
Abstract
Key message
HvBGlu3, a β-glucosidase enzyme gene, negatively influences β-glucan content in barley grains by mediating starch and sucrose metabolism in developing grains.
Abstract
Barley grains are rich in β-glucan, an important factor affecting end-use quality. Previously, we identified several stable marker-trait associations (MTAs) and novel candidate genes associated with β-glucan content in barley grains using GWAS (Genome Wide Association Study) analysis. The gene HORVU3Hr1G096910, encoding β-glucosidase 3, named HvBGlu3, is found to be associated with β-glucan content in barley grains. In this study, conserved domain analysis suggested that HvBGlu3 belongs to glycoside hydrolase family 1 (GH1). Gene knockout assay revealed that HvBGlu3 negatively influenced β-glucan content in barley grains. Transcriptome analysis of developing grains of hvbglu3 mutant and the wild type indicated that the knockout of the gene led to the increased expression level of genes involved in starch and sucrose metabolism. Glucose metabolism analysis showed that the contents of many sugars in developing grains were significantly changed in hvbglu3 mutants. In conclusion, HvBGlu3 modulates β-glucan content in barley grains by mediating starch and sucrose metabolism in developing grains. The obtained results may be useful for breeders to breed elite barley cultivars for food use by screening barley lines with loss of function of HvBGlu3 in barley breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Barley (Hordeum vulgare L.) is the fourth largest cereal crop in the world. Like oats, barley (3–10%) is characterized by high grain β-glucan content compared with other cereal crops (less than 1%) including rice, maize and wheat (Havrlentova and Kraic 2006; Marcotuli et al. 2019). β-glucan is an important non-cellulosic polysaccharide affecting the end use of barley. When barley is used as materials for malting and feed processing, low grain β-glucan content is desirable as it has a negative impact on malt extract and nutrient intake in animals, respectively (Jacob and Pescatore 2014; Kumar et al. 2022; Perera et al. 2022). In the brewing industry, a high content of β-glucan in barley may lead to insufficient degradation of cell walls, which reduces malt extract (Bamforth 1982). Residual β-glucan can also cause filtration difficulties, deteriorating beer quality (Bamforth 1982). On the other hand, β-glucan has positive roles in human nutrition and health as it can lower blood cholesterol level, blunt the postprandial rise in blood glucose, and increase vitamin bio-availability (Din et al. 2018; Geng et al. 2022; Hughes and Grafenauer 2021).
β-glucan mainly exists in sub-aleurone layers and cell walls of cereal grains. It is synthesized by members of the Cellulose synthase-like (CslF and CslH) gene families (Burton et al. 2006; Doblin et al. 2009; Love et al. 2014). HvCslF6 was considered a main gene directly involved in β-glucan synthesis in barley grains (Burton et al. 2011; Garcia-Gimenez et al. 2020). However, several studies failed to identify CslF6 gene in QTL analysis and Genome Wide Association Study (GWAS) using different genetic populations (Houston et al. 2014; Oziel et al. 1996; Panozzo et al. 2007). Thus, the exploration of genes controlling β-glucan synthesis in addition to CslF6 is imperative.
Previously, we performed GWAS on grain β-glucan content of 100 accessions from International Barley Core Selected Collection (IBCS) planted in different environments (Geng et al. 2021). Unexpectedly, we failed to identify the CslF6 gene, but found some novel genetic loci and candidate genes associated with β-glucan content. Among them, a gene HORVU3Hr1G096910, encoding β-glucosidase 3, named HvBGlu3, was identified to be involved in the regulation of β-glucan content in barley grains. β-glucosidases have been well characterized and classified into glycoside hydrolase (GH) families GH1, GH3, GH5, GH9, and GH30 (see CAZy database www.cazy.org), with GH1 being most extensively investigated (Cairns and Esen 2010).
Glycosyl hydrolase (GH) family enzymes are widely present in prokaryotes and eukaryotes (Cantarel et al. 2009; Henrissat 1991). GH, also referred to as glycoside hydrolase or glycosidase, is a highly functional class of enzymes that can hydrolyze glycosidic bonds connecting two or more carbohydrates, or between carbohydrates and non-carbohydrates (Chen et al. 2020; Sathya and Khan 2014). Carbohydrates have unique functions in biological systems, including energy storage, signal transduction and intracellular transportation. They are also the main final products of plant primary production, accounting for the majority of plant fixed carbon (Lichtenthaler 2007). β-glucan is one of the most abundant carbohydrates in barley grains. There should be a certain correlation between β-glucan and other carbohydrates, including starch, sucrose and glucose, as they are synthesized through sugar metabolism. However, little is known about the catalytic and regulatory effects of GH enzyme on β-glucan.
In this study, CRISPR/Cas9 gene editing technology was used to obtain HvBGlu3 knockout mutants to confirm the hypothesis that GH family genes are involved in β-glucan synthesis and accumulation in barley grains.
