
Abstract
The barley shrunken grain mutant M292 has a novel high-amylose starch phenotype caused by a mutation in the starch synthase IIa gene (SsIIa) located at the starch excess-6 (sex6) locus on chromosome 7H of barley. The loss of SSIIa enzyme activity leads to a decrease in amylopectin synthesis to less than 20% of the levels found in wild-type grains. Detailed composition analysis indicates that the contents of protein, non-starch polysaccharides, lipid, sucrose and hexoses, and fructo-oligosaccharides are increased in mature M292 grain compared to wild type. Using a microarray analysis, we characterize the differences between the transcription profiles of wild-type and mutant barley endosperms at mid-grain fill. The expression changes include genes involved in carbon storage, stress-related genes, and a number of transcripts with unassigned function. The changes in gene expression are discussed in terms of the altered grain composition of the mutant seed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Starch, a complex carbohydrate, is an important compact storage form for sugars produced during a plant’s growth. For the plant, it provides a carbon and energy source during diurnal cycles in the short term and for seed germination and early growth in the longer term. Starch also serves as the primary carbohydrate component in the diet of humans. The starch synthase IIa (ssIIa) mutant M292 provides a novel form of starch with benefits for human health (Morell et al. 2003; Topping et al. 2003; Bird et al. 2004a, b). An understanding of the molecular and biochemical changes involved in the accumulation of this novel starch may provide further knowledge for the modification of starch and other grain carbohydrates to improve human health.
Starch is composed of glucose molecules which form complex carbohydrate polymers that occur in two main forms, amylose and amylopectin. Starch synthesis is initiated by ADP-glucose pyrophosphorylase which catalyzes the reaction of glucose-1-phosphate, with ATP, to form ADP-glucose. Starch synthase enzymes form polymers from these ADP-glucose molecules by linking glucose residues via α-1,4-glycosidic bonds to form amylose. Branches are introduced into the chain by starch branching enzymes which cleave approximately 5% of these α-1,4-glycosidic bonds and replace them with α-1,6-glycosidic bonds to create amylopectin. These branch linkages are not random, forming clusters of branches along the chain. This architecture allows the amylopectin units to align in parallel arrays of double helices, enabling the dense packaging of glucose units and giving rise to the crystalline structure of starch (Myers et al. 2000). Defining this relatively simple pathway of starch synthesis is complicated by the presence of multiple isoforms of the above enzymes. The relative contribution of these isoforms to starch biosynthesis remains largely unknown. Comprehensive reviews of starch biosynthesis and the enzymes involved have been published recently (Kossmann and Lloyd 2000; Rahman et al. 2000; Morell et al. 2001, 2006; Smith 2001; Ball and Morell 2003; James et al. 2003; Tetlow et al. 2004).
The ssIIa mutation created a stop codon in the SsIIa gene leading to premature termination of translation of the transcript, and consequently no evidence of the SSIIa protein is found in the endosperm of this mutant (Morell et al. 2003). The loss of SSIIa activity leads to an 80% decrease in amylopectin, and the remaining amylopectin polymers in general have altered chain length distribution. Although not significantly altering the expression levels of starch synthase I or branching enzymes IIa and IIb, the incorporation of their respective protein products within the starch granules in M292 is abolished. Based on the above observations, the hypothesis was formulated that changes in the level of the SSIIa protein in the developing endosperm has pleiotropic effects on the expression of genes in a broader transcriptional network, particularly the network associated with the assembly of the starch granule.
A comparison of gene expression profiles provides a basis for examining the above hypothesis by providing information on transcriptional networks, and this may allow testing of our hypothesis which may, in turn, lead to an alternative view. Microarray data reveal the relative expression levels of large numbers of genes. As such, they provide a platform on which to establish the “differentially expressed” genes among mutant and wild-type plants. As a source of target probes, the wheat and barley complimentary DNA (cDNA) libraries (Clarke et al. 2000; Clarke and Rahman 2005) provide an extensive range of genes for this type of comparison.
In this paper, we present a comparison of the differential gene expression, of endosperm tissue at mid-grain fill, between the barley ssIIa mutant (M292), and the wild-type barley variety “Himalaya”.
