
Abstract
Key message
A large-effect QTL was fine mapped, which revealed 79 gene models, with 10 promising candidate genes, along with a novel inversion.
Abstract
In commercial maize breeding, doubled haploid (DH) technology is arguably the most efficient resource for rapidly developing novel, completely homozygous lines. However, the DH strategy, using in vivo haploid induction, currently requires the use of mutagenic agents which can be not only hazardous, but laborious. This study focuses on an alternative approach to develop DH lines—spontaneous haploid genome duplication (SHGD) via naturally restored haploid male fertility (HMF). Inbred lines A427 and Wf9, the former with high HMF and the latter with low HMF, were selected to fine-map a large-effect QTL associated with SHGD—qshgd1. SHGD alleles were derived from A427, with novel haploid recombinant groups having varying levels of the A427 chromosomal region recovered. The chromosomal region of interest is composed of 45 megabases (Mb) of genetic information on chromosome 5. Significant differences between haploid recombinant groups for HMF were identified, signaling the possibility of mapping the QTL more closely. Due to suppression of recombination from the proximity of the centromere, and a newly discovered inversion region, the associated QTL was only confined to a 25 Mb region, within which only a single recombinant was observed among ca. 9,000 BC1 individuals. Nevertheless, 79 gene models were identified within this 25 Mb region. Additionally, 10 promising candidate genes, based on RNA-seq data, are described for future evaluation, while the narrowed down genome region is accessible for straightforward introgression into elite germplasm by BC methods.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Doubled haploid (DH) technology has become an increasingly important tool to produce completely homozygous lines in shorter time and to speed up breeding cycles (Geiger and Gordillo 2009; Prigge et al. 2012; Jacquier et al. 2020). Moreover, DH populations were shown to have an increased usefulness compared to F2-derived populations (Mayor and Bernardo 2009). Genetic gain (ΔG) is dependent on the amount of phenotypic variance present in the population (σ2P), heritability, selection intensity (i), and the number of generations between cycle intervals (Moose and Mumm 2008). Genomic selection relies on genotypic and phenotypic values, rather than phenotypic values alone, to obtain reliable genomic estimated breeding values (GEBVs) (Meuwissen et al. 2001). Combining genomic selection with DH technology would allow plant breeders to obtain more reliable estimates, while simultaneously reducing the time between breeding cycle intervals (Heffner et al. 2009; Gaynor et al. 2017; Hickey et al. 2017; Ren et al. 2017).
The in vivo DH process requires that kernels with a haploid embryo are induced via in vivo maternal haploid induction with a male inducer. Haploid seed then needs to be identified, sown in a glasshouse, carefully exposed to chemical doubling agents, and finally transplanted into the field as seedlings (Vanous et al. 2017). The process of DH line development routinely uses chemical doubling agents, the most common being colchicine, to induce genome doubling (Prigge and Melchinger 2012; Molenaar and Melchinger 2019). The process of treating and transplanting haploids is very laborious and expensive. The final step in the DH process is the self-pollination of fertile haploid plants (Aboobucker et al. 2022). Only a fraction of haploid plants restore fertility to produce DH seed where seed set in the D0 generation is generally low—requiring an additional generation for seed increase before being utilized in a breeding program.
Instead of utilizing hazardous chemicals to induce genome doubling, plant breeders can use a genetic mechanism, known as spontaneous haploid genome doubling (SHGD). SHGD encompasses two key traits: haploid male fertility (HMF) and haploid female fertility (HFF). Both traits must be present simultaneously to successfully produce DH progeny. It has been previously reported that HMF is the limiting factor for successful SHGD derived DH lines (Chase 1952; Chalyk 1994; Geiger et al. 2006). There are other benefits in using SHGD compared to traditional methods. In particular, the ability to directly sow haploid seed instead of transplanting treated haploids substantially reduces the costs for DH line production. A major constraint is the limited availability of HMF in maize (Zea mays L.) (Kleiber et al. 2012; Ma et al. 2018; Chaikam et al. 2019a). Therefore, identification of novel genotypes with high levels of HMF and underlying candidate genes is a ‘needle in a haystack’ approach. However, some genotypes exhibited HMF levels greater than 50% (Geiger and Schönleben 2011; Kleiber et al. 2012; Wu et al. 2017; Ma et al. 2018; Chaikam et al. 2019b; De La Fuente et al. 2020; Ren et al. 2020; Trampe et al. 2020;). Various QTL for HMF or SHGD identified by genome-wide association (GWAS), or QTL mapping studies, have been reported in maize (Boerman et al. 2020), and the first genes affecting HMF were identified in Arabidopsis (Aboobucker et al. 2023).
A region on chromosome 5 was found to have a strong effect on SHGD (Ren et al. 2020; Trampe et al. 2020; Verzegnazzi et al. 2021; Santos et al. 2022). Ren et al. (2020) utilized three inbreds with high (A427), moderate (CR1Ht), and poor (Wf9) SHGD to construct mapping populations. Three QTL related to SHGD were identified—qshgd1, qshgd2 and qshgd3. Within the same region as qshgd1, Trampe et al. (2020) identified a large effect QTL explaining approximately 40% of the phenotypic variance using the SHGD donor A427 crossed with CR1Ht to create haploid families from 220 F2:3 families. Verzegnazzi et al. (2021) utilized A427-derived offspring to determine, whether qshgd1 could be detected via a case–control GWAS. Four populations derived from temperate-adapted germplasm with poor SHGD, BS39 (Hallauer and Carena 2016), or the cross between BS39 and A427, were compared. Their analysis suggested that qshgd1 enabled SHGD in DH lines obtained without colchicine treatment. Finally, Santos et al. (2022) showed that the presence of qshgd1 from A427 did not impact the overall testcross performance for agronomic traits, suggesting no undesired linkage drag effects with this QTL in BS39 background.