Materials and methods
Barley material and growth conditions
Both knockout mutants and wild-type Gold Promise (GP) were cultivated in plant growth chambers of Zhejiang University. The growth conditions were set as light 14 h/22 °C and darkness 10 h/18 °C. Both wild type and mutant lines of T2 generation were planted in pots with one seedling in each pot. Four seedlings from each genotype or line were taken as a biological replicate, and there were three biological replicates. After heading, grains were collected from the middle of the spike on the 7th day post-anthesis (DPA). Then, they were quick-frozen in liquid nitrogen and stored in a freezer at − 80 °C until further use. At maturity, grains of mutant and wild-type plants were harvested, dried in an oven at 37 °C for 5 days, and stored in a cold storage at 0 °C for subsequent experiments.
Protein domain analysis and phylogenetic analysis
The protein sequence of HvBGlu3 was downloaded from the Ensemble database (http://plants.ensembl.org/index.html). The protein domain analysis was carried out using InterPro online website (http://www.ebi.ac.uk/interpro/scan.html).
Homologous protein sequences of HvBGlu3 obtained from 1KP database (https://db.cngb.org/onekp/) and Ensembl databases were used to conduct phylogenetic analysis. Protein sequences were aligned using MAFFT online website (https://mafft.cbrc.jp/alignment/server/index.html). A phylogenetic tree was structured with FastTree using the maximum likelihood. Finally, ITOL (Interactive Tree Of Life) online website (https://itol.embl.de) was used to beautify the evolutionary tree.
Tissue-specific expression of HvBGlu3
To determine tissue expression of HvBGlu3, different tissues at different developmental stages of Golden Promise (GP) were sampled. The tissues include radicles and seeds of germinated barley grains; roots and leaves of barley seedlings at two-leaf stage; roots, leaves, first tillers and shoot tips of barley plants at tillering stage; nodes, internodes and leaves of barley plants at jointing stage; nodes, internodes, leaves and spikes of barley plants at booting stage; and grains at 7 DPA, 14 DPA, 21 DPA and 28 DPA of barley plants at grain filling stage. The total RNA of samples was extracted by the MiniBEST kit (Takara, Japan). cDNA was synthesized using PrimeScript™ RT reagent kit (Takara, Japan), and quantitative real-time PCR (qRT-PCR) was performed using TB Green Premix Ex Taq (Tli RNase H Plus) (Takara, Japan) on LightCycler 480. The expression of HvBGlu3 was calculated using the 2–ΔΔCT method with GAPDH (glyceraldehyde 3-phosphate dehydrogenase), actin and α-tubulin used as housekeeping genes. The primers used in the study are listed in Table S1. There were three biological replicates and three technical replicates for each expression analysis.
Subcellular localization of HvBGlu3
To determine the subcellular localization of HvBGlu3, a translational HvBGlu3-sGFP (super-folder green fluorescent protein) fusion was constructed. The coding region of HvBGlu3 without the stop codon was amplified by PCR. Then, the cloned product was fused into a frame before sGFP driven by the 35S promoter in the pCAMBIA-1300-35S-sGFP-Nos vector by the homologous recombination cloning method with ClonExpress® II One Step Cloning Kit (Vazyme). The constructs of HvBGlu3-sGFP or sGFP alone, co-transformed with RFP (a biofilm-localized marker), were transformed into barley leaf protoplasts (Feng et al. 2020). The plasmids of HvBGlu3-sGFP, sGFP, and biofilm-localized RFP marker were transformed into Agrobacterium tumefaciens strain EHA105. Finally, these two plasmids were mixed with RFP marker separately to infect 4-week-old tobacco (Nicotiana benthamiana) leaves. Fluorescence signals were observed under a LSM780 confocal microscope (Zeiss). The primers used are listed in Table S1.
Generation of HvBGlu3 knockout barley plants
The coding sequence (CDS) of HvBGlu3 was put into targetDesign online website (http://skl.scau.edu.cn/targetdesign/) to design the sgRNA sequences. Then, off-target detection was conducted on Cas-OFFinder (http://www.rgenome.net/cas-offinder/). Finally, two sgRNA sequences were obtained, i.e., gRNA1: ACACATATTGCAATCCTTCC GGG; gRNA2: CCTATCTATGTGCAAGAGAA TGG. The sgRNAs were cloned into the pRGEB32 vector (Figure S1) (Xie et al. 2015). The knockout construct was transformed into AGL1 Chemical Competent Cell (Shanghai Weidi Biotechnology Co., Ltd.), and the genetic transformation of barley cultivar GP was performed according to Ye et al. (Ye et al. 2019). Genomic DNA from the transgenic plants was extracted, and the knockout mutations were identified by PCR amplification and DNA sequencing. The plants of wide type (GP) were used as controls. The primers used are listed in Table S1.