Materials and methods
cDNA microarrays
The New South Wales Centre for Agricultural Genomics (NSWCAG) array contains 19,635 wheat clones and 1,613 barley clones of which 16,000 are unique (unique = less than 80% sequence similarity at the nucleotide level). The design of this array and the cDNA libraries that contributed to its construction are detailed in Clarke and Rahman (2005). The complete set of EST sequences for the clones used to construct this array are available on the GENA database at http://www.pi.csiro.au/gena.
Plant material
The M292 mutant was obtained after sodium azide treatment of the barley variety “Himalaya” (Morell et al. 2003). Both wild-type Himalaya and the mutant M292 were grown in a controlled growth cabinet at day and night temperatures of 18°C and 12°C, respectively, with a 12-h day length. Barley spikes were labeled as at anthesis 2 days after the awns first appeared through the top of the flag leaf containing the enclosed spike. Developing seeds were harvested at 20 days post-anthesis (DPA) and, after removal of the embryo, the developing endosperm was extruded through the cut surface and stored at −80°C.
Comparison of RNA transcription between wild-type Himalaya and M292
For this analysis, total RNA from developing endosperm was used. The comparison was made between the wild-type “Himalaya” (control) and mutant “M292”.
Seeds at the same age and morphological stage of development were selected from a single spike, and six to eight endosperms were dissected and pooled from these seeds to represent a sample. Seeds collected from a different spike, from either the same plant or a different plant, represented a biological replicate sample.
Extraction of total RNA, labeling, and slide hybridization
The procedures for the RNA extraction and labeling with the Cy3 and Cy5 dyes as well as slide hybridization and washing were as detailed in Clarke and Rahman (2005).
The control plant, “Himalaya” was labeled with Cy3 in the un-swapped replicates. The dye labeling of the samples was reversed for two of the replicates to minimize any bias in cDNA incorporation and photo-bleaching of the fluorescent dyes.
Statistical analysis of microarray data
The slides were scanned with a GenePix 4000A microarray scanner (Axon Instruments, Union, CA, USA). The analysis of the microarray data files was carried out using functions contained in tRMA (tools for R microarray analysis; Wilson et al. 2003). The process of analysis using tRMA is detailed in Clarke and Rahman (2005). Using the median values of the normalized log base 2 ratios for each gene in each replicate, differentially expressed genes were determined using the “FindDiffExpGenes” function at a stringency level of 1e-10. All the microarray results pertaining to these experiments are on the following web sites: http://www.pi.csiro.au/gena and http://www.ncbi.nlm.nih.gov/geo under the accession number: GSE5368.
RNA electrophoresis, probe preparation, and hybridization conditions
For the Northern analysis, a new pool of RNA was made using endosperm from a number of spikes, but selecting seeds at the same developmental stage as those used for the microarrays. For the Northern analysis, 10 μg of total RNA was used per lane. The electrophoresis conditions for the separation of RNA and the transfer to a nylon membrane, as well as the protocol for amplification and labeling of the probes for hybridization, are detailed in Clarke and Rahman (2005). The clone pTa250.2, containing the coding sequences of the ribosomal genes (Appels and Dvorak 1982), was used to estimate the uniformity of loading for the RNA from Himalaya and M292 onto the gels.
The hybridized membrane was exposed using a Fujifilm FLA-5000 series fluorescent image analyzer system, and the image obtained was analyzed using the Multi Gauge version-2 analysis software (Fuji Photo Film, 26-30 Nishiazabu, 2-Chome Minato-ku Tokyo, 106-8620, Japan). The variation in hybridization intensities between the Himalaya and M292 RNA samples was measured (pixels per square millimeter) using the Multi-Gauge software, and a correction was made for the level of background hybridization to the gel lanes.
The analysis of grain composition
Grain texture: The single-kernel characterization system 4100 (SKCS; Perten Instruments, Springfield, IL, 62707, USA) using the RACI Cereal Chemistry official testing method 12-01. Seed weight and moisture content: Seed weight as an average of 100 seeds. Seed moisture content was measured using the air oven method (AOAC Method no. 925.10, 2000). Milling: Seeds were ground to whole meal that would pass through a 0.5-mm sieve using a cyclonic mill (Cyclotec 1093, Tecator, Sweden). Total starch analysis: Megazyme method (AACC76.13) using 20 mg of whole meal for each of three replicate samples. β-glucan analysis: Megazyme method (AACC32.23) using 20 mg of whole meal for each of three replicate samples.