The overall objective of this study was to fine-map the chromosomal region associated with the large-effect QTL, qshgd1, by (i) scoring A427 x Wf9 haploid recombinant groups for HMF, (ii) determining the position of this QTL using composite interval mapping, (iii) and identify positional candidate genes underlying this QTL, based on the available A427 genomic data.
Materials and methods
Plant materials
A BC1F1 was generated with A427 as the donor parent and Wf9 as the recurrent parent. A427 is an inbred line developed by the University of Minnesota during the early 1970s (Roth et al. 1970; Moss et al. 1971; Findley 1972). A427 has been shown to have a rate of HMF which was consistently greater than 65% (De La Fuente et al. 2020; Ren et al. 2020; Trampe et al. 2020). The second parental inbred line, Wf9, was developed at Purdue University during the 1930s (Patch and Bottger 1937; Gethi et al. 2002). In contrast to A427, Wf9 has near 0% HMF (De La Fuente et al. 2020; Ren et al. 2020).
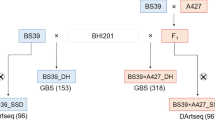
The subsequent work associated with genotyping, marker selection and haploid induction was equally shared between KWS and Limagrain companies. The collaborating companies grew out 4,500 seed each in the field and screened for recombinants in the chromosomal area of interest. The resulting 232 recombinant BC1F2 families represented 24 recombination groups. They served as donor populations in haploid induction and individuals were again screened with the same set of marker. Induction in the field was accomplished by sowing & induction of 100K per BC1F2 families (4 rows X 25 plants), using inducer having R1-nj color maker, sorting done at mature seed level based on absence of coloration, at the same time genotyping to identify the homo recombinants. For each family, two contrasting groups of haploids were selected—those derived from individuals completely homozygous for the recurrent parent alleles and haploids derived from completely homozygous recombinant individuals. Haploid sets derived from a single BC1F2 family with at least 100 potential haploid seed for control and recombinant haploids were used for the screenings at KWS and LG, whereas for the final screening at ISU, haploids from BC1F2 families assigned the same recombination group were pooled (Fig. 1).

Diagram of breeding scheme for deriving haploid recombinant groups
The rationale was (1) to use the BC1F2 families with the recurrent parent allele fixed as an overall baseline for HMF to control for any QTL present in the A427 genome outside of the chromosome 5 region of interest, and (2) to be able to induce multiple recombinant plants to get sufficient haploid seed for subsequent trials.
Marker screening during plant material development
Genotyping was conducted as described by Gilles et al. (2017) , where DNA was isolated from five young leaf punches (diameter 6 mm). Leaf discs were collected in plates with 96 deep wells and ground with two metallic balls (diameter 4 mm). Genomic DNA was then extracted using a CTAB type lysis buffer and a magnetic bead-based purification.
The markers in this study originated from a public 50k Illumina array chip (Ganal et al. 2011). A combination of both published and company-specific markers were used to target a 45 Mb region on chromosome 5. We applied Kompetitive Allele-Specific PCR (KASP) assays, which utilize allele-specific forward primers, along with a common reverse primer, to detect single nucleotide polymorphisms (He et al. 2014). Using this method, we were able to deploy 17 SNP markers (Table S1; Table S2) within our 45 Mb target region, to differentiate alleles originating from either the A427 or Wf9 parental genotype to create a chromosome 5-specific fine-mapping population. Physical coordinates of markers are based on the B73 genome assembly (RefGen_v4) (Jiao et al. 2017).
Maternal haploid induction and haploid identification
Selected individuals from BC1F2 families were induced via in vivo haploid induction using a male inducer bulk, developed by Iowa State University (BHI301, BHI305, BHI306, BHI310), which harbors the dominant R1-navajo (R1-nj) anthocyanin marker. Haploids were manually selected using the R1-nj anthocyanin marker. Hybrid kernels with a purple/red embryo were discarded and kernels with a colorless haploid embryo were used for further analyses. Moreover, false-positive diploid plants were rogued in experimental trials (Vanous et al 2017). A total of 232 A427 recombinant BC1F2 families were produced utilizing this method. The 232 resulting A427 recombinant BC1F2 haploid families were assigned to 24 haploid recombinant groups based upon the recombination breakpoint within the 45 Mb region, using 17 markers within this region (Fig. 2). There were two classes of A427 recombinants, with 14 recombinant groups in ‘Class 1’ and 10 recombinant groups in ‘Class 2’ (Fig. 2). The first class carrying the donor region from the distal end to the recombination break point, and the second carrying the donor region from the proximal end to the recombination break point (Fig. 2).

Allelic codes for each of the marker positions, given in megabases (Mb), for the A427 BC1F2 haploid recombinant groups
The first class of recombinant groups were made by recombining the donor region from the proximal end to the break point and the second by recombing the donor region from the distal end to the break point.