Determination of β-glucan, total starch, total dietary fiber and protein contents
Mature grains (around 20 g) of both T2 knockout mutant and GP plants (T3 grains) were milled into flour for quantification of β-glucan, total starch, total dietary fiber and protein. β-glucan content was determined according to Houston et al. (2014) using a modified version of Megazyme mixed-linkage β-glucan Assay Kit (K-BGLU). Total starch content was measured using Megazyme Total Starch Assay Kit (K-TSTA). Total dietary fiber was determined by enzymatic–gravimetric method (McCleary et al. 2012). Protein content was measured using the method described previously (Ye et al. 2016). Three biological replicates were performed for each measurement.
Transcriptome analysis
Total RNA of grains at 7 DPA was extracted from both mutant line hvbglu3-m2 and WT plants to construct RNA libraries. RNA was sequenced using the Illumina NovaSeq 6000 (Illumina, USA). Clean reads were mapped to the barley reference genome using the Hisat2 software (Kim et al. 2019). The details of reads mapping are shown in Table S2. The expression levels of genes (FPKM, fragments per kilobase million) were normalized, and the fold change (FC) between hvbglu3-m2 and WT was calculated according to Love et al. (Love et al. 2014). Differently expressed genes (DEGs) were selected with the criterion of |FC| =|log2(hvbglu3-m2 FPKM/WT FPKM)|≥ 1 (adjusted P value ≤ 0.05). GO and KEGG analysis of DEGs were performed using the clusterProfiler R package based on the GO database (http://geneontology.org/) and KEGG database (http://www.genome.jp/kegg).
qRT-PCR analysis
To verify the effect of base insertions or deletions in knockout mutations, qRT-PCR validation was performed on the DEGs involved in starch and sucrose metabolism in all mutant lines. qRT-PCR was performed as described in the tissue-specific expression of HvBGlu3. The primers used are listed in Table S1.
Quantification of sugar content
Sugar content in grains at 7 DPA was quantified by HPLC and GC–MS based on the Agilent 8890-5977B platform, according to the methods described by Zheng et al. (2016).
Statistical analysis
Statistical analysis was conducted through one-way ANOVA by using SPSS software (IBM SPSS Statistics 20.0). Significant and highly significant differences were set at P < 0.05 and P < 0.01, respectively. Data visualization of the bar plots and heat map was created using GraphPad Prism v8.0.1 and TBtools v1.064, respectively.
Results
Phylogenetic analysis on HvBGlu3 protein family
Conserved domain analysis demonstrated that the amino acid sequence, ranging from 25 to 487th position in HvBGlu3, exhibited a distinct region of the Glyco_hydro_1 domain, a distinctive feature of glycoside hydrolase family 1. Thus, HvBGlu3 is classified as GH1, encoding β-glucosidase (Fig. 1A).

Phylogenetic tree and protein analysis of HvBGlu3. A The structure of HvBGlu3 protein. Different colors of bars indicate domains of the protein. Glyco_hydro_1, Glycoside hydrolase family 1; Glycoside_hydrolase_SF, Glycoside hydrolase superfamily; Glyco_hydro_1_N_CS, Glycosyl hydrolases family 1, N-terminal conserved site. B Phylogenetic tree of HvBGlu3 proteins in 67 representative species of the major lineage of green plants. The maximum likelihood was used to construct the tree. C Phylogeny of the HvBGlu3 protein family. A phylogenetic tree was built using amino acid sequences of predicted proteins from Arabidopsis, tomato, maize, sorghum, wheat, B. distachyon, barley and rice using the maximum likelihood
Totally, 67 homologous protein sequences of HvBGlu3 were obtained from the 1KP and Ensemble database. Phylogenetic analysis of these homologous protein sequences showed that HvBGlu3 homologous genes are widely distributed in various green plants and algae (Fig. 1B). HvBGlu3 likely originates from the ancestors of the moss Neckera douglasii. Meanwhile, evolutionary analysis was also carried out on the HvBGlu3 homologous protein sequences from eight common plants, i.e., barley, corn, rice, wheat, Arabidopsis, soybean, tomato and sorghum. The results showed that the homologous genes from wheat genome shared the closest genetic relationship with HvBGlu3. Moreover, another homologous gene in barley genome, namely HORVU.MOREX.r3.3HG0312790, shared 79.6% similarity to HvBGlu3 in the amino acid sequence (Fig. 1C), indicating partial conservation of HvBGlu3 protein family.
Expression patterns and subcellular localization of HvBGlu3
The qPCR results showed that HvBGlu3 was expressed during the whole development of barley. The relatively high expression level of HvBGlu3 was found in leaves at various development stages (germination, seedling, tillering, jointing, booting) and in spikes at booting stage (Fig. 2A). Particularly, the highest expression level was detected in leaves at tillering stage, and the expression level was relatively lower in grains.