Estimation of amylose content by Sepharose CL-2B gel filtration
Approximately 10 mg of total starch was dissolved in 3.0 ml of 1 M NaOH and separated by molecular weight using a Sepharose CL-2B column (Regina et al. 2006). The separated fractions of starch were assayed using the starch assay kit (Sigma).
Lipid analysis
Total lipid content was assayed by nuclear magnetic resonance (NMR; Oxford 4000 NMR Magnet, Oxford Analytical Instruments Limited). For each sample, 1 g of seeds was dried at 38.8°C for 64 h. The dried seeds were measured using NMR and compared against a pure barley oil control extracted from the Himalaya mutant M292.
Protein analysis
Protein content was estimated by determining the total nitrogen content of the seed using the method of Dumas (RACI Method 02-03, 2003 ) and expressing the result as “protein” by multiplying the value obtained by a factor of 5.7. For each sample, 10 mg of whole meal was used, and the nitrogen content was determined by mass spectrometry.
Total dietary fiber assay
The gravimetric method of Prosky et al. (1985; AOAC 985.29) was used to determine total dietary fiber of whole meal flour. Duplicate samples were assayed.
Non-starch polysaccharide assay
Total neutral non-starch polysaccharides (NSP) were measured by a modification of the gas chromatographic procedure of Theander et al. (1995). The modification involved a 2-h hydrolysis with 1 M sulfuric acid followed by centrifugation to remove insoluble NSP and a further hydrolysis of the supernatant using 2 M trifluoroacetic acid for soluble NSP.
Resistant starch assay
An in vitro procedure (Bird AR CSIRO Health Sciences and Nutrition, Kintore Avenue, Adelaide, SA, 5000, Australia) was used to determine resistant starch content. Triplicate whole meal from barley mature grains, along with appropriate standards, were mixed with artificial saliva, and the resultant bolus was incubated with pancreatic and gastric enzymes at physiological pH and temperature. The amount of residual starch in the digesta was determined using conventional enzymatic and spectrophotometric techniques, and the resistant starch content of the sample was expressed as a percentage of sample weight or total starch content.
Quantification of sucrose, hexoses, and fructo-oligosaccharides
Total sugars were extracted from whole meal following the method of Lunn and Hatch (1995) with the following modification. Barley whole meal (100 mg) was extracted three times with 10 ml of 80% ethanol (v/v) in a boiling water bath for 10 min. The supernatant from each extraction was pooled and freeze-dried and resuspended in 2 ml of milliQ water. The quantities of sucrose, glucose, and fructose were measured using a colorimetric microtiter plate enzymatic assay as described (Campbell et al. 1999; Ruuska et al. 2006). Sugars and fructo-oligosaccharides were also analyzed by high-performance anion exchange chromatography (HPAEC) as described in Ruuska et al. (2006); both methods resulted in comparable values.
Maltose assay
Total sugars, extracted from barley whole meal, and maltose standard solution were assayed as described by Bernfeld (1955). Total sugars were diluted 10 to 100 times. Maltose dilutions (ten tubes) were prepared as 0.3 to 5 μmol/ml. One milliliter of each dilution of maltose (in total sugars or maltose dilutions) was mixed with 1 ml of dinitrosalicylic acid color reagent. The sugar solution was incubated at 100°C for 5 min and cooled to room temperature. Ten milliliters of reagent-grade water was added to each tube and mixed well. The samples were measured at A540 with a spectrophotometer. Maltose was also determined by HPAEC as above.
Results
Grain composition and functional parameters
To provide a basis on which to examine changes in gene expression, a detailed analysis of the chemical composition of the mature grain from both wild type (Himalaya) and the mutant (M292) was undertaken (Table 1). Individual grain weight for M292 was 80% of the weight for the Himalaya grain and the components determined in each case (excluding water) accounted for approximately 90% of this weight. The moisture was 10.2% for M292 and 10.6% for Himalaya as measured by the air oven method. There is a 1–2% difference between total seed weight and the measurements of total components, including moisture, in the barley grains.
