
Abstract
Aims/hypothesis
In type 1 diabetes, abnormalities of both glucose and lipoprotein metabolism are seen. The relationship between these factors is not understood, but studies indicate that hyperglycaemia may increase hepatic VLDL-triacylglycerol (VLDL-TG) secretion and reduce VLDL-TG fatty acid oxidation, which could lead to the development of dyslipidaemia. The aim of this study was to determine the isolated effect of hyperglycaemia on VLDL-TG and NEFA kinetics in men with type 1 diabetes.
Methods
VLDL-TG and palmitate kinetics were measured in eight men with type 1 diabetes using ex vivo labelled VLDL-TG and palmitate tracers. A 2.5 h basal period (plasma glucose 5 mmol/l) was followed by a 4 h hyperglycaemic period (plasma glucose 16 mmol/l). Steady-state VLDL-TG kinetics (VLDL-TG secretion, clearance and oxidation rates) were assessed by an isotope dilution technique using an intravenous primed-constant infusion of ex vivo labelled [1-14C]VLDL-TG in combination with sampling of blood and expired air. Palmitate turnover was measured using [9,10-3H]palmitate.
Results
The VLDL-TG secretion rate (36.0 ± 9.6 vs 30.8 ± 6.1 μmol/min, NS) and clearance rate (209 ± 30.4 vs 197 ± 41.7 ml/min, NS) were unchanged during the basal and hyperglycaemic periods, resulting in unchanged VLDL-TG concentrations (0.25 ± 0.11 μmol/l vs 0.28 ± 0.10 μmol/l, NS). In addition, VLDL-TG fatty acid oxidation and palmitate flux were not changed during hyperglycaemia.
Conclusions/interpretation
Four hours of acute hyperglycaemia (16 mmol/l) without a concomitant increase in insulin does not alter VLDL-TG and NEFA kinetics in men with type 1 diabetes.
Clinical trial registry number: NCT01178957
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 1 diabetes is associated with abnormalities of glucose and lipid metabolism as well as an increased risk of cardiovascular disease [1]. A positive association between a higher than optimum blood glucose concentration and risk of CHD has been established in both healthy individuals and patients with diabetes [2, 3], and the intensive treatment of type 1 diabetes has been shown to decrease the risk of CHD [4]. In addition, diabetic dyslipidaemia, a cardinal risk factor for CHD in type 2 diabetes [5], is also present in type 1 diabetes [6]. This consists of increased plasma triacylglycerols, an increased number of small dense LDL particles and low HDL-cholesterol concentrations. Here, a hepatic overproduction of VLDL particles is thought to play a key role in generating the other lipid alterations [7]. Dyslipidaemia in type 1 diabetes can be partly corrected by improved insulin treatment and blood glucose control [8, 9].
A possible link between hyperglycaemia and dyslipidaemia could be a stimulatory effect of hyperglycaemia per se on the hepatic production and secretion of VLDL-tyiacylglycerol (VLDL-TG). Previous studies have shown that hyperglycaemia may have the ability to increase VLDL-TG synthesis by: (1) increasing hepatic glucose oxidation at the expense of fatty acids (FAs), resulting in a diversion of FAs towards triacylglycerol synthesis [10, 11]; (2) the direct stimulation of transcription factors involved in VLDL-TG production [12–15]; and (3) the stimulation of apolipoprotein B-100 (ApoB-100) production [16].
The ability to assess the effect of isolated acute hyperglycaemia on VLDL-TG and NEFA kinetics is, however, impeded by the simultaneous increase in endogenous insulin secretion, which independently suppresses both VLDL-TG secretion and lipolysis [10, 17–19] and has even been found to lower VLDL-TG secretion in healthy lean individuals [19, 20]. To our knowledge, no studies have attempted to determine the acute effect of overt hyperglycaemia on VLDL-TG kinetics either in type 1 diabetes or in experimental settings that effectively circumvent the concomitant increase in insulin in healthy humans. Thus, somatostatin administration was insufficient to prevent postprandial insulin secretion in a study of VLDL-TG secretion following a high-carbohydrate diet [18]. With respect to lipolysis, we found only two studies that had been performed in patients with type 1 diabetics, in which moderate hyperglycaemia (11–12 mmol/l) did not alter lipolysis [21, 22]. We found no studies in humans examining the effect of more pronounced hyperglycaemia (>15 mmol/l) on VLDL kinetics or lipolysis.