Expression pattern analysis and subcellular localization of HvBGlu3. A Relative expression of HvBGlu3 in developing barley plants. Data were means (± SD), n = 3. B Subcellular localization of HvBGlu3 in barley leaf protoplasts. Scale bars are 5 μm. C Subcellular localization of HvBGlu3 in leaf epidermal cells of N. benthamiana. Scale bars are 50 um
The results of subcellular localization showed that fluorescence signals of sGFP and RFP were observed in the biomembrane (including cell membrane and cell nuclear membrane) of mesophyll protoplasts in barley leaves when GFP and RFP were co-expressed (Fig. 2B). In addition, when HvBGlu3-sGFP and RFP were co-expressed in epidermal cells of Nicotiana benthamiana leaves, GFP signals could be detected in the nucleus membrane and plasma membrane, overlapping with marker RFP (Fig. 2C). These results indicated that HvBGlu3 is mainly localized in cell membranes.
CRISPR-targeted mutations of HvBGlu3
CRISPR/Cas9-based gene editing technology was used to examine the roles of HvBGlu3 in regulating β-glucan content in barley grains. There were 13 exons and 12 introns in HvBGlu3 (Fig. 3A). According to the PAM searching result, two gRNAs were designed in the 11th exon. CRISPR-targeted mutations were identified by Sanger sequencing with GP used as control. A total of 29 plants were obtained from T0 generation. According to DNA sequencing, several plants were detected in the heterozygous mutation state. Then, heterozygous mutant plants were planted and self-cross-screened to identify homozygous mutants in T1 generation. Finally, three homozygous CRISPR-targeted mutants were identified and they differed in the sizes of base insertion or deletion at two sites. As a result, three homozygous mutants, named hvbglu3-m1, hvbglu3-m2 and hvbglu3-m3, respectively, were obtained. For hvbglu3-m1, there were a 1-bp deletion at site 1 and site 2, a 48-bp insertion at site 1; for hvbglu3-m2, a 1-bp deletion was found at site 1 and site 2, respectively. For hvbglu3-m3, there was a 4-bp deletion at site 2. All three mutants caused the early termination of HvBGlu3 translation (Fig. 3B).

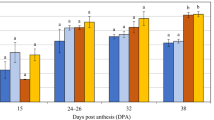
Schematic diagram of HvBGlu3 and targeted mutants induced by CRISPR/Cas9. A Gene structure of HvBGlu3 with two targeting sites. Exon is represented by the black box. The white boxes represent 5 ‘-UTR and 3’-UTR, respectively. The red dotted box indicates target sites. B Partial amino acid alignment of three mutant lines and WT. C Morphology of mature grains in mutants and WT; D Thousand grain weight (g); E β-glucan content (%); F Total starch content (%); G Protein content (%). Statistical significant difference was determined by one-way ANOVA (P < 0.05; n = 3; error bars indicate SD)
Loss function of HvBGlu3 increased β-glucan content in barley grains
Grain traits of T2 mutants and wide-type (GP) plants were analyzed (Table S3). Thousand grain weight of three mutants was significantly lower than WT (Fig. 3D), while three mutants had significantly higher β-glucan content in mature grains than WT, with hvbglu3-m1, hvbglu3-m2 and hvbglu3-m3 being 4.26%, 4.18% and 4.46%, respectively, increasing by 7.69%,5.56% and 9.44% compared with WT (3.96%), indicating that loss function of HvBGlu3 increases β-glucan content (Fig. 3E). In addition, the contents of total dietary fiber, total starch and protein in mature grains were also determined. The result showed that total starch content in grains of three mutants decreased slightly relative to WT, but the difference was only significant between mutant line hvbglu3-m1 and WT (Fig. 3F). Total dietary fiber content was slightly higher in three mutants than WT, but the difference was not significant (Table S3). Protein content of three mutants was significantly higher than WT (Fig. 3G). In short, HvBGlu3 negatively affects β-glucan content in barley grains, and its loss function significantly increases β-glucan content in barley mature grains, accompanied by reduced thousand grain weight and total starch content, and increased protein content.
Loss function of HvBGlu3 alters carbohydrate metabolism in developing grains
RNA-seq analysis was conducted to compare the transcriptome profiles of developing grains between mutant line hvbglu3-m2 and WT. In total, 4573 differentially expressed genes (DEGs) were identified in the mutant line compared with WT, with. 2426 DEGs being up-regulated and 2147 DEGs being down-regulated (Figure S2). KEGG analysis showed that the up-regulated DEGs were significantly enriched in starch and sucrose metabolism (Fig. 4A). The details of these up-regulated DEGs involved in starch and sucrose metabolism are shown in Table S4. Obviously, HvBGlu3 knockout did not alter the expression level of CslF family genes, except for CslF4 (HORVU2Hr1G042300). To verify the consistency of three mutant lines, qRT-PCR validation was performed on DEGs involved in starch and sucrose metabolism in all mutant lines. The results showed that these DEGs were also up-regulated in all mutant lines compared with those in WT, being consistent with the results obtained in RNA-seq analysis on mutant hvbglu3-m2 (Fig. 4B). Therefore, it may be suggested that the loss function of HvBGlu3 alters the gene expression level involved in starch and sucrose in the developing grains, but has a slight effect on the expression of CslF family genes.