Experimental design
For this study, haploid recombinant groups were grown in three different environments, a KWS 2020/21 winter nursery in Chile, a Limagrain 2020/21 winter nursery in Chile, and an Iowa State 2021 summer nursery at the Agricultural Engineering and Agronomy Research Farm in Boone, Iowa, USA. Depending on haploid seed availability, each A427 BC1F2 haploid recombinant group was evaluated, alongside its Wf9 BC1F2 haploid baseline pair, at a minimum of two (2) environments. Different experimental designs were utilized for the project with a minimum of two replications for each represented recombinant group in each environment. The experimental design at the Limagrain and KWS winter nursey was a split-plot design and the experimental design at the ISU summer nursey was a randomized incomplete block design. Within each environment, replications of recombinant classes were combined as sample sizes for recombinant groups were often low. In addition, haploids of parents A427 and Wf9 were replicated in each environment. In total, 22 of the 24 haploid recombinant groups were evaluated in the KWS winter nursery, whereas all 24 haploid recombinant groups were evaluated in the Limagrain winter nursery, and the Iowa State summer nursery.
Phenotypic scoring
HMF was evaluated by scoring each tassel, on a daily basis, for the presence of fertile anthers and for the capacity of those fertile anthers to shed pollen as in Aboobucker et al. (2022). Scores were expected to fluctuate from day to day. In this circumstance, the highest recorded score is the reported score for each trait.
Different combinations of fertile scores were created for each trait. Scores of 1–5 encompass all tassels showing any level of fertility. However, scores of 2–5 are more interesting as the likelihood of misclassifying a sterile haploid plant as being fertile is reduced. Therefore, four trait/score combinations were utilized for calculating HMF: (i) scores ranging 1–5, for pollen shedding capacity (PSC); (ii) scores ranging 1–5, for fertile anther emergence (FAE); (iii) scores ranging 2–5, for PSC; and (iv) scores ranging 2–5 for FAE. HMF rates were calculated as:
Statistical analysis
Data analysis was conducted using R software version 4.2.2 (R Core Team 2022) in RStudio (RStudio Team 2022) with the tidyverse package (Wickham et al. 2019). Logit transformed best linear unbiased estimates (BLUEs) were calculated for each haploid recombinant group, where haploid recombinant groups were considered as fixed effects and environmental effects were also considered fixed. The response variable, HMF, was logit transformed to obtain normality of residuals with the following equation from Trampe et al. (2020):
A linear model was implemented to calculate each of the logit transformed BLUE values using the following equation:
where Yik is the phenotypic response of ith recombinant group in the kth environment, µ is the overall mean, Gi is the effect of the ith recombinant group, Ek is the effect of the kth environment and εik is the residual effect.
A linear mixed effect model, where haploid recombinant groups and environmental effects were both considered random, was utilized to obtain the variance components for calculating entry-mean based heritability estimates using the following formula:
where σg2 is the variance component of the haploid recombinant groups, σr2 is the variance of the residuals and \(\overline{n}_{e}\) is the harmonic mean of the number of environments (Schmidt et al. 2019).
Linkage map construction
The linkage map was constructed based upon 232 recombinant BC1s of a ca. 9,000 BC1 population utilizing the MAP function in QTL IciMapping version 4.2 (Meng et al. 2015). The Kosambi mapping function was used to determine distance, in centimorgans (cM), based on the recombination rate between a set of markers. Markers within linkage groups were ordered using a window size of five and rippled using the recombination frequency matrix between markers (Fig. 3).

Comparative map of markers utilized for QTL analysis. Genetic position, in centimorgans, (right) and physical position, in megabase pairs, (left) are provided
QTL mapping
QTL mapping was performed using the R/qtl package (Broman et al. 2003) in R. Kosambi was the selected mapping function (Kosambi 2016) using the composite interval mapping (CIM) method with an error probability of p = 0.0001, a window size of 0.5 centimorgan (cM) and one marker covariate—M13 (Table S1). The logarithm of odds (LOD) threshold for QTL detection was set based on 1,000 permutation tests using a Type I error rate of p = 0.05. Because genotypic data for calculating genetic probability was based on haploid recombinant groups, this information was coded as either ‘A’ or ‘B’, for a given marker, with ‘A’ signifying the allele originated from the Wf9 parental genotype and ‘B’ signifying the allele originated from the A427 parental genotype (Table S1). The only markers that were utilized were markers lying within the targeted region. Preceding mapping studies have shown that qshgd1 had by far the strongest effect with 50% of the genetic variance being contributed by qshgd1 alone (Trampe et al. 2020). For this reason, we focused on fine-mapping this major QTL region and did not conduct a genome-wide mapping study.
Candidate gene identification
At the time of publication, annotated gene models for A427 were not available. However, high quality PacBio genomic data of A427 were available (Seetharam et al. in prep.). To find candidate genes associated with qshgd1, alignments of A427 against B73, IL14H, and Ia453-sh2 were made using Mugsy 1.2.3 (Angiuoli and Salzberg 2011). The Ia453-sh2 genome region was most similar among sequenced maize genomes (Fig. S1). Gene models of Ia453-sh2 were then compared with the B73v5 reference genome using standalone BLAST for Windows (Tao 2010). Resulting gene models were filtered for only those gene models found on chromosome 5 within the genomic region associated with qshgd1 from CIM. Publicly available data (GO term, GO Domain and RNA-Seq) were used to filter the most promising candidate genes in this genomic region.