The present study was performed to assess the effect of acute overt hyperglycaemia (16 mmol/l) on VLDL-TG and palmitate kinetics in men with type 1 diabetes. We chose to study men with type 1 diabetes as a patient model in order to evaluate the isolated effects of hyperglycaemia on hepatic VLDL-TG secretion and oxidation without the potential impact of concomitant perturbations in endogenous insulin secretion. The patients were studied during the infusion of a replacement dose of insulin to mimic fasting insulinaemia in combination with ex vivo labelled VLDL-TG and palmitate tracers. We hypothesised that acute short-term hyperglycaemia, in the absence of hyperinsulinaemia, would increase VLDL-TG secretion and reduce VLDL-TG clearance, but have no impact on lipolysis. As a secondary objective, we hypothesised that hyperglycaemia would decrease the oxidation of VLDL-TG-associated FAs.
Methods
The study was approved by the local Ethics Committee and carried out in accordance with the principles of the Declaration of Helsinki as revised in 2008. Informed consent was obtained from all participants.
Participants
Eight men with type 1 diabetes mellitus (aged 18–50 years) with an HbA1c ≤9% (75 mmol/mol) were recruited from the outpatient clinic. Potentially eligible patients visited the Clinical Research Centre (CRC) after a 10 h overnight fast, where a medical history was taken and a physical examination performed. A normal lipid profile, blood count and chemistry panel were documented for all participants. The C-peptide concentration was below the lower level of normal in all patients. All patients were treated with a combination of long-acting and rapid-acting insulin. In addition, all were non-smokers and did not suffer from other diseases unrelated to diabetes. One patient was taking prophylactic simvastatin and another was receiving enalapril treatment, both of which were paused 2 weeks before the study day. All the patients were recreationally active, but none was elite-trained. The patients were instructed not to perform high-intensity exercise for 3 days prior to the study day.
Protocol
One week before the study day, a 72 ml blood sample was drawn for VLDL-TG ex vivo labelling after an overnight fast. The patients were then interviewed by a dietitian, who estimated their daily caloric intake. A weight-maintaining diet (55% carbohydrate, 15% protein and 30% fat) was provided by the hospital kitchen during the 3 days preceding the study day in order to secure a constant macronutrient intake. Long-acting insulin was stopped 24 h before the study day, and the patients were instructed to take their rapid-acting insulin as usual with the addition of extra doses based on self-measurements of blood glucose five times during the 24 h period prior to the study day: at 07:30, 11:30, 17:30, 22:00 and 03:00 hours. If the blood glucose level exceeded 10 mmol/l or 12 mmol/l, patients were instructed to take an additional 2 or 4 U of rapid-acting insulin, respectively.
Study day
The participants were admitted to the CRC at 22:00 hours on the day before the study for an overnight stay. From this time until the end of the study day, they remained in bed with the exception of voiding, and fasted with the exception of tap water. The following morning, catheters were placed in an antecubital vein for [1-14C]triolein-labelled VLDL, 0.9% saline and 20% glucose infusion, and in a contralateral heated hand vein for sampling of arterialised blood, and basal blood samples were collected.