Total starch content in M292 was 38% of that found in Himalaya with an amylose and amylopectin content of 75 and 16%, respectively. The results showed that the reduction in total starch comprised an 84% reduction in amylopectin and 25% reduction in amylose on a per grain basis, resulting in a high amylose content starch (>70%) in M292.
The contents of protein, total NSP, and lipid (measured as milligram per grain), which together make up about half the grain weight of M292, were increased in absolute amounts and as a percentage of total grain weight, by a 1.2-, 1.3-, and 1.7-fold change, respectively. The levels of free sugars were increased in M292 to an even greater extent, by about eightfold in total, with individual sugar levels increasing between 6- and 18-fold. Of the carbohydrate components which may be particularly significant in a nutritional context, β-glucan, arabinoxylans, and fructo-oligosaccharides were increased in the mutant by, 1.3-, 1.2-, and 42.0-fold, respectively. The resistant starch (the proportion of the total starch that is resistant to digestion in the human small intestine) was also increased in M292 by 4.3-fold change, milligram per grain basis. The total content of carbohydrates (including starch, total NSP, free sugar, and fructan) in M292 reduces to 22.8 from 33.5 mg present in wild-type Himalaya, milligram per grain basis.
The composition of the soluble carbohydrate components was further examined by HPAEC to study the changes in the oligosaccharides. The chromatography profiles (Fig. 1) confirm the increased content of sucrose, maltose, and hexoses in M292 as well as higher levels of a range of fructo-oligosaccharides of increasing degree of polymerization (DP) up to about 12. The wild-type Himalaya contains negligible components of fructo-oligosaccharides above a DP of about 6.

Soluble carbohydrate profile of mutant “M292” and wild-type “Himalaya” using equal amounts of material from mature barley grains. HPAE chromatographic elution profile of grain extracts indicating increased levels of hexose, sucrose, maltose, and fructo-oligosaccharides in the mutant M292 compared to wild-type Himalaya grain. Glc Glucose, Fru fructose, Suc sucrose, Mal maltose, 1-K 1-kestose; numerals indicate tentative degree of polymerization (DP) of fructo-oligosaccharides
The level of grain hardness was also measured using the SKCS to obtain an average hardness index (HI) based on measurements from 300 grains. For M292, the HI value was 109 ± 15, and the HI value for Himalaya was 84 ± 17, indicating an increase in grain hardness for the mutant.
Identification of differentially expressed genes using a cDNA microarray
Four biological replicate experiments for M292 and the control Himalaya were used to compare the transcription profiles of developing endosperm at 20 DPA. The differentially expressed genes were identified as those genes showing a twofold or greater level of differential expression at a stringency of 1e-10 using the median data of the four microarray experiments. This level of stringency was determined empirically from many microarray experiments carried out in this laboratory as that which gives the least number of false positive results when confirming the microarray results using Northern blot analysis. From the tRMA analysis, 42 clones were identified as being differentially expressed, 20 were up-regulated, and 22 were down regulated in M292 compared to Himalaya. All the clones identified were verified by sequence analysis from both the 3′ and 5′ ends to ensure that the annotation of the clone was correct. These results showed that one clone was a chimera and was not analyzed further. Using sequence data from the clones, a sequence homology search was made against the TIGR (http://www.tigr.org) tentative consensus sequence (TC) data base using either the barley or wheat gene index depending on the origin of the gene on the microarray. From this comparison, the remaining 41 cDNA clones could be associated with 23 different genes. A verification of the expression changes was undertaken using one cDNA clone as a representative for each TC using a Northern blot analysis in which RNA was isolated from a fifth replicate endosperm sample.
A subset of the results from the Northern analysis is shown in Fig. 2. The change in expression for six clones was not confirmed, and these clones were not analyzed further. Seventeen clones passed the quality control conditions, their TC sequence identity and the expression change, relative to Himalaya, for both the microarray and the Northern blot analyses are listed in Table 2. In Table 2, the genes have been grouped into three categories: carbohydrate-related, stress responsive, and genes for which a biological function has not yet been established.