Results
Phenotypic variation and heritability
The parents, A427 and Wf9, as well as the Wf9 BC1F2 haploid baseline and the A427 BC1F2 haploid recombinant families showed considerable variation across trait/score combinations for calculating HMF (Table 1; Fig. 4). A427 parental haploids had a higher mean than haploids derived from the Wf9 parent. A427 haploids averaged 80.4–88.0% HMF and Wf9 haploids averaged 1.2–4.7% HMF (Table 1). Haploids derived from the BC1F2 generation that were fixed for the recurrent parent (Wf9) allele in the 45 Mb region averaged 3.3–8.8% HMF. In contrast, across all A427 recombinant haploids derived from the BC1F2 generation the average HMF rate was 14.9–19.8% (Table 1).

Distribution of haploid male fertility percentage across haploid recombinant groups. The number of haploid plants evaluated for each recombinant group is provided above the distribution bar
For haploids from A427 BC1F2 haploid recombinant families, the highest means of haploids from BC1F2 families for HMF via PSC scores 1–5 and HMF via FAE scores 1–5 were found at the KWS winter nursery research station in Chile at 19.3 and 38.0%, respectively (Table 1). The highest means for HMF via PSC scores 2–5 were found at the Iowa State Agronomy/Agricultural Engineering Research Farm in Iowa, USA at 15.6% (Table 1). Finally, the highest means for HMF via FAE scores 2–5 were found at the Limagrain winter nursery research station in Chile at 18.5% (Table 1).
Correlations between environments, specifically for HMF via PSC scores of 2–5 and HMF via FAE scores of 2–5, were significant with values ranging from 0.65 to 0.73 and 0.48 to 0.76, respectively (Fig. S2). The highest correlation between environments, for HMF via FAE scores 2–5, was found between ISU and KWS (0.76; Fig. S2). Furthermore, the highest correlation between environments, for HMF via PSC scores 2–5, was found between ISU and LG (0.73; Fig. S2). Correlations between traits and scores were also calculated (Fig. S3). Heritabilities were 0.81, 0.68, 0.87, and 0.87 for HMF for PSC 1–5, HMF for FAE scores 1–5, HMF for PSC 2–5, and HMF for FAE scores 2–5, respectively (Table 2). Finally, analysis of variance calculations revealed significant variation, at the α = 0.001 significant level, for recombinant groups (Table 3).
Linkage map
The region for constructing our linkage map was approximately 45 Mb in physical distance and 2.4 cM in genetic distance (Fig. 3). The linkage map shows a large non-recombing region from marker 11 (96.01 Mb) to marker 14 (119.77 Mb) (Fig. 3; Table S1). This ‘dead zone’ for recombination includes both the centromere and the pericentromere. In addition, the parental line, A427, contains a 600 kb inversion region, near to the centromere on chromosome 5, that is not present in B73v5 (Fig. S4; Seetharam et al. in prep.), which could also contribute to high physical linkage of markers in this region.
QTL analysis
The mapping region of interest was approximately 2.4 cM in length where 17 SNP markers were utilized (Fig. 3). For HMF based on either PSC or FAE, CIM results showed similar results—a significant association was found at 102,458,115 bp (Fig. 5), which corresponds to a genetic position of 2.0 cM on the linkage map (Fig. 3). The QTL was found at this chromosomal position, regardless of the scoring system used for the traits (Fig. 5).

QTL detection on chromosome 5 by composite interval mapping
Candidate gene identification
The sequence alignment and filtering of gene models revealed 79 potential candidate genes dispersed over the 96–120 Mb region closest associated with qshgd1 on chromosome 5 (Table S3). Of those gene models, ten promising candidate genes for qshgd1 were identified (Table 2) based on available RNA-seq data (Dataset S1). Because qshgd1 confers the ability of haploid plants to spontaneously double their genome to the diploid state, RNA-seq expression of genes associated with mitotic or meiotic division were considered most promising.
Most promising candidate genes were selected as being highly expressed in plant tissues in which mitosis or meiosis are actively occurring, such as candidate gene Zm00001eb234730 (Table 4). Zm00001eb235700 codes an actin-related protein of the SWR1 complex, which is required for essential life processes, such as vegetative and reproductive development (Blessing et al. 2004; Dion et al. 2010; Nie and Wang 2021). As final example, a homolog of Zm00001eb235200 has been reported in Arabidopsis thaliana (L.) Heynh. as an augmin-like complex (AUG6) which plays an essential role in microtubule organization during sexual reproduction (Oh et al. 2016).
Discussion
Phenotyping
To critically assess the quality of our experiments, correlations between environments were calculated (Figs. S2, S3) and were found to be significant and moderate to high. High entry-mean based heritabilities (0.81–0.87) for three HMF measures (Table 2) were consistent with previous studies (Chaikam et al. 2019b; Molenaar et al. 2019; Trampe et al. 2020). Only HMF via FAE scores of 1–5 showed a lower heritability (0.68). One possible explanation is that sterile haploid plants can produce needle-like anthers, which do not contain pollen. In such a case, a haploid plant could be misclassified as ‘fertile’ with a score of 1 when it is ‘sterile’. Differences found between environments are most likely attributed to differences between scorers. It was initially planned that a single investigator scores all three trials. However, travel restrictions due to the COVID-19 pandemic made it necessary that phenotypic values were gathered by a different researcher at each location. Despite developing and exchanging detailed scoring guidelines, classifying haploids with low levels (‘1’), is likely to have differed between locations. This is also consistent with our finding that means for each of the HMF methods between environments were consistent, except for the calculated means between environments for HMF via FAE scores of 1–5 (Table 1). For this reason, we primarily focused further analyses on HMF via FAE scores and PSC scores of 2–5.