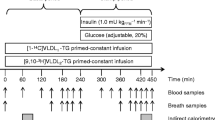
At 07.30 hours, before the start of the study, an insulin (Actrapid; Novo Nordisk, Bagsvaerd, Denmark) infusion of 1.0 mU kg−1 min−1 was administered for 30–85 min in order stabilise the plasma glucose level at approximately 5 mmol/l, after which the infusion rate was reduced to a replacement dose of 0.15 mU kg−1 min−1 for the duration of the study period. Hence, the study consisted of a 2.5 h basal period (0–150 min) followed by a 4 h hyperglycaemic period (150–390 min), during both of which insulin was infused at 0.15 mU kg−1 min−1. At time = 150 min, the plasma glucose level was raised to approximately 16 mmol/l by an intravenous infusion of 20% glucose. The plasma glucose was measured every 10 min and kept stable by adjusting the glucose infusion. A primed-constant infusion of [1-14C]triolein-labelled VLDL-TG (priming with 20% of the dose) was given throughout the study day (Fig. 1). Blood samples for measurements of VLDL-TG concentration and specific activity (sp. act.) were drawn prior to infusion, at time = 105, 120, 135, 150 min, and every half hour thereafter. Breath samples were collected prior to infusion and at time = 60, 120, 135, 150, 180, 240, 300, 330, 360 and 390 min for determination of CO2 production and 14CO2 sp. act. Palmitate turnover was measured using a 1 h infusion of [9,10-3H]palmitate during the last hour of each period. Blood samples for determination of palmitate concentration and sp. act. were drawn prior to infusion and every 10 min during the last half hour of the palmitate infusion. Insulin concentration was measured prior to insulin infusion, at time = 120 and 150 min, and once every hour thereafter. C-peptide level was measured at time = 0 and 390 min. NEFA were measured prior to insulin infusion and at time = 120, 150, 360 and 390 min. Indirect calorimetry was performed for 30 min from time = 30 min and time = 360. At time = 390 min, all the infusions were stopped, the catheters were removed, and the patients were then served lunch and discharged.

Study protocol. The white bars represent periods of insulin, glucose and [1-14C]VLDL infusion, and the patterned bars periods of [9,10-3H]palmitate infusion and indirect calorimetry. The arrows illustrate time points of blood and breath samples
VLDL-TG tracer preparation
VLDL-TG labelling was performed as previously described with minor modifications [23]. In brief, serum from a 72 ml blood sample was mixed with 1.48 MBq [1-14C]triolein (PerkinElmer, Waltham, MA, USA) dissolved in 40 μl of ethanol and sonicated gently at 4°C for 2 h. The labelled serum was then transferred to sterile 5.9 ml polyallomer quick-seal centrifuge tubes (Beckman Coulter, Brea, CA, USA) in 3.0 ml fractions under a saline solution (density = 1.006 g/ml), and centrifuged in a Ti 50.3 rotor (Beckman Coulter, CA, Brea, USA) for 18 h at 115,046 g and 10°C. The supernatant fraction containing the labelled VLDL particles was removed using a sterile Pasteur pipette, filtered and stored at 4°C. Samples were tested for bacterial growth to ensure sterility.
Plasma VLDL-TG concentration and sp. act
VLDL was isolated from approximately 3 ml of plasma by ultracentrifugation as described above. The supernatant fraction containing the VLDL fraction (approximately 1.2 ml) was obtained by tube slicing (Beckman Coulter, Brea, CA, USA) and transferred to a scintillation vial. A small sample (300 μl) was obtained and analysed for TG concentration, after which the quantity (in μmol) of VLDL-TG in the sample and the plasma concentration (in mmol/l) were calculated. The exact volume of the remaining sample was recorded by weighing the scintillation vials. Scintillation fluid (Optiphase HiSafe 2; PerkinElmer, Waltham, MA, USA) was then added, and 14C activity was counted to a <2% counting error. VLDL-TG sp. act. was expressed as disintegrations per minute per μmol.
Breath 14CO2 activity
Breath samples were collected in IRIS breath bags (Wagner Analysen Technik, Bremen, Germany). The exhaled air was passed through a solution containing benzethonium hydroxide (Sigma-Aldrich, St. Louis, MO, USA) with thymolphthalein (Sigma-Aldrich, St. Louis, MO, USA) in a scintillation vial. A colour change occurred when exactly 0.25 mmol CO2 had been trapped. Scintillation fluid was added, and 14C activity was measured by liquid scintillation counting to a <2% counting error.
Palmitate turnover
Systemic palmitate flux was measured using the isotope dilution technique during a 1 h constant infusion of [9,10-3H]palmitate (0.3 μCi/min) (Department of Nuclear Medicine, Aarhus University Hospital, Aarhus, Denmark). The plasma palmitate concentration and sp. act. were measured by HPLC using [2H31]palmitate as the internal standard. Steady-state sp. act. was verified for each individual. Palmitate flux (μmol/min) was calculated as the [9,10-3H]palmitate infusion rate divided by the steady-state palmitate sp. act.