Transcriptomic analysis of the grains at 7 DPA in HvBGlu3 knockout line. A KEGG pathway enrichment analysis of up-regulated DEGs between the hvbglu3-m2 and WT. Red pentacles indicate significantly enriched KEGG pathways. B Heat map of fold changes (FC) for DEGs involved starch and sucrose metabolism in all mutant lines
Accordingly, we assume that loss function of HvBGlu3 might change sugar metabolism of developing grains in the mutant lines. Furthermore, we measured the contents of 12 sugars in 7 DPA grains of both mutant lines and WT (Table S5). The result showed that there were 6 and 2 sugars with mutant lines being significantly higher and lower than WT, respectively (Fig. 5). Obviously, the loss function of HvBGlu3 alters the content of the individual sugar by affecting starch and sucrose metabolisms in developing grains, also leading to the change of β-glucan accumulation in mature grains.

Fold changes (FC) of grain sugar levels in knockout mutant lines compared to WT at 7 DPA. Statistical significant difference was determined by one-way ANOVA (P < 0.05; n = 3)
Discussion
β-Glucosidase, an enzyme hydrolyzing glycosidic bonds and releasing non-reducing terminal glucosyl residues from glycosides and oligosaccharides, exists universally in bacteria, fungi and eukaryotes, and plays a variety of biological functions in plants, including defense, symbiosis, cell wall catabolism and lignification, signaling and secondary metabolism (Cairns and Esen 2010). It has been well characterized and classified into glycoside hydrolase (GH) families GH1, GH3, GH5, GH9 and GH30 (see CAZy database www.cazy.org), with GH1 being most extensively investigated (Brunner et al. 2002; Cairns and Esen 2010; Cantarel et al. 2009; Henrissat 1991; Opassiri et al. 2007). According to the protein domain analysis in this study, the 25–487 amino acid sequence of HvBGlu3 protein is Glyco_hydro_1 domain, belonging to GH1 family. Therefore, it may be assumed that HvBGlu3 belonging to GH1 family encodes a β-glucosidase.
Plant cell wall is a reservoir of many carbohydrates, and one of them is β-linked glucosyl residues. Therefore, it is conceivable that β-glucosidases may play an important role in the development of cell wall. To date, some β-glucosidases that hydrolyze cell-wall-derived oligosaccharides have been identified in barley (Hrmova et al. 2006, 1996; Leah et al. 1995) and rice (Opassiri et al. 2006). It is also reported that β-glucosidases hydrolyze cellobiose to glucose, a crucial step in glucose synthesis from any cellulose (Hill and Reilly 2008). However, little has been known about the specific function of β-glucosidases in regulating β-glucan synthesis and accumulation, although β-glucan is one of major polysaccharides in barley cell wall.
β-Glucan in plant cell wall is mainly controlled by synthetic genes, while it is also affected by remodeling and degradation of polysaccharides. In fact, the genes encoding hydrolyzing enzymes were also highly expressed during the synthesis of cell wall polysaccharides (Fincher 2009). In addition, some members of the GH family (including GH1, GH5, GH9, GH16 and GH17) were found to be associated with β-glucan content in barley grains (Houston et al. 2014). Szyjanowicz et al. found that mutations in the KORRIGAN (KOR) gene encoding β,1–4 endoglucanase affected the secondary cell wall of Arabidopsis thaliana (Szyjanowicz et al. 2004). It was suggested that the synthesis of β-glucan involved many other enzymes besides CslF and CslH (Burton et al. 2010). Based on two independent QTL and GWAS studies of β-glucan content in barley grains, HvCslF9 and HvGlbI are co-located at the same association peak, at 48.1 cM and 54.4 cM of 1H, respectively (Han et al. 1995; Houston et al. 2014). However, neither overexpression nor loss function of CslF9 affects β-glucan content in barley grains (Burton et al. 2011; Garcia-Gimenez et al. 2020). Therefore, it may be suggested that β-glucan content in barley grains is mainly attributed to HvGlbI rather than HvCslF9. In addition, it was found that β-glucan content in barley grains is controlled by the genes encoding corresponding synthase and endohydrolase enzymes (Garcia-Gimenez et al. 2019). In short, the current results indicate that hydrolases play an important role in determining β-glucan content in barley grains.