It should be noted that in this study, plants were classified as either ‘fertile’ or ‘sterile’ based solely on the quantity of anthers emerging from the tassel and the quantity of pollen shed from those anthers. Essentially, if any pollen was shed from anthers, the pollen was assumed to be fertile. However, given more time and resources, the pollen could be further differentiated into aborted and non-aborted pollen (Peterson et al. 2010). In addition, recorded scores were categorical and with some subjectivity of scores. With deep learning and image-based phenotyping, quantitative scores could be obtained, which would increase accuracy of the study (Pound et al. 2017). This method has yet to be reported for the traits of interest in this study. Moreover, heritabilities for our traits were sufficiently high for reliable mapping studies.
Although not every A427 recombinant haploid contained the region associated with qshgd1, the difference in BLUEs found between these 24 groups suggests that the QTL is located within the 96–120 Mb region (Figs. 3, 5). To support these findings, recombinant groups sharing common genetic markers were combined to create sets of recombinant classes (Table S4). The rationale of this approach was to locate the QTL either upstream, directly upstream, or far downstream of the centromere. BLUEs for each recombinant set were obtained using the previously described methods (Table S5). There was no increase in HMF until the A427 sequence was incorporated at 102,458,115 bp, regardless of the direction of recovering the A427 sequence, for HMF determined by PSC or FAE, using only scores of 2–5. This was consistent with QTL mapping across all recombinant groups (Tables S4, S5; Fig. 5). Nearly all recombinant sets from the combined approach that lack the A427 allele at 102,458,115 bp were not different from the Wf9 BC1F2 haploid baseline, which further indicates that the HMF phenotype from A427 is largely controlled by a single, large-effect gene, or > 1 tightly linked genes (Tables 1, S4, S5).
Fine-mapping results
The QTL position at 102,458,115 bp lies in a large non-recombining region. For the mapping population of this study, suppression of recombination resulted in very few novel recombinant groups. Specifically, recombination from 96,007,337 bp to 119,766,475 bp was limited, which led to close linkage between markers (Fig. 3). Of the ca. 9,000 BC1 plants, only one individual had a recombination event within this large genomic region. Given the large size of our mapping population, finding no recombinant in a > 20 Mb region is not encouraging for systematic map-based cloning of the underlying genes for qshgd1. However, we were able to further reduce the qshgd1 QTL region from 45 Mb to a region of ca. 25 Mb.
Suppressed recombination in the qshgd1 region can be due to different phenomena. First, pericentromeric regions near a chromosome’s centromeres have been shown to exhibit low recombination rates (Slotman et al. 2006). Unfortunately, the centromere on chromosome 5 is located within the qshgd1 region, which helps to explain the observed low recombination rates. Our observed recombination rate was similar to the recombination rate reported for the NAM haplotype map for this region on chromosome 5, where this region was shown to be a ‘dead zone’ for recombination (Gore et al. 2009). In addition, suppressed recombination could be influenced by a 600 kb inversion found downstream of the centromere in A427 relative to B73 (Fig. S4). Most sequenced lines in the NAM population are like B73 and do not carry the inversion (Seetharam et al. in prep.). Haploids of other three NAM genotypes (CML333, P39, IL14H) carryied a similar inversion like A427, did not exhibit HMF (data not shown). This observation suggests that the inversion itself is not causative for HMF.
Candidate genes
An intriguing attribute of plant pericentromeric regions is the presence of functional genes that are conserved across plant species (Wang and Bennetzen 2012). Additionally, highly repetitive segments, such as centromeric and pericentromeric regions, of eukaryotic genomes commonly include transposable elements and satellite DNA (Biscotti et al. 2015). Therefore, satellite DNAs are highly concentrated in centromeric and pericentromeric regions of chromosomes (Jagannathan et al. 2018). Although much is still unknown about centromeric regions, the overabundant presence of satellite DNA in this region, in comparison to the rest of the genome, signifies their importance in centromere function (Hartley and O’neill 2019). Extensive presence of sequence repeats in pericentromeric regions can help to explain why our methods for identifying potential candidate genes only produced a total of 79 gene models for a large genomic region (~ 25 Mb). Given that the maize genome is approximately 2.4 gigabases (Gb) (Haberer et al. 2005), and with an estimated 59,000 genes (Liu et al. 2007), one should expect approximately 600 genes for every 25 Mb region, more than sevenfold of the amount found.
We used a reasonable method of identifying conserved genes by evaluating, whether the B73v5 gene name had a homolog in Sorghum (Andorf 2022). Using this method, six of the 10 candidate genes have syntenic homologs in Sorghum (Table S6). Two of the three previously described gene models, Zm00001eb234730 and Zm00001eb235700, contain Sorghum homologs and are highly likely to be functional genes. While the remaining described gene model, Zm00001eb235200, does not appear to contain a homolog in Sorghum and does not appear to be conserved across species.