Indirect calorimetry
Resting energy expenditure (REE) and respiratory exchange rates (RQ) were measured by indirect calorimetry (Deltatrac monitor; Datex Instruments, Baldwin Park, CA, USA). The protein oxidation rate was estimated from the urinary urea excretion, and the net lipid and glucose oxidation rates were calculated from indirect calorimetry measurements with correction for protein oxidation [24].
Laboratory procedures
Plasma glucose concentrations were measured immediately using an YSI 2300 STAT Plus glucose analyser (YSI, Yellow Springs, OH, USA). Blood samples were placed on ice and separated as quickly as possible by centrifugation (2,753 g at 4°C for 10 min). Aliquots of plasma were stored at 4°C for isolation of VLDL after completion of the study day. The remaining samples were stored at −80°C for later analysis. TG concentrations were analysed using a COBAS c 111 analyser (Roche, Basel, Switzerland). Serum insulin concentrations were measured with an AutoDELFIA insulin immunoassay kit (PerkinElmer, Waltham, MA, USA), and plasma C-peptide was measured using a C-peptide ELISA kit (ALPCO, Salem, NH, USA). Serum NEFA concentrations were determined by a colorimetric method (Wako Pure Chemical Industries, Osaka, Japan).
Calculations
The steady state for VLDL-TG was effectively reached during the last hour of the basal and the hyperglycaemic periods. The VLDL-TG secretion rate was calculated by dividing the tracer infusion rate by the steady-state sp. act. in each period:
The VLDL-TG clearance rate was calculated by dividing the secretion rate by the average VLDL-TG concentration (CVLDL-TG) in each period:
The fractional oxidation (%) of the infused [1-14C]VLDL-TG was calculated as follows:
where k is the volume of 1 mole of CO2 at 20°C and 1 atm pressure (22.4 l/mol), Ar is the fractional acetate carbon recovery factor for breath CO2, and F is the tracer infusion rate. Sidossis et al have previously estimated Ar to be 0.56 during basal resting conditions [25]. Published data on the increment in 14CO2 recovery during a prolonged infusion of [14C]acetate in healthy normoglycaemic participants were used to visually compare the increase in sp. act. between 4 and 8 h of infusion (approximately 50%) [26].
The total VLDL-TG oxidation rate was calculated as:
Statistics
Data are presented as mean ± SEM or range. Comparisons of changes in the VLDL-TG kinetic variables (basal-to-clamp) were performed using the Student’s t test for paired comparisons or Wilcoxon’s test. A p value below 0.05 was considered significant.
Results
The patients’ characteristics are presented in Table 1. The morning pre-study plasma glucose level averaged 12.4 mmol/l, and the serum insulin was below the limits of detection in all patients. Patients who took additional insulin at 03:00 hours all used insulin aspart (B28Lys,B29Pro human insulin; Novo Rapid, Novo Nordisk, Bagsvaerd, Denmark). The plasma glucose level was stable during the basal period (approximately 5 mmol/l) and plateaued as intended around 16 mmol/l during the hyperglycaemic period (Fig. 2a). As seen in Fig. 2b, the low-dose insulin infusion resulted in stable serum insulin concentrations with no evidence of additional endogenous insulin secretion during hyperglycaemia. This was confirmed by the measurement of C-peptide, which remained unchanged and well below normal levels during the basal and hyperglycaemic periods (70.5 pmol/l ±62.7 vs 80.6 ± 88.3 pmol/l, NS).

