox/lox mice (p = 0.06) (Fig. 3g). Finally, the extent to which endogenous glucose production was suppressed by insulin tended to be higher in the HGKO mice (Fig. 3g). These results indicate that hepatic GPX1 deficiency enhances insulin sensitivity by potentiating the suppression of gluconeogenic enzymes and by promoting hepatic glucose storage.
Insulin signalling in HGKO mice
In the liver insulin promotes glycogen synthesis by activating Akt, which phosphorylates and inhibits glycogen synthase kinase-3 that phosphorylates and inhibits glycogen synthase (GS). Akt also phosphorylates and inhibits forkhead box protein O1 (FOXO1), which promotes PEPCK and G6P and inhibits GCK expression [30]. We monitored for IR activation and downstream PI3K/Akt signalling in liver, epididymal fat and muscle in mice that had been fasted (4 h) and administered a bolus of insulin. Hepatic GPX1 deficiency enhanced basal IR-Y1162/Y1163 phosphorylation in fasted mice, but not insulin-induced IR phosphorylation (Fig. 4a). Similarly GPX1 deficiency enhanced basal PI3K/Akt signalling, as assessed by Akt-S473 phosphorylation, but had no effect on insulin-induced Akt phosphorylation (Fig. 4a). The activation status of other signalling pathways in fasted mice, including those mediated by STAT-3 (signal transducer and activator of transcription-3), ERK1/2 (extracellular signal-regulated kinases 1/2) and c-Jun N-terminal kinases (JNK) were not altered (ESM Fig. 3a) and there were no overt changes in general tyrosine phosphorylation (ESM Fig. 3b). In keeping with the increased basal PI3K/Akt signalling, FOXO1 Thr-24 phosphorylation was higher and GS Ser-640/Ser-641 phosphorylation lower in fasted HGKO mice (Fig. 4b). The increased IR phosphorylation and PI3K/Akt signalling in 4 h fasted HGKO mice suggests that insulin signalling may be prolonged by GPX1 deficiency. In keeping with this, 12 h fasting IR and Akt phosphorylation were not altered (ESM Fig. 3c). No differences were evident in basal or insulin-induced IR or Akt phosphorylation in fat or muscle (ESM Fig. 3d). To reaffirm the enhanced insulin signalling we compared responses in hepatocytes from lox/lox and HGKO mice. Hepatocyte basal and insulin-induced H2O2 generation was exacerbated by GPX1 deficiency (Fig. 4c). Moreover, insulin-induced IR-Y1162/Y1163 and Akt-S473 phosphorylation was enhanced (Fig. 4d). These results indicate that GPX1 deficiency enhances hepatic insulin-induced H2O2 and insulin-induced IR activation and PI3K/Akt signalling to repress HGP and promote liver glucose storage or utilisation.

Insulin signalling. (a, b) Ten-week-old chow-fed lox/lox (black bars) and HGKO (white bars) mice were fasted (4 h) and injected (intraperitoneally) with PBS or insulin and liver extracts were immunoblotted for Y1162/Y1163-phosphorylated IR (p-IR) and S473-phosphorylated Akt (p-Akt) (a) or T24-phosphorylated FOXO1 (p-FOXO1) and S640/S641-phosphorylated GS (p-GS) (b). p-IR, p-Akt, p-FOXO1 and p-GS were quantified by densitometry. (c, d) Serum-starved hepatocytes were stimulated with PBS or insulin (100 nmol/l); H2O2 was measured in live cells (n = 5) (c) or cells were processed for immunoblotting (d). Representative and quantified (n = 3) results are shown. Quantified results are means ± SE. AU, arbitrary units
PTP oxidation in HGKO mice
One mechanism by which the increased H2O2 levels associated with GPX1 deficiency might enhance IR signalling and thereby improve hepatic glucose metabolism is through the oxidation and inactivation of PTPs involved in IR dephosphorylation. PTP oxidation was examined in livers from lox/lox and HGKO mice by immunoblot analysis (Fig. 5a,b) and immunoblots were reprobed with antibodies to PTPs with corresponding molecular masses (Fig. 5c). The oxidation status of PTPs (Fig. 5a,b), including those co-migrating with tyrosine-protein phosphatase non-receptor type 2 (TCPTP), PTP1B and small heterodimer partner (SHP)-1, implicated previously in IR desphosphorylation [31, 32], was enhanced by GPX1 deficiency (Fig. 5b,c). These results are consistent with the elevated H2O2 in GPX1-deficient hepatocytes promoting the oxidation and inactivation of PTPs and hence insulin signalling and glucose homeostasis.