Given limitations of fine mapping of our target region due to suppressed recombination, isolating respective genes will likely require alternative approaches. This includes studying candidate genes using the Arabidopsis thaliana DH system, which has been successfully used by Aboobucker et al. (2023), assuming conservation of gene function. Alternatively, the Cre-lox system could be utilized to precisely target, and delete, DNA sequences. Using this method, small, sequential regions within the large 25 Mb region could be targeted for deletion. Mutants with a loss of function for HMF would signify a gene responsible for the phenotype lies within the deleted region. The primary limitation to this approach is the extensive resources needed to cover the entire 25 Mb region. Successfully targeted deletions have been reported in studies of a region smaller than 70 kb (Ullrich and Schüler 2010; Shaw et al. 2021).
Implications for breeding
Because the effect of qshgd1 is quite large (Trampe et al. 2020), breeding and selection for this QTL is likely to be effective. Breeding and selection using PSC scores of 2–5 would provide the best means for trait assessment as this trait/score combination provides the most consistency in quantifying the HMF trait. Incorporating qshgd1 into elite germplasm could be accomplished by phenotypic or marker-assisted backcrossing. In any attempt to incorporate qshgd1, tracking the QTL through scoring for PSC 2-5 would be preferred over FAE 2-5 as there is more consistency in quantifying the trait for a categorical score. With incorporating such a large non-recombining region, linkage drag of other undesirable genes could be expected. Thus far, testcross trials did not show negative impacts when incorporating qshgd1, but were limited to BS39 background (Santos et al. 2022). Therefore, we recommend further testing to assess the extent of linkage drag, if any, in other genetic backgrounds.
Linkage drag is not the only potential limitation with incorporating qshgd1 into elite maize germplasm. For example, qshgd1 has been mainly associated with restored male fertility in haploids and restored female fertility for qshgd1 remains unstudied. Without both restored fertile for both the male and female reproductive systems, DH-progeny derived from SHGD is not possible. Nonetheless, reports of haploid female fertility, with rates > 90%, indicate that HFF should not be a limiting factor (Chalyk 1994). Finally, including qshgd1 into all available elite germplasm would result in fixed genome regions in hybrids and thus likely cause suboptimal performance through the loss of natural genetic variation. It would be preferable to utilize qshgd1 in only a single heterotic group and rely on other SHGD QTL (Boerman et al. 2020) in complementary heterotic groups to maximize hybrid performance.
Data availability
The datasets generated and the figures/tables created with R software have been made available in the following public Github repository: https://github.com/Tfost1994/Fine-mapping-qshgd1-for-SHGD-in-maize.
References
Aboobucker SI et al (2023) Haploid male fertility is restored by parallel spindle genes in Arabidopsis thaliana. Nature Plants 9(2):214–218. https://doi.org/10.1038/s41477-022-01332-6
Aboobucker SI, et al (2022) Protocols for in vivo doubled haploid (DH) technology in maize breeding: from haploid inducer development to haploid genome doubling. In: Plant gametogenesis: methods and protocols. Springer, New York, pp 213–235
Andorf CM (2022) NAM_subgenomes_vs_Sorghum_from_Hufford-2021.txt. Available at: https://ars-usda.app.box.com/v/maizegdb-public/folder/186350887665.
Angiuoli SV, Salzberg SL (2011) Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics 27(3):334–342. https://doi.org/10.1093/bioinformatics/btq665
Biscotti MA et al (2015) Repetitive DNA in eukaryotic genomes. Chromosome Res 23(3):415–420. https://doi.org/10.1007/s10577-015-9499-z
Blessing CA et al (2004) Actin and ARPs: action in the nucleus. Trends Cell Biol 14(8):435–442. https://doi.org/10.1016/j.tcb.2004.07.009
Boerman NA et al (2020) Impact of spontaneous haploid genome doubling in maize breeding. Plants. https://doi.org/10.3390/plants9030369
Broman KW et al (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19(7):889–890. https://doi.org/10.1093/bioinformatics/btg112
Chaikam V et al (2019a) Doubled haploid technology for line development in maize: technical advances and prospects. Theor Appl Genet 132(12):3227–3243. https://doi.org/10.1007/s00122-019-03433-x
Chaikam V et al (2019b) Genome-wide association study to identify genomic regions influencing spontaneous fertility in maize haploids. Euphytica 215(8):1–14. https://doi.org/10.1007/s10681-019-2459-5
Chalyk ST (1994) Properties of maternal haploid maize plants and potential application to maize breeding. Euphytica 79(1–2):13–18. https://doi.org/10.1007/BF00023571
Chase SS (1952) Production of homozygous diploids of maize from monoploids. Agron J 44(5):263–267. https://doi.org/10.2134/agronj1952.00021962004400050010x
De La Fuente GN et al (2020) A diallel analysis of a maize donor population response to in vivo maternal haploid induction: II. Haploid male fertility. Crop Sci 60(2):873–882. https://doi.org/10.1002/csc2.20021
Dion V et al (2010) Actin-related proteins in the nucleus: life beyond chromatin remodelers. Curr Opin Cell Biol 22(3):383–391. https://doi.org/10.1016/j.ceb.2010.02.006
Findley WR (1972) Maize dwarf mosaic ratings of corn strains grown near Portsmouth, Ohio, in 1970 and 1971. Ohio Agricultural Research and Development Center, Wooster, Ohio; Research Circular, 190
Ganal MW et al (2011) A large maize (Zea mays L.) SNP genotyping array: development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE. https://doi.org/10.1371/journal.pone.0028334