Levels of (a) plasma glucose, (b) serum insulin, (c) serum NEFA, (d) plasma TG (black circles) and VLDL-TG (white circles) during the basal and clamp periods. Data are mean ± SEM
NEFA kinetics
Serum NEFA concentrations did not differ significantly between the basal and hyperglycaemic periods (0.23 ± 0.04 mmol/l vs 0.31 ± 0.07 mmol/l, NS) (Fig. 2c). No significant difference was observed in palmitate flux between the basal and hyperglycaemic periods (175 ± 25.8 μmol/min vs 190 ± 34.3 μmol/min, NS). Plasma palmitate concentrations increased slightly, but not significantly, during hyperglycaemia compared with the basal period (68 ± 11.3 μmol/l vs 103 ± 24 μmol/l, p = 0.13 for the basal period and hyperglycaemia, respectively).
VLDL-TG kinetics
Plasma total TG and VLDL-TG concentrations remained unchanged during the basal and hyperglycaemic periods (Fig. 2d). Steady-state plasma VLDL-TG sp. act. was effectively reached in the last hour of the basal period and the last 2 h of the hyperglycaemic period (Fig. 3a). Individual changes in VLDL-TG secretion were seen, but the secretion rate in all participants was not changed by experimental hyperglycaemia (36.0 ± 9.6 μmol/min vs 30.8 ± 6.1 μmol/min, NS) (Fig. 3b). The participants who showed a pronounced change in secretion rate did not differ from the other participants. Similarly, the VLDL-TG clearance rate was not significantly different between the basal and hyperglycaemic periods (209 ± 30.4 vs 197 ± 41.7 ml/min, NS) (Fig. 3c).

(a) [14C]VLDL-TG sp. act. (disintegrations per minute per μmol) as a percentage of the maximum. The individual time series of patient VLDL-TG sp. act. data are normalised by the maximum value for each patient, yielding the relative changes. The mean ± SEM of the relative changes for all patients is depicted at each sample point in time. (b) Individual changes in VLDL-TG secretion rate (μmol/min) and (c) clearance rate (ml/min), comparing the kinetics from the last hour of the basal and clamp periods. The mean (black square) ± SEM is also depicted. (d) 14CO2 sp. act. expressed as the proportion of infused tracer oxidised during the study day (fractional recovery). Data are mean ± SEM
Breath 14CO2 sp. act. increased progressively during the study day and reached a steady state at the end of the hyperglycaemic period (Fig. 3d). A comparison of breath 14CO2 sp. act. curves in the present study with published data [26] on the increment in acetate recovery during prolonged [14C]acetate infusion revealed comparable increments (approximately 50%) in 14CO2 sp. act. between 4 and 8 h of infusion, indicating that no major difference in VLDL-TG FA oxidation occurred after the initiation of experimental hyperglycaemia.
REE and substrate oxidation
REE was stable during the study day (basal REE 8190 ± 284 kJ/24 h vs hyperglycaemia REE 7871 ± 326 kJ/24 h). RQ was slightly greater during the basal period compared with the hyperglycaemic period (0.85 ± 0.01 mg kg−1 min−1 vs 0.81 ± 0.01 mg kg−1 min−1, p = 0.035). Similarly, the glucose oxidation rate was greater during the basal period (1.58 ± 0.18 mg kg−1 min−1 vs 1.06 ± 0.11 mg kg−1 min−1, p = 0,017), whereas lipid oxidation did not differ significantly (0.66 ± 0.12 mg kg−1 min−1 vs 0.84 ± 0.07 mg kg−1 min−1, NS).
Discussion
This study was performed to determine the independent effect of overt acute hyperglycaemia (16 mmol/l) on VLDL-TG and palmitate kinetics in men with type 1 diabetes. We found that the VLDL-TG secretion rate, clearance rate and concentrations remained unchanged during 4 h of hyperglycaemia compared with normoglycaemia when insulin was clamped at a concentration comparable to fasting levels in healthy men. Furthermore, we found no effect of overt hyperglycaemia on lipolysis as measured by palmitate flux.
Our findings do not support previous reports suggesting that isolated hyperglycaemia may acutely increase VLDL-TG secretion [10, 18]. The study by Sidossis et al, which had a primary aim of studying the effect of insulin on VLDL-TG secretion, used somatostatin to suppress postprandial endogenous insulin secretion following 5 days on a carbohydrate enriched diet compared with a normal diet. Complete inhibition was, however, not achieved, resulting in different postprandial insulin increments in the groups [18]. In the second study, plasma NEFA levels were substituted using an intralipid infusion in order to account for the concomitant insulin-mediated suppression of plasma NEFA during hyperglycaemia [10]. Although this approach may account for a NEFA-mediated impact on VLDL-TG secretion, no attempt was made to avoid hyperinsulinaemia.