This study highlights that HvBGlu3 may regulate β-glucan content in barley grains by co-working with other enzymes involved in remodeling or degradation of cell wall polysaccharides. Accordingly, transcriptome analysis was performed on the developing grains of mutant line hvbglu3-m2 and WT. The results showed that the up-regulated DEGs were significantly enriched in starch and sucrose metabolism pathways. In particular, some genes encoding starch synthetase (HORVU7Hr1G038420, HORVU7Hr1G012380) and starch branching enzyme (HORVU2Hr1G077120, HORVU0Hr1G022790, HORVU7Hr1G082830) were significantly up-regulated in the HvBGlu3 knockout mutant, indicating a regulatory link between starch and β-glucan. In fact, the close association between starch and β-glucan content has been confirmed recently. Barley with low starch content, such as high-amylose barley or waxy barley, usually shows high β-glucan content (Izydorczyk et al. 2000). It was also reported that β-glucan content in barley grains was obviously changed in the mutants of starch biosynthesis gene (Clarke et al. 2008; Munck et al. 2004). Over-expression of CslF6 gene under the control of an endosperm-specific promoter resulted in almost doubling of grain β-glucan content, accompanied by a significant reduction of starch content (Burton et al. 2011). Interestingly, β-glucan content in grains of Brachypodium distachyon was up to 40% and starch content is only about 6%, which provides additional support for a regulatory link between starch and β-glucan synthesis (Guillon et al. 2011; Trafford et al. 2013). The link between β-glucan and starch synthesis is possibly related to the regulation of sugar nucleotide levels, as ADP-Glc and UDP-Glc are glucose donors for starch and β-glucan synthesis, respectively (Trafford et al. 2013). In conclusion, knockout of HvBGlu3 enhances starch and sucrose metabolism in developing grains, thus affecting sugar metabolism and finally increasing β-glucan content in mature grains.
Unfortunately, the current results failed to make a comprehensive understanding of how β-glucosidase is involved in β-glucan regulation, as the detected sugars in developing grains are quite limited in this study. More sugars should be detected using more advanced technology or methods in future research in order to reveal the influence of β-glucosidase on the composition of sugars as well as β-glucan content. In addition, as carbohydrate metabolism is dramatically changing during grain development, the transcriptome and sugar metabolism of whole grain development should be analyzed to reveal the regulation mechanism of β-glucosidase on β-glucan content. On the other hand, most available research is focused on the genes of β-glucan synthesis (Garcia-Gimenez et al. 2020), and little attention has been paid on β-glucan-degrading related genes. In our understanding, this is the first report about the gene negatively regulating β-glucan content, confirming that β-glucosidase is involved in the regulation of β-glucan accumulation in barley grains. It is suggested that the genes encoding this enzyme can be used for breeding to increase the content of β-glucan in grains of barley and other cereal crops. Particularly, it is desirable to breed elite barley cultivars for food use by screening barley lines with loss of function of HvBGlu3, as the negative influences of the gene on β-glucan content in barley grains.
References
Bamforth CW (1982) Barley β-glucans: their role in malting and brewing. Brew Dig 3:22–35
Brunner F, Wirtz W, Rose JKC, Darvill AG, Govers F, Scheel D, Nürnberger T (2002) A β-glucosidase/xylosidase from the phytopathogenic oomycete, Phytophthora infestans. Phytochemistry 59:689–696
Burton RA, Wilson SM, Hrmova M, Harvey AJ, Shirley NJ, Medhurst A, Stone BA, Newbigin EJ, Bacic A, Fincher GB (2006) Cellulose synthase-like CslF genes mediate the synthesis of cell wall (1,3;1,4)-β-D-glucans. Science 311:1940–1942
Burton RA, Gidley MJ, Fincher GB (2010) Heterogeneity in the chemistry, structure and function of plant cell walls. Nat Chem Biol 6:724–732
Burton RA, Collins HM, Kibble NAJ, Smith JA, Shirley NJ, Jobling SA, Henderson M, Singh RR, Pettolino F, Wilson SM, Bird AR, Topping DL, Bacic A, Fincher GB (2011) Over-expression of specific HvCslF cellulose synthase-like genes in transgenic barley increases the levels of cell wall (1,3;1,4)-β-d-glucans and alters their fine structure. Plant Biotechnol J 9:117–135
Cairns J, Esen A (2010) β-Glucosidases. Cell Mol Life Sci 67:3389–3405