Gaynor RC et al (2017) A two-part strategy for using genomic selection to develop inbred lines. Crop Sci 57(5):2372–2386. https://doi.org/10.2135/cropsci2016.09.0742
Geiger HH, Gordillo GA (2009) Doubled haploids in hybrid maize breeding. Maydica 54(4):485–499
Geiger HH, Schönleben M (2011) Incidence of male fertility in haploid elite dent maize germplasm. Maize Genet Coop Newsl 85:22–32
Geiger HH, et al (2006) Variation for female fertility among haploid maize lines. Maize Genetics Cooperation Newsletter
Gethi JG et al (2002) SSR variation in important U.S. maize inbred lines. Crop Sci 42(3):951–957. https://doi.org/10.2135/cropsci2002.9510
Gilles LM et al (2017) Loss of pollen-specific phospholipase NOT LIKE DAD triggers gynogenesis in maize. EMBO J 36(6):707–717
Gore MA et al (2009) A first-generation haplotype map of maize. Science 326(5956):1115–1117. https://doi.org/10.1126/science.1177837
Haberer G et al (2005) Structure and architecture of the maize genome. Plant Physiol 139(4):1612–1624. https://doi.org/10.1104/pp.105.068718
Hallauer AR, Carena MJ (2016) Registration of BS39 Maize Germplasm. J Plant Regist 10(3):296–300. https://doi.org/10.3198/jpr2015.02.0008crg
Hartley G, O’neill RJ (2019) Centromere repeats: hidden gems of the genome. Genes. https://doi.org/10.3390/genes10030223
He C, et al (2014) SNP genotyping: the KASP assay. In: Methods in molecular biology, pp 75–86. https://doi.org/10.1007/978-1-4939-0446-4_1
Heffner EL et al (2009) Genomic selection for crop improvement. Crop Sci 49(1):1–12. https://doi.org/10.2135/cropsci2008.08.0512
Hickey JM et al (2017) Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery. Nat Genet 49(9):1297–1303. https://doi.org/10.1038/ng.3920
Hufford MB et al (2021) De novo assembly, annotation, and comparative analysis of 26 diverse maize genomes. Science 373(6555):655–662. https://doi.org/10.1126/science.abg5289
Jacquier NMA et al (2020) Puzzling out plant reproduction by haploid induction for innovations in plant breeding. Nature Plants 6(6):610–619. https://doi.org/10.1038/s41477-020-0664-9
Jagannathan M et al (2018) A conserved function for pericentromeric satellite DNA. Elife 7:1–19. https://doi.org/10.7554/eLife.34122
Jiao Y et al (2017) Improved maize reference genome with single-molecule technologies. Nature 546(7659):524–527
Kleiber D et al (2012) Haploid fertility in temperate and tropical maize germplasm. Crop Sci 52(2):623–630. https://doi.org/10.2135/cropsci2011.07.0395
Kosambi DD (2016) The estimation of map distances from recombination values. In: Kosambi DD (ed) Selected works in mathematics and statistics, pp 125–130. https://doi.org/10.1007/978-81-322-3676-4
Liu R et al (2007) A genetrek analysis of the maize genome. Proc Natl Acad Sci USA 104(28):11844–11849. https://doi.org/10.1073/pnas.0704258104
Ma H et al (2018) Genome-wide association study of haploid male fertility in maize (Zea mays L.). Front Plant Sci. https://doi.org/10.3389/fpls.2018.00974
Mayor PJ, Bernardo R (2009) Genomewide selection and marker-assisted recurrent selection in doubled haploid versus F2 populations. Crop Sci 49(5):1719–1725. https://doi.org/10.2135/cropsci2008.10.0587
Meng L et al (2015) QTL IciMapping: integrated software for genetic linkage map construction and quantitative trait locus mapping in biparental populations. Crop J 3(3):269–283. https://doi.org/10.1016/j.cj.2015.01.001
Meuwissen THE et al (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819–1829. https://doi.org/10.1093/genetics/157.4.1819
Molenaar WS et al (2019) Haploid male fertility and spontaneous chromosome doubling evaluated in a diallel and recurrent selection experiment in maize. Theor Appl Genet 132(8):2273–2284. https://doi.org/10.1007/s00122-019-03353-w
Molenaar WS, Melchinger AE (2019) Production of doubled haploid lines for hybrid breeding in maize. In: Advances in breeding techniques for cereal crops. Burleigh Dodds Science Publishing, pp 143–172
Moose SP, Mumm RH (2008) Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiol 147(3):969–977. https://doi.org/10.1104/pp.108.118232
Moss DN et al (1971) CO2 compensation concentration in maize (Zea mays L.) genotypes. Plant Physiol 47(6):847–848. https://doi.org/10.1104/pp.47.6.847
Nie W, Wang J (2021) Actin-related protein 4 interacts with pie1 and regulates gene expression in arabidopsis. Genes. https://doi.org/10.3390/genes12040520
Nunes-Nesi A et al (2016) Natural genetic variation for morphological and molecular determinants of plant growth and yield. J Exp Bot 67(10):2989–3001
Oh SA et al (2016) Analysis of gemini pollen 3 mutant suggests a broad function of AUGMIN in microtubule organization during sexual reproduction in Arabidopsis. Plant J: Cell Mol Biol 87(2):188–201. https://doi.org/10.1111/tpj.13192
Patch LH, Bottger GT (1937) Investigations of the varietal resistance of field corn to the European corn borer in 1936. U.S. Dep. Agric. Bur. Ent. PI Quarantine, p 11
Peterson R et al (2010) A simplified method for differential staining of aborted and non-aborted pollen grains. Int J Plant Biol 1(2):66–69. https://doi.org/10.4081/pb.2010.e13
Pound MP et al (2017) Deep machine learning provides state-of-the-art performance in image-based plant phenotyping. GigaScience 6(10):1–10. https://doi.org/10.1093/gigascience/gix083
Prigge V et al (2012) New insights into the genetics of in vivo induction of maternal haploids, the backbone of doubled haploid technology in maize. Genetics 190(2):781–793. https://doi.org/10.1534/genetics.111.133066
Prigge V, Melchinger AE (2012) Production of haploids and doubled haploids in maize. In: Plant cell culture protocols, 3rd edn., pp 161–172
R Core Team (2022) R: A language and environment for statistical computing, R Foundation for Statistical Computing.