In rodents, however, studies have suggested a VLDL stimulatory effect of isolated hyperglycaemia during a clamp of only short duration. In rats, blood glucose was clamped at 17 mmol/l for 2 h in combination with either somatostatin or a blockade of insulin signalling through transcription factor FoxO1 [27] to create hyperglycaemic–hypoinsulinaemic conditions. This led to a 4.7-fold and 4.6-fold increase in serum VLDL levels, respectively. The insulin levels attained in these experiments were very low or insulin signalling was blocked, and perhaps such near-complete insulin inhibition, more than hyperglycaemia per se, could explain the pronounced increase in VLDL levels.
We found no evidence to suggest that overt hyperglycaemia alters lipolysis or NEFA concentrations. Thus, palmitate flux did not change significantly during hyperglycaemia, which corresponds to studies in men with type 1 diabetes that compared palmitate flux during euglycaemic and hyperglycaemic clamping under comparable stable insulinaemia [21, 22]. Compared with these studies, we chose a somewhat higher glucose concentration (16 mmol/l vs 11–12 mmol/l). Thus, our results confirm and extend these previous findings to include more pronounced hyperglycaemia as well.
Hyperglycaemia has been reported to decrease whole-body fat oxidation as well as reduce hepatic fat oxidation and increase hepatic TG synthesis in healthy individuals [10]. In the absence of hyperinsulinaemia, we found no evidence for a shift towards a whole-body preference for glucose oxidation during hyperglycaemia. Instead we found RQ to decrease slightly during the study day, probably reflecting the prolonged fasting state, although no significant increase in lipid oxidation could be detected. In accordance with this, we observed increments in breath 14CO2 sp. act. that were similar to the available data for long-term postabsorptive acetate recovery in breath [26]. Thus, the lack of a clear change in the increment in 14CO2 sp. act., which we would have taken as supportive evidence that hyperglycaemia alters VLDL-TG oxidation, refutes our hypothesis that short-term hyperglycaemia alters VLDL-TG FA oxidation. Collectively, our results do not support the notion that isolated overt hyperglycaemia acutely invokes increased VLDL-TG secretion or suppression of lipolysis, and we interpret our results as an indication of unchanged VLDL-TG FA oxidation following the induction of experimental hyperglycaemia.
The use of a systemic, as opposed to portal, insulin infusion may result in a greater redistribution of insulin action from the liver to the peripheral tissues [28], which may have affected our results. In dogs, Sindelar et al found that the hepatic glucose output was similar but was delayed by approximately 1 h when equivalent amounts of insulin were infused peripherally compared with into the portal vein. The authors suggested that the delayed response involved an indirect (non-hepatic) effect on gluconeogenic precursors, for example NEFA. We are unaware of published studies using similar insulin infusion approaches to evaluate the effects of portal vs peripheral insulin on VLDL-TG secretion. In the present study, insulin was infused for 150 and 240 min in the basal and hyperglycaemic periods, which we believe would ameliorate a time-dependent effect on the liver. In addition, peripheral subcutaneous insulin administration is the treatment of choice in patients with type 1 diabetes. A more elaborate design including portal insulin infusion would be necessary to account for the difference in insulin distribution encountered in healthy individuals.
Importantly, we deliberately confined our study to men with type 1 diabetes in order to evaluate the isolated effects of hyperglycaemia on hepatic VLDL-TG secretion and oxidation without the potential impact of concomitant perturbations in endogenous insulin secretion, and we administered intravenously to minimise the impact of intestinally derived factors, for example chylomicrons and glucagon-like peptide-1 (GLP-1). In accordance, we were not able to elicit detectable insulin secretion during hyperglycaemia. We did not measure plasma insulin aspart concentration, which was used by the patients to control their blood glucose during the evening and night before the study day. We believe that any effect of residual insulin aspart on lipid kinetics 4–10 h after the last injection would, if anything, promote a greater suppression of VLDL-TG and palmitate in the basal period compared with the hyperglycaemic period, that is, it would promote changes in favour of our hypothesis. Since, however, we did not find such changes, we believe the effect to be irrelevant.