Cantarel BI, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B (2009) The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucl Acids Res 37:D233–D238
Chen H, Jin X, Zhu L, Lu Y, Ma Z, Liu S, Chen X (2020) Glycosyl hydrolase catalyzed glycosylation in unconventional media. Appl Microbiol Biot 104:9523–9534
Clarke B, Liang R, Morell MK, Bird AR, Jenkins CLD, Li Z (2008) Gene expression in a starch synthase IIa mutant of barley: changes in the level of gene transcription and grain composition. Funct Integr Genomics 8:211–221
Din A, Chughtai MFJ, Khan MRK, Shahzad A, Khaliq A, Nasir MA (2018) Nutritional and functional perspectives of barley β-glucan. Int Food Res J 25:1773–1784
Doblin MS, Pettolino FA, Wilson SM, Campbell R, Burton RA, Fincher GB, Newbigin E, Bacic A (2009) A barley cellulose synthase-like CSLH gene mediates (1,3;1,4)-β-D- glucan synthesis in transgenic Arabidopsis. P Natl Acad Sci USA 106:5996–6001
Feng X, Liu W, Qiu CW, Zeng F, Wang Y, Zhang G, Chen ZH, Wu F (2020) HvAKT2 and HvHAK1 confer drought tolerance in barley through enhanced leaf mesophyll H+ homoeostasis. Plant Biotechnol J 18:1683–1696
Fincher GB (2009) Exploring the evolution of (1,3;1,4)-β-d-glucans in plant cell walls: comparative genomics can help! Curr Opin Plant Biol 12:140–147
Garcia-Gimenez G, Russell J, Aubert MK, Fincher GB, Burton RA, Waugh R, Tucker MR, Houston K (2019) Barley grain (1,3;1,4)-β-glucan content: effects of transcript and sequence variation in genes encoding the corresponding synthase and endohydrolase enzymes. Sci Rep 9(1):17250
Garcia-Gimenez G, Barakate A, Smith P, Stephens J, Khor SF, Doblin MS, Hao P, Bacic A, Fincher GB, Burton RA, Waugh R, Tucker MR, Houston K (2020) Targeted mutation of barley (1,3;1,4)-β-glucan synthases reveals complex relationships between the storage and cell wall polysaccharide content. Plant J 104:1009–1022
Geng L, Li M, Xie S, Wu D, Ye L, Zhang G (2021) Identification of genetic loci and candidate genes related to β-glucan content in barley grain by genome-wide association study in international barley core selected collection. Mol Breed 41:1–12
Geng L, Li M, Zhang G, Ye L (2022) Barley: a potential cereal for producing healthy and functional foods. Food Quality and Safety 6:fyac012
Guillon F, Bouchet B, Jamme F, Robert P, Quéméner B, Barron C, Larré C, Dumas P, Saulnier L (2011) Brachypodium distachyon grain: characterization of endosperm cell walls. J Exp Bot 62:1001–1015
Han F, Ullrich SE, Chirat S, Menteur S, Jestin L, Sarrafi A, Hayes PM, Jones BL, Blake TK, Wesenberg DM, Kleinhofs A, Kilian A (1995) Mapping of β-glucan content and β-glucanase activity loci in barley grain and malt. Theor Appl Genet 91:921–927
Havrlentova M, Kraic J (2006) Content of β-D-glucan in cereal grains. J Food Nutr Res-Slov 45:97–103
Henrissat B (1991) A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J 280:309–316
Hill AD, Reilly PJ (2008) Computational analysis of glycoside hydrolase family 1 specificities. Biopolymers 89:1021–1031
Houston K, Russell J, Schreiber M, Halpin C, Oakey H, Washington JM, Booth A, Shirley N, Burton RA, Fincher GB, Waugh R (2014) A genome wide association scan for (1,3;1,4)-β- glucan content in the grain of contemporary 2-row Spring and Winter barleys. BMC Genomics 15:1–15
Hrmova M, Harvey AJ, Wang J, Shirley NJ, Jones GP, Stone BA, Høj PB, Fincher GB (1996) Barley β-D-glucan exohydrolases with β-D-glucosidase activity: purification, characterization, and determination of primary structure from a cDNA clone. J Biol Chem 271:5277–5286
Hrmova M, Burton RA, Biely P, Lahnstein J, Fincher GB (2006) Hydrolysis of (1,4)-β-D-mannans in barley (Hordeum vulgare L.) is mediated by the concerted action of (1,4)-β-D-mannan endohydrolase and β-D-mannosidase. Biochem J 399:77–90
Hughes J, Grafenauer S (2021) Oat and barley in the food supply and use of beta glucan health claims. Nutrients 13:2556
Izydorczyk MS, Storsley J, Labossiere D, MacGregor AW, Rossnagel BG (2000) Variation in total and soluble β-glucan content in hulless barley: effects of thermal, physical, and enzymic treatments. J Agric Food Chem 48:982–989
Jacob JP, Pescatore AJ (2014) Barley β-glucan in poultry diets. Ann Transl Med 2:20
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol 37:907–915
Kumar D, Narwal S, Venkatesh K, Verma RPS, Singh GP (2022) Grain beta-glucan as selection criteria for wort beta-glucan in malt barley improvement. J Cereal Sci 107:103519