Hepatic PTP oxidation. Livers from lox/lox and HGKO mice were processed for assessment of total PTP oxidation by immunoblotting with PTPox antibody to detect oxidised PTPs (a, b) and reprobed as indicated (c). PTPox species increased in HGKO livers are marked by arrows; those co-migrating with PTP1B, SHP-1 and TCPTP are marked by coloured arrows. PTPox species co-migrating with PTP1B, SHP-1 and TCPTP were quantified. Black bars, lox/lox mice; white bars, HGKO mice. Representative and quantified (means ± SE) results are shown
Oxidative stress in obese HGKO mice
Obesity is characterised by oxidative stress and insulin resistance [5–7]. Accordingly, we determined whether hepatocyte GPX1 deficiency might exacerbate oxidative stress and insulin resistance in mice fed an obesogenic diet. Hepatic GPX1 deficiency did not alter body weight, adiposity (ESM Fig. 4a–c), food intake, energy expenditure or ambulatory activity (ESM Fig. 4d) after 12 weeks of high-fat feeding. Liver-specific GPX1-deficient mice exhibited systemic oxidative stress as indicated by increased blood GSSG levels and decreased GSH:GSSG ratios (Fig. 6a). When compared with lox/lox mice, no differences were evident in hepatic H2O2 detoxification and redox gene or protein levels (ESM Fig. 5a,b) and there were no differences in GSH or GSSG (Fig. 6b), GPX1, catalase or PRDX1 in muscle (ESM Fig. 5c). Interestingly, GSSG levels in the liver were decreased so that the GSH:GSSG ratio was increased (Fig. 6c). Peroxide detoxification by GPX1 is reliant on GSH and results in the generation of GSSG. Hence the decrease in GSSG is in keeping with GPX1 being one of the major enzymes that uses GSH as an electron donor to generate GSSG. Despite the decreased liver GSSG, H2O2 was markedly increased in hepatocytes isolated from high-fat-fed (HFF) HGKO vs lox/lox mice; this increase far exceeded that normally seen in response to insulin (Fig. 6d). These results are consistent with hepatocyte GPX1 deficiency exacerbating hepatocyte-derived H2O2 and promoting systemic oxidative stress in HFF mice.

Oxidative stress in HFF mice. Ten-week-old male lox/lox (black bars, n = 9) and HGKO (white bars, n = 10) mice were fed with a high-fat diet for 12 weeks. (a–c) total GSH (tGSH), GSSG and normalised GSH:GSSG ratios in blood, liver and muscle. (d) Hepatocytes were stimulated with PBS or insulin (100 nmol/l) and H2O2 production measured in live cells (n = 3). Quantified results are means ± SE
Glucose homeostasis in obese HGKO mice
We determined whether the systemic oxidative stress evident in HFF HGKO mice might exacerbate the development of insulin resistance and glucose intolerance. When comparing HGKO mice with lox/lox mice, although no differences were evident in blood glucose levels, fasted insulin levels were reduced (Fig. 7a), consistent with improved insulin sensitivity. Moreover, although no differences were evident in insulin tolerance tests (Fig. 7b), pyruvate responses were attenuated (Fig. 7c) and the expression of G6pc and Pck1 in the livers of fasted mice was reduced (Fig. 7d), consistent with decreased gluconeogenesis. In keeping with this, GPX1 deficiency was accompanied by significantly increased insulin-induced phosphorylation of IR and Akt Ser-473 in the liver (Fig. 7e). These results point towards hepatic GPX1 deficiency improving liver glucose homeostasis even in the context of obesity and systemic oxidative stress.