This study has, however, limitations. First, the participants had high blood glucose levels at the beginning of the study day, and a potential carry-over effect leading to increased basal VLDL-TG secretion rates may have affected our findings. However, we find this unlikely, since VLDL-TG sp. act. was constant during the basal period. Second, a greater initial insulin infusion was necessary in order to obtain an adequate control of plasma glucose level. This could have resulted in an untoward suppression of VLDL-TG secretion and palmitate flux in the basal period, leading to a rebound effect in the hyperglycaemic period. However, as this was not observed, we expect that any effect of extra insulin was negligible. A control group was not included. Caruso et al previously demonstrated that no difference was found in palmitate flux when comparing patients with type 1 diabetes during a control (saline) and a hyperglycaemic day [21]. Thus, any effect of palmitate on VLDL-TG secretion seems unlikely. Third, participants were studied in a highly experimental setting with 4 h of constant hyperglycaemia. However, since we did not observe any effect in these circumstances, we find it unlikely that real-life short-term excursions in plasma glucose will affect VLDL-TG secretion rate. Finally, our results cannot be extended to women or to individuals without diabetes.
We conclude that short-term hyperglycaemia in the absence of increased insulinaemia does not have any independent effect on short-term VLDL-TG or NEFA kinetics in men with type 1 diabetes. Whether similar results apply to healthy individuals, insulin-resistant obese people or patients with type 2 diabetes is currently unknown.
Abbreviations
- ApoB-100:
-
Apolipoprotein B-100
- CRC:
-
Clinical Research Centre
- FA:
-
Fatty acid
- REE:
-
Resting energy expenditure
- sp. act.:
-
Specific activity
- VLDL-TG:
-
VLDL-triacylglycerol
References
Morrish NJ, Wang SL, Stevens LK, Fuller JH, Keen H (2001) Mortality and causes of death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia 44(Suppl 2):S14–S21
Danaei G, Lawes CM, Vander HS, Murray CJ, Ezzati M (2006) Global and regional mortality from ischaemic heart disease and stroke attributable to higher-than-optimum blood glucose concentration: comparative risk assessment. Lancet 368:1651–1659
Khaw KT, Wareham N, Bingham S, Luben R, Welch A, Day N (2004) Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann Intern Med 141:413–420
Nathan DM, Cleary PA, Backlund JY et al (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 353:2643–2653
Hokanson JE, Austin MA (1996) Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk 3:213–219
Duca L, Sippl R, Snell-Bergeon JK (2013) Is the risk and nature of CVD the same in type 1 and type 2 diabetes? Curr Diab Rep 13:350–361
Taskinen MR (2003) Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia 46:733–749
Azad K, Parkin JM, Court S, Laker MF, Alberti KG (1994) Circulating lipids and glycaemic control in insulin dependent diabetic children. Arch Dis Child 71:108–113
Maahs DM, Ogden LG, Dabelea D et al (2010) Association of glycaemia with lipids in adults with type 1 diabetes: modification by dyslipidaemia medication. Diabetologia 53:2518–2525
Sidossis LS, Mittendorfer B, Walser E, Chinkes D, Wolfe RR (1998) Hyperglycemia-induced inhibition of splanchnic fatty acid oxidation increases hepatic triacylglycerol secretion. Am J Physiol 275:E798–E805
Parks EJ, Hellerstein MK (2000) Carbohydrate-induced hypertriacylglycerolemia: historical perspective and review of biological mechanisms. Am J Clin Nutr 71:412–433
Yamashita H, Takenoshita M, Sakurai M et al (2001) A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc Natl Acad Sci U S A 98:9116–9121
Koo SH, Dutcher AK, Towle HC (2001) Glucose and insulin function through two distinct transcription factors to stimulate expression of lipogenic enzyme genes in liver. J Biol Chem 276:9437–9445