Results from the Northern blot hybridizations of some of the selected genes identified from the microarray analysis. RNA loaded: left Himalaya, right M292, for each membrane shown. Various exposure times were used for each probe, the ratio of differential expression between the tracks is listed in Table 2
An unexpected outcome from these array experiments was the observation that relatively few genes involved in the starch biosynthetic pathway were differentially expressed between the mutant and wild-type seed. It is therefore of interest to note those genes on the microarray that are involved in starch biosynthesis that did not show significant differential expression in this analysis. These genes are listed in Table 3 along with their expression values, and a possible explanation for the reason that these genes were not differentially expressed is addressed in “Conclusion”.
The complete datasets from the microarray experiments used for the analyses are on the GENA database at http://www.pi.csiro.au/gena/ and http://www.ncbi.nlm.nih.gov/geo.
Discussion
Changes of grain composition in barley mutant M292
An understanding of the genes involved in the synthesis and accumulation of storage products in the developing grain is of fundamental importance in cereal crop research. Knowledge of how a plant responds to particular gene mutations provides information on the genes involved in specific pathways and how plants compensate with the perturbations these mutations cause.
The protein SSIIa is responsible for the elongation of short-chain glucose polymers with DP5-10 to form longer chains with DP11-24 (Ohdan et al. 2005). In the M292 mutant, there is no evidence of SSIIa protein activity, and this mutant has a shrunken grain in which the seed weight is reduced from an average of 46 mg in Himalaya to 36 mg in M292 (Table 1). In this study, we have extended the characterization of this mutant to determine the pleiotropic effects arising from the ssIIa mutation on the biochemical composition of the grain and the expression of the genes involved in endosperm development.
The changes to seed composition of the M292 mutant include a decrease in starch content and a modified starch composition. The reduction in starch content is coupled with significantly increased levels of free sugars, fructo-oligosaccharides, and β-glucan. Other components that have changed in the grain include an increased level of protein, total NSP, and lipid. The higher fructo-oligosaccharide levels determined here may also be important in the context of beneficial dietary outcomes. Fructo-oligosaccharides in immature durum wheat meal have been implicated, in rat feeding studies, to positively modify lipid profiles leading to decreased triglyceride and cholesterol levels (Merendino et al. 2006).
Gene expression analysis
The aim of this microarray study was to determine the gene expression changes caused by the loss of activity of the SSIIa enzyme and how this impacts on the process of grain-fill and in particular starch granule formation.
The decision to compare the transcript variation between mutant and wild-type endosperm at 20 DPA and to assume that expression at this time point would reflect final grain composition was made on the basis that the accumulation of storage compounds in the developing barley grain takes place between 8 and 25 DPA (Wobus et al. 2004). Other studies have also looked at gene expression over the grain-fill period in related cereals. Ohdan et al. (2005) profiled 27 rice genes involved in starch synthesis and found that most of these genes were highly expressed between 7 and 20 DPA. In addition, a transcription profile of the developing wheat caryopsis by Laudencia-Chingcuanco et al. (2007) found that the genes associated with starch and protein biosynthesis had maximum expression between 14 and 21 DPA.
Although transcript level is not necessarily associated with protein abundance, Giroux et al. (1994), looking at a range of mutants affecting the endosperm of maize, observed that in mutants where transcript abundance was not increased, there was no increase in the protein levels. The relatively low level of change in gene expression observed in the present array experiments corresponds with levels seen in previous work. Doehlert and Kuo (1994) examined a number of different endosperm mutants in maize and the effects of these mutations on eight different genes. Their results showed that each gene mutation had an affect on each of the eight genes examined and that the expression varied from a tenfold reduction to a threefold increase in expression compared to wild type. This level of change, measured using radiological imaging, is similar to the results presented here.
We used Northern analysis rather than reverse transcription–polymerase chain reaction (RT-PCR) to confirm the microarray expression changes because the wheat probes, used in the array, would hybridize to the homoeologous sequences in the barley RNA. Conversely, specific primers made to the wheat sequences might fail to amplify from barley cDNA in an RT-PCR. A final point on the expression changes observed in the microarray results as compared to those obtained in the Northern blot analysis relates to the fact that in Table 2, the median value of four biological replicate samples are presented for the microarray results but the Northern value is the result of one comparison. As RNAs are not stable molecules and the physiological state of the cells within and among replicates varies, this will lead to some degree of difference in gene expression among replicate samples. This variation may be reflected in the differences observed between the microarray and Northern results presented in Table 2.