Glucose homeostasis and insulin signalling in HFF mice. Ten-week-old male lox/lox (black bars or squares, n = 8–11) and HGKO (white bars or circles, n = 8–12) mice were fed a high-fat diet for 12 weeks. (a) Fed and fasted (6 h) blood glucose and plasma insulin levels. (b) Insulin (0.65 mU/g) and (c) pyruvate (2 mg/g) tolerance tests. (d) Livers were processed for quantitative PCR. (e) Fasted mice (4 h) were administered PBS or insulin and livers were extracted for immunoblotting. Quantified results are means ± SE. AU, arbitrary units
Reduced inflammation and NASH in HGKO mice
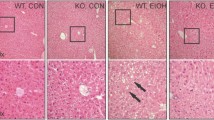
Hepatic oxidative stress has been linked with the development of NAFLD and NASH, the latter characterised by lymphocyte infiltrates, inflammation and liver damage [4, 8–11]. We found that hepatocyte-specific GPX1-defiency in HFF mice was not associated with overt differences in steatohepatitis, as assessed by histology (Fig. 8a) or the expression of lipogenic genes (Fig. 8b). Surprisingly, the enhanced hepatocyte-derived H2O2 in HFF HGKO mice resulted in the decreased expression of genes associated with inflammation (Fig. 8c). Since HFF C57BL/6 mice develop fatty liver, but not NASH, we sought to further explore the impact of GPX1 deficiency on the development of NASH and the associated inflammation and fibrosis in mice fed a CDAA diet for 12 weeks. The CDAA diet promotes weight gain, insulin resistance and key features of NASH, including steatosis, lymphocytic infiltrates and fibrosis [8]. Hepatic GPX1 deficiency did not alter body weight in CDAA diet-fed mice (ESM Fig. 6a) and had no obvious effect on hepatic steatosis as assessed by histology (Fig. 8a), or the expression of lipogenic genes (Fig. 8b). Strikingly, GPX1 deficiency resulted in reduced lymphocytic infiltration in the liver and repressed hepatic inflammation, as assessed by histology (Fig. 8d; ESM Fig. 6b) and expression of acute-phase and pro-inflammatory genes (Fig. 8c). Moreover, GPX1 deficiency markedly reduced fibrosis, as assessed by histology (PicroSirius Red collagen staining; Fig. 8e and ESM Fig. 6b) and expression of fibrogenic genes (Fig. 8f). Finally CDAA-fed HGKO mice exhibited a significant reduction in systemic inflammation, as assessed by reduced circulating levels of IFNγ, IL-6 and TNF (Fig. 8g). Therefore, GPX1 deficiency represses lymphocytic infiltration, inflammation and fibrosis in the liver, key diagnostic and pathological features of NASH.

Inflammation and NASH. Ten-week-old male lox/lox (black bars or circles, n = 5–8) and HGKO (white bars or circles, n = 5–9) mice were fed a high-fat diet or a CDDA diet for 12 weeks. (a–f) Livers were processed for histology (a, d, e) (scale bar, 50 μm in a; 100 μm in d and e) or quantitative PCR (b, c, f). (g) Plasma IFNγ, IL-6 and TNF levels were determined using a BD Cytometric Bead Array; for TNF, p = 0.06 for lox/lox vs HGKO mice fed a CDAA diet. Representative and quantified (means ± SE) results are shown
Discussion
Global GPX1-deficient mice fed an obesogenic diet are protected from the development of steatohepatitis and ensuing liver damage [25]. Although this was previously attributed to decreased pancreatic insulin secretion and attenuated insulin-mediated hepatic lipogenesis [25], a hepatocyte-intrinsic contribution could not be excluded. Here, we confirm that hepatocyte GPX1 deficiency does not alter the development of steatohepatitis in mice fed either an obesogenic diet that promotes NAFLD or a CDAA diet that promotes NASH. Strikingly, despite the unaltered steatosis, hepatic GPX1 deficiency was associated with decreased hepatic and systemic inflammation and reduced liver lymphocytic infiltration and fibrosis in mice fed a CDAA diet. Although the molecular mechanism by which hepatocyte GPX1 deficiency may decrease inflammation and ensuing NASH remains to be resolved, one possibility is that this may be linked to increased PTP1B oxidation, since hepatic PTP1B deficiency represses hepatocyte endoplasmic reticulum stress and inflammation [33, 34].