Chong MF, Hodson L, Bickerton AS et al (2008) Parallel activation of de novo lipogenesis and stearoyl-CoA desaturase activity after 3 d of high-carbohydrate feeding. Am J Clin Nutr 87:817–823
Schwarz JM, Linfoot P, Dare D, Aghajanian K (2003) Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am J Clin Nutr 77:43–50
Brown AM, Wiggins D, Gibbons GF (1999) Glucose phosphorylation is essential for the turnover of neutral lipid and the second stage assembly of triacylglycerol-rich ApoB-containing lipoproteins in primary hepatocyte cultures. Arterioscler Thromb Vasc Biol 19:321–329
Sondergaard E, Sorensen LP, Rahbek I, Gormsen LC, Christiansen JS, Nielsen S (2012) Postprandial VLDL-triacylglycerol secretion is not suppressed in obese type 2 diabetic men. Diabetologia 55:2733–2740
Ginsberg HN, Jacobs A, Le NA, Sandler J (1982) Effect of somatostatin-induced suppression of postprandial insulin response upon the hypertriglyceridemia associated with a high carbohydrate diet. J Clin Invest 70:1225–1233
Durrington PN, Newton RS, Weinstein DB, Steinberg D (1982) Effects of insulin and glucose on very low density lipoprotein triglyceride secretion by cultured rat hepatocytes. J Clin Invest 70:63–73
Lewis GF, Uffelman KD, Szeto LW, Weller B, Steiner G (1995) Interaction between free fatty acids and insulin in the acute control of very low density lipoprotein production in humans. J Clin Invest 95:158–166
Caruso M, Divertie GD, Jensen MD, Miles JM (1990) Lack of effect of hyperglycemia on lipolysis in humans. Am J Physiol 259:E542–E547
Cersosimo E, Coppack S, Jensen M (1993) Lack of effect of hyperglycemia on lipolysis in humans. Am J Physiol 265:E821–E824
Gormsen LC, Jensen MD, Nielsen S (2006) Measuring VLDL-triglyceride turnover in humans using ex vivo-prepared VLDL tracer. J Lipid Res 47:99–106
Frayn KN (1983) Calculation of substrate oxidation rates in vivo from gaseous exchange. J Appl Physiol 55:628–634
Sidossis LS, Coggan AR, Gastaldelli A, Wolfe RR (1995) A new correction factor for use in tracer estimations of plasma fatty acid oxidation. Am J Physiol 269:E649–E656
Mittendorfer B, Sidossis LS, Walser E, Chinkes DL, Wolfe RR (1998) Regional acetate kinetics and oxidation in human volunteers. Am J Physiol 274:E978–E983
Wu K, Cappel D, Martinez M, Stafford JM (2010) Impaired-inactivation of FoxO1 contributes to glucose-mediated increases in serum very low-density lipoprotein. Endocrinology 151:3566–3576
Sindelar DK, Balcom JH, Chu CA, Neal DW, Cherrington AD (1996) A comparison of the effects of selective increases in peripheral or portal insulin on hepatic glucose production in the conscious dog. Diabetes 45:1594–1604
Acknowledgements
The authors acknowledge the excellent technical assistance of D. Wulff and H. Petersen of the Medical Research Laboratories, Aarhus University Hospital, Denmark.
Funding
This study was supported by grants from the Novo Nordisk Foundation and the Augustinus Foundation. No additional funding was received for this study.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
RFJ performed acquisition of the data and data analysis, and wrote the manuscript. ES wrote the protocol and reviewed/edited the manuscript. BN performed data acquisition and reviewed the manuscript. LPS and JSC contributed with interpretation of the data and reviewed the manuscript. SN designed the study, reviewed and edited the manuscript and is the guarantor of the study as he had full access to all data and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors approved the final version to be published.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Johansen, R.F., Søndergaard, E., Sørensen, L.P. et al. Isolated hyperglycaemia does not increase VLDL-triacylglycerol secretion in type 1 diabetic men. Diabetologia 58, 355–362 (2015). https://doi.org/10.1007/s00125-014-3422-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-014-3422-6