Ren J et al (2017) Novel technologies in doubled haploid line development. Plant Biotechnol J 15(11):1361–1370. https://doi.org/10.1111/pbi.12805
Ren J et al (2020) Mapping of QTL and identification of candidate genes conferring spontaneous haploid genome doubling in maize (Zea mays L.). Plant Sci 293:110337. https://doi.org/10.1016/j.plantsci.2019.110337
Roth LS et al (1970) Genetic variation of quality traits in maize (Zea mays L.) forage. Crop Sci 10(4):365–367. https://doi.org/10.2135/cropsci1970.0011183x001000040014x
RStudio Team (2022) RStudio: integrated development environment for R. http://www.rstudio.com/
Santos IG et al (2022) Usefulness of temperate-adapted maize lines developed by doubled haploid and single-seed descent methods. Theor Appl Genet 135(6):1829–1841. https://doi.org/10.1007/s00122-022-04075-2
Schmidt P et al (2019) Estimating broad-sense heritability with unbalanced data from agricultural cultivar trials. Crop Sci 59(2):525–536. https://doi.org/10.2135/cropsci2018.06.0376
Shaw D et al (2021) LoxTnSeq: random transposon insertions combined with cre/lox recombination and counterselection to generate large random genome reductions. Microb Biotechnol 14(6):2403–2419. https://doi.org/10.1111/1751-7915.13714
Slotman MA et al (2006) Reduced recombination rate and genetic differentiation between the M and S forms of Anopheles gambiae s.s. Genetics 174(4):2081–2093. https://doi.org/10.1534/genetics.106.059949
Tao T (2010) Standalone BLAST Setup for Windows PC, National Center for Biotechnology Information. https://www.ncbi.nlm.nih.gov/books/NBK52637/
Trampe B et al (2020) QTL mapping of spontaneous haploid genome doubling using genotyping-by-sequencing in maize (Zea mays L.). Theor Appl Genet 133(7):2131–2140. https://doi.org/10.1007/s00122-020-03585-1
Ullrich S, Schüler D (2010) Cre-lox-based method for generation of large deletions within the genomic magnetosome island of magnetospirillum gryphiswaldense. Appl Environ Microbiol 76(8):2439–2444. https://doi.org/10.1128/AEM.02805-09
Vanous K et al (2017) Generation of maize (Zea mays) doubled haploids via traditional methods. Curr Protocols Plant Biol. https://doi.org/10.1002/cppb.20050
Verzegnazzi AL et al (2021) Major locus for spontaneous haploid genome doubling detected by a case–control GWAS in exotic maize germplasm. Theor Appl Genet 134(5):1423–1434. https://doi.org/10.1007/s00122-021-03780-8
Wang H, Bennetzen JL (2012) Centromere retention and loss during the descent of maize from a tetraploid ancestor. Proc Natl Acad Sci USA 109(51):21004–21009. https://doi.org/10.1073/pnas.1218668109
Wickham H et al (2019) Welcome to the Tidyverse. J Open Sour Softw 4(43):1686. https://doi.org/10.21105/joss.01686
Wu P et al (2017) New insights into the genetics of haploid male fertility in maize. Crop Sci 57(2):637–647. https://doi.org/10.2135/cropsci2016.01.0017
Funding
Funding for this work was provided by USDA’s National Institute of Food and Agriculture (NIFA) Project, No. IOW04314, IOW01018, and IOW05510; NIFA award 2018–51181-28419; and the National Science Foundation under Grant No. DGE-1545453. Funding for this work was also provided by the R.F. Baker Center for Plant Breeding, Plant Sciences Institute, and K.J. Frey Chair in Agronomy at Iowa State University.
Author information
Authors and Affiliations
Contributions
Project conceptualization: L.G., M.K.M., U.K.F., and T.H.L.; Data collection: T.H.L, L.G., and M.K.M.; Data analysis: T.L.F.; Data interpretation: T.L.F., S.P., S.D., Y.C., A.S.S, M.B.H.; Manuscript writing: T.L.F and T.H.L with input from the other authors.
Corresponding author
Ethics declarations
Conflict of interest
The authors have not disclosed any competing interests. The authors have no relevant financial or non-financial interests to disclose.
Additional information
Communicated by Annaliese S Mason.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Foster, T.L., Kloiber-Maitz, M., Gilles, L. et al. Fine mapping of major QTL qshgd1 for spontaneous haploid genome doubling in maize (Zea mays L.). Theor Appl Genet 137, 117 (2024). https://doi.org/10.1007/s00122-024-04615-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-024-04615-y