Altered oxidative phosphorylation and increased ROS levels are reported in patients with NASH [4, 11] and there is compelling evidence from rodent models linking oxidative stress with NAFLD or NASH [10, 35, 36]. Hepatocyte-specific SOD1-deficiency or combined SOD1/2 deficiencies increase steatohepatitis in HFF mice [37, 38]. This is consistent with superoxide being important in the promotion of steatohepatitis and liver disease. In keeping with this, NOXs are elevated in models of fibrogenic disease [39], whereas NOX1/4 inhibition or hepatocyte NOX4 deficiency protect mice against NASH [8, 40]. In contrast, a recent study has shown that the deletion of the haem oxygenase-1 gene in hepatocytes promotes the generation of H2O2 and attenuates high-fat diet-induced insulin resistance and hepatic inflammation and damage [41]. Our findings also challenge the concept that hepatocyte-derived H2O2 per se is a driver of liver disease.
We previously reported that global GPX1 deficiency promotes hepatic insulin signalling and repression of HGP in the obese state [25]. However, it was unclear whether these effects were intrinsic to hepatocytes or reflected reduced pancreatic insulin secretion [25], since hyperinsulinaemia can drive insulin resistance [42–44] and reduction in circulating insulin can protect mice from diet-induced obesity, insulin resistance and steatosis [45]. In this study we found that insulin-induced H2O2 generation and insulin signalling were increased in hepatocytes from HGKO mice, accompanied by increased hepatic IR activation and signalling even in the context of obesity. Why does GPX1 deficiency in HFF mice not promote insulin resistance? One argument could be that a sufficient level of H2O2 was not achieved in hepatocytes. However, H2O2 generation by hepatocytes isolated from HFF HGKO mice far exceeded levels normally achieved in response to insulin. We propose that either H2O2 per se is not detrimental or that hepatic oxidative stress and the promotion of insulin resistance may also be reliant on contributions from non-parenchymal cells. In keeping with the latter premise, mice overexpressing PRDX4, a secreted enzyme that scavenges ROS, are protected from steatohepatitis [46].
Reactive and potentially modulatory cysteines exist in many proteins [14, 15]. However, the low thiol pKa of the catalytic cysteine in PTPs renders them highly susceptible to oxidation and inactivation [18]. In this study we report that the oxidation of select hepatic PTPs, including PTP1B, SHP-1 and TCPTP, is increased in HGKO mice. Previous studies have established that PTP1B and SHP-1 can dephosphorylate the IR to regulate hepatic insulin sensitivity [31, 33, 47]. Similarly, heterozygous TCPTP deficiency enhances hepatocyte IR phosphorylation and represses HGP [28, 48]. We speculate that the oxidation of such PTPs would promote insulin signalling to regulate hepatic glucose metabolism, although we cannot exclude the contribution of other pathways given the growing number of metabolic proteins that can be oxidised, including pyruvate kinase M2 [49] and pyruvate kinase 2 [50].
Our findings warrant a redress of the contributions of ROS such as H2O2 to hepatic pathophysiology in obesity and suggest that increases in hepatocyte H2O2 may in fact represent a compensatory and beneficial response to attenuate disease progression.