Leah R, Kigel J, Svendsen I, Mundy J (1995) Biochemical and molecular characterization of a barley seed β-glucosidase. J Biol Chem 270:15789–15797
Lichtenthaler FW (2007) Carbohydrates as renewable raw materials: a major challenge of green chemistry. Willey, pp 23–63
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:1–21
Marcotuli I, Colasuonno P, Cutillo S, Simeone R, Blanco A, Gadaleta A (2019) β-glucan content in a panel of Triticum and Aegilops genotypes. Genet Resour Crop Ev 66:897–907
McCleary BV, DeVries JW, Rader JI, Cohen G, Prosky L, Mugford DC, Champ M, Okuma K (2012) Determination of insoluble, soluble, and total dietary fiber (CODEX definition) by enzymatic-gravimetric method and liquid chromatography: collaborative study. J Aoac Int 95:824–844
Munck L, Møller B, Jacobsen S, Søndergaard I (2004) Near infrared spectra indicate specific mutant endosperm genes and reveal a new mechanism for substituting starch with (1→3,1→4)-β-glucan in barley. J Cereal Sci 40:213–222
Opassiri R, Pomthong B, Onkoksoong T, Akiyama T, Esen A, Ketudat Cairns JR (2006) Analysis of rice glycosyl hydrolase family 1 and expression of Os4bglu12 β-glucosidase. Bmc Plant Biol 6:1–19
Opassiri R, Pomthong B, Akiyama T, Nakphaichit M, Onkoksoong T, Ketudat Cairns M, Ketudat Cairns JR (2007) A stress-induced rice (Oryza sativa L.) β-glucosidase represents a new subfamily of glycosyl hydrolase family 5 containing a fascin-like domain. Biochem J 408:241–249
Oziel A, Hayes PM, Chen FQ, Jones B (1996) Application of quantitative trait locus mapping to the development of winter-habit malting barley. Plant Breed 115:43–51
Panozzo JF, Eckermann PJ, Mather DE, Moody DB, Black CK, Collins HM, Barr AR, Lim P, Cullis BR (2007) QTL analysis of malting quality traits in two barley populations. Aust J Agric Res 58:858–866
Perera WNU, Abdollahi MR, Zaefarian F, Wester TJ, Ravindran V (2022) Barley, an undervalued cereal for poultry diets: limitations and opportunities. Animals 12:2525
Sathya TA, Khan M (2014) Diversity of glycosyl hydrolase enzymes from metagenome and their application in food industry. J Food Sci 79:2149–2156
Szyjanowicz PMJ, McKinnon I, Taylor NG, Gardiner J, Jarvis MC, Turner SR (2004) The irregular xylem 2 mutant is an allele of korrigan that affects the secondary cell wall of Arabidopsis thaliana. Plant J 37:730–740
Trafford K, Haleux P, Henderson M, Parker M, Shirley NJ, Tucker MR, Fincher GB, Burton RA (2013) Grain development in Brachypodium and other grasses: possible interactions between cell expansion, starch deposition, and cell-wall synthesis. J Exp Bot 64:5033–5047
Xie K, Minkenberg B, Yang Y (2015) Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc Natl Acad Sci USA 112:3570–3575
Ye L, Huang Y, Li M, Li C, Zhang G (2016) The chemical components in malt associated with haze formation in beer. J Inst Brew 122:524–529
Ye L, Wang Y, Long L, Luo H, Shen Q, Broughton S, Wu D, Shu X, Dai F, Li C, Zhang G (2019) A trypsin family protein gene controls tillering and leaf shape in barley. Plant Physiol 181:701–713
Zheng H, Zhang Q, Quan J, Zheng Q, Xi W (2016) Determination of sugars, organic acids, aroma components, and carotenoids in grapefruit pulps. Food Chem 205:112–121
Acknowledgements
We thank Professor Kabin Xie from Huazhong Agricultural University for providing the pRGEB32 plasmid vector and the technical support in sugar determination from MetWare Co., Ltd. (http://www.metware.cn/, Wuhan, China)
Funding
This work was funded by the Key Research Foundation of Science and Technology Department of Zhejiang Province of China (2021C02057, 2020C02002, 2021C02064-3), the National Natural Science Foundation of China (32171917), the Science and Technology Program of Zhejiang Province of China (LGN20C130007) and China Agriculture Research System (CARS-05).
Author information
Authors and Affiliations
Contributions
GZ and LY developed the experimental design and conceived the project. LG, ML, SX, XH, HW and NS performed the experiments. LG, ML and SX conducted and validated the phenotypic experiments. LG, LY and GZ wrote the manuscript. All authors discussed the results and contributed to the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Communicated by Kevin Smith.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Geng, L., Li, M., Xie, S. et al. HvBGlu3, a GH1 β-glucosidase enzyme gene, negatively influences β-glucan content in barley grains. Theor Appl Genet 137, 14 (2024). https://doi.org/10.1007/s00122-023-04517-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-023-04517-5