Abbreviations
- CDAA:
-
Choline-deficient amino-acid-defined
- FOXO1:
-
Forkhead box protein O1
- GCK:
-
Glucokinase
- GPX:
-
Glutathione peroxidase
- GS:
-
Glycogen synthase
- GSSG:
-
Oxidised glutathione
- GSH:
-
Reduced glutathione
- G6P:
-
Glucose 6-phosphatase
- HFF:
-
High-fat-fed
- HGKO:
-
Hepatocyte-specific Gpx1-knockout
- HGP:
-
Hepatic glucose production
- IR:
-
Insulin receptor
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- NOX:
-
NADPH oxidase
- PEPCK:
-
Phosphoenolpyruvate carboxykinase
- PI3K:
-
Phosphatidylinositol 3-kinase
- PRDX:
-
Peroxiredoxin
- PTP:
-
Protein tyrosine phosphatase
- ROS:
-
Reactive oxygen species
- SHP:
-
Small heterodimer partner
- SOD:
-
Superoxide dismutase
- TCPTP:
-
Tyrosine-protein phosphatase non-receptor type 2
References
Johnson AM, Olefsky JM (2013) The origins and drivers of insulin resistance. Cell 152:673–684
Samuel VT, Shulman GI (2012) Mechanisms for insulin resistance: common threads and missing links. Cell 148:852–871
Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414:799–806
Rolo AP, Teodoro JS, Palmeira CM (2012) Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic Biol Med 52:59–69
Furukawa S, Fujita T, Shimabukuro M et al (2004) Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 114:1752–1761
Houstis N, Rosen ED, Lander ES (2006) Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440:944–948
Anderson EJ, Lustig ME, Boyle KE et al (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119:573–581
Bettaieb A, Jiang JX, Sasaki Y et al (2015) Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology 149:468–480
Satapati S, Kucejova B, Duarte JA et al (2015) Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest 125:4447–4462
Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI (2015) Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347:1253–1256
Seki S, Kitada T, Yamada T, Sakaguchi H, Nakatani K, Wakasa K (2002) In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J Hepatol 37:56–62
Park EJ, Lee JH, Yu GY et al (2010) Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 140:197–208
Gurzov EN, Tran M, Fernandez-Rojo MA et al (2014) Hepatic oxidative stress promotes insulin-STAT-5 signaling and obesity by inactivating protein tyrosine phosphatase N2. Cell Metab 20:85–102
Finkel T (2011) Signal transduction by reactive oxygen species. J Cell Biol 194:7–15
Fisher-Wellman KH, Neufer PD (2011) Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol Metab 23:142–153
Mahadev K, Motoshima H, Wu X et al (2004) The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol Cell Biol 24:1844–1854
Frijhoff J, Dagnell M, Augsten M, Beltrami E, Giorgio M, Ostman A (2014) The mitochondrial reactive oxygen species regulator p66Shc controls PDGF-induced signaling and migration through protein tyrosine phosphatase oxidation. Free Radic Biol Med 68:268–277
Tiganis T (2011) Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends Pharmacol Sci 32:82–89
Seo JH, Ahn Y, Lee SR, Yeol Yeo C, Chung Hur K (2005) The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol Biol Cell 16:348–357
Meng TC, Buckley DA, Galic S, Tiganis T, Tonks NK (2004) Regulation of insulin signaling through reversible oxidation of the protein-tyrosine Phosphatases TC45 and PTP1B. J Biol Chem 279:37716–37725
Mahadev K, Zilbering A, Zhu L, Goldstein BJ (2001) Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem 276:21938–21942
Loh K, Deng H, Fukushima A et al (2009) Reactive oxygen species enhance insulin sensitivity. Cell Metab 10:260–272
Ho YS, Magnenat JL, Bronson RT et al (1997) Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem 272:16644–16651
de Haan JB, Bladier C, Griffiths P et al (1998) Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J Biol Chem 273:22528–22536
Merry TL, Tran M, Stathopoulos M et al (2014) High-fat-fed obese glutathione peroxidase 1-deficient mice exhibit defective insulin secretion but protection from hepatic steatosis and liver damage. Antioxid Redox Signal 20:2114–2129
Passonneau JV, Lauderdale VR (1974) A comparison of three methods of glycogen measurement in tissues. Anal Biochem 60:405–412
Karisch R, Fernandez M, Taylor P et al (2011) Global proteomic assessment of the classical protein-tyrosine phosphatome and redoxome. Cell 146:826–840
Fukushima A, Loh K, Galic S et al (2010) T cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes 59:1906–1914
Postic C, Magnuson MA (2000) DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26:149–150
Zhang W, Patil S, Chauhan B et al (2006) FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem 281:10105–10117
Dubois MJ, Bergeron S, Kim HJ et al (2006) The SHP-1 protein tyrosine phosphatase negatively modulates glucose homeostasis. Nat Med 12:549–556
Tiganis T (2013) PTP1B and TCPTP – nonredundant phosphatases in insulin signaling and glucose homeostasis. FEBS J 280:445–458
Delibegovic M, Zimmer D, Kauffman C et al (2009) Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced ER stress. Diabetes 58:590–599
Miraldi ER, Sharfi H, Friedline RH et al (2013) Molecular network analysis of phosphotyrosine and lipid metabolism in hepatic PTP1b deletion mice. Integr Biol (Camb) 5:940–963
Tomita K, Teratani T, Suzuki T et al (2012) p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J Hepatol 57:837–843
Xu Z, Chen L, Leung L, Yen TS, Lee C, Chan JY (2005) Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc Natl Acad Sci U S A 102:4120–4125
Wang L, Jiang Z, Lei XG (2012) Knockout of SOD1 alters murine hepatic glycolysis, gluconeogenesis, and lipogenesis. Free Radic Biol Med 53:1689–1696
Uchiyama S, Shimizu T, Shirasawa T (2006) CuZn-SOD deficiency causes ApoB degradation and induces hepatic lipid accumulation by impaired lipoprotein secretion in mice. J Biol Chem 281:31713–31719
Aoyama T, Paik YH, Watanabe S et al (2012) Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology 56:2316–2327
Jiang JX, Chen X, Serizawa N et al (2012) Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic Biol Med 53:289–296
Jais A, Einwallner E, Sharif O et al (2014) Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell 158:25–40
Cusin I, Terrettaz J, Rohner-Jeanrenaud F, Jeanrenaud B (1990) Metabolic consequences of hyperinsulinaemia imposed on normal rats on glucose handling by white adipose tissue, muscles and liver. Biochem J 267:99–103
Koopmans SJ, Ohman L, Haywood JR, Mandarino LJ, DeFronzo RA (1997) Seven days of euglycemic hyperinsulinemia induces insulin resistance for glucose metabolism but not hypertension, elevated catecholamine levels, or increased sodium retention in conscious normal rats. Diabetes 46:1572–1578
Gray SL, Donald C, Jetha A, Covey SD, Kieffer TJ (2010) Hyperinsulinemia precedes insulin resistance in mice lacking pancreatic β-cell leptin signaling. Endocrinology 151:4178–4186
Mehran AE, Templeman NM, Brigidi GS et al (2012) Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab 16:723–737
Nabeshima A, Yamada S, Guo X et al (2013) Peroxiredoxin 4 protects against nonalcoholic steatohepatitis and type 2 diabetes in a nongenetic mouse model. Antioxid Redox Signal 19:1983–1998
Xu E, Charbonneau A, Rolland Y et al (2012) Hepatocyte-specific Ptpn6 deletion protects from obesity-linked hepatic insulin resistance. Diabetes 61:1949–1958
Galic S, Klingler-Hoffmann M, Fodero-Tavoletti MT et al (2003) Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol Cell Biol 23:2096–2108
Anastasiou D, Poulogiannis G, Asara JM et al (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334:1278–1283
Hurd TR, Collins Y, Abakumova I et al (2012) Inactivation of pyruvate dehydrogenase kinase 2 by mitochondrial reactive oxygen species. J Biol Chem 287:35153–35160
Acknowledgements
We thank E. N. Gurzov (St Vincent’s Institute, Australia) for reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the National Health and Medical Research Council of Australia (TT and SA) and the Diabetes Australia Research Trust (TT).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript
Contribution statement
All authors contributed to the study conception and design, acquisition of data or analysis and interpretation of data. TT supervised all aspects of the study and drafted the manuscript. All authors participated in the critical revision of the manuscript and approved the final version. TT is the guarantor of this work.
Additional information
Troy L. Merry and Melanie Tran contributed equally to this paper.
An erratum to this article is available at http://dx.doi.org/10.1007/s00125-016-4124-z.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM
(PDF 848 kb)
Rights and permissions
About this article

Cite this article
Merry, T.L., Tran, M., Dodd, G.T. et al. Hepatocyte glutathione peroxidase-1 deficiency improves hepatic glucose metabolism and decreases steatohepatitis in mice. Diabetologia 59, 2632–2644 (2016). https://doi.org/10.1007/s00125-016-4084-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4084-3