Genes involved in the metabolism of carbohydrates
The principal changes in the M292 grain are the production of starch with high amylose content but a decreased amount of starch. Surprisingly, we have identified only four differentially expressed genes that have been linked to the storage of carbon through sucrose uptake in the endosperm. Of these four genes, two were up-regulated in M292 (encoding β-d-glucan exohydrolase and sucrose synthase), and two (encoding β-amylase and ssIIa) were down-regulated.
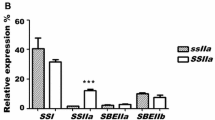
The reduction in transcription of ssIIa in the microarray analysis was confirmed by the absence of a transcript in the Northern blot analysis of M292 (Fig. 2). The smaller reduction of ssIIa transcript observed in the microarray experiment, compared to the Northern blot result, is probably due to cross-hybridization, on the microarrays, of transcripts from genes encoding other members of the starch synthase family (see Table 3 and discussions in Poole et al. 2007 on cross-hybridization among homoeologous and paralogous genes on cDNA arrays).
Why there is a reduction in β-amylase activity, and how this might relate to the disruption of starch synthesis, is unknown. However, in the developing seeds of oilseed rape, da Silva et al. (1997) have demonstrated that a plastidial form of β-amylase is active during starch synthesis, and these authors propose that changes in starch content represents a net balance between the synthetic and degradative capacity in the amyloplast.
Sucrose synthase (SuS) was up-regulated in M292 endosperm. The conversion of sucrose and UDP to form UDP-glucose and fructose is carried out by SuS in a reversible reaction. The UDP-glucose produced by SuS can be converted, via the combined action of UDP-glucose pyrophosphorylase and phosphoglucomutase, to Gluc-1-P and Gluc-6-P, which can enter glycolysis or be used in starch synthesis (Winter and Huber 2000). Alternatively, UDP-glucose can provide the substrate for the synthesis of cellulose, callose, or other cell wall polysaccharides.
Increased expression of the gene β-d-glucan exohydrolase was evident in M292. This exohydrolase has a broad range of substrate specificity which presents a problem in assigning a specific target molecule for this enzyme (Hrmova and Fincher 2001). It has been suggested that the β-d-glucan exohydrolases can be classified as polysaccharide exohydrolases because they can hydrolyze a range of polysaccharides and oligosaccharides and are usually found in situations where cell wall degradation or modification is occurring (Harvey et al. 2001; Hrmova and Fincher 2001). A possible role for this enzyme in M292 is the degradation of those endosperm cells that are not filled in the shrunken mutant grain for recycling of glucose.
Genes related to stress
In vivo protein folding is mediated by an array of proteins that act as either “foldases” or “molecular chaperones”. The stress-related genes identified in this study are all up-regulated in M292 (t-complex, HSP 70 and 80, dnaK-type, and prohibitin) and fall within the “molecular chaperone” class. The t-complex and HSP80 are active in the cytoplasm, the major site of protein synthesis in the cell (Boston et al. 1996). The changes noted above in the composition of the M292 grain, particularly the storage of large amounts of osmotically active sugars, would potentially result in changes of osmotic pressure and water potential in the grain. Part of the adjustment to this increase of vacuolar sugars may be the increase in synthesis of proteins involved in stress responsiveness, as is seen at the berry-ripening stage in grapes (Davies and Robinson 2000).
Recent studies of the mitochondrial proteome of Arabidopsis (Millar et al. 2001) revealed that the proteins of the prokaryotic dnaK-type gene (most likely the mitochondrial equivalent of HSP70) and prohibitin were localized in this structure. A study by Nijtmans et al. (2000) has shown that human prohibitins act in a protein complex as membrane-bound chaperones to stabilize mitochondrial proteins.
Genes of indeterminate function in the endosperm
Of the eight genes in this category, four were up-regulated (annexin, S-adenosylmethionine decarboxylase, O.s r40g2 protein, and a Rubber elongation factor). Four genes (clone CV060362, hordoindolin-a, serpin, and α-amylase inhibitor) had reduced expression in M292 compared to the wild-type parent.
The annexins are a family of calcium-dependent phospholipid-binding proteins found in most, if not all, eukaryotes. A study by Andrawis et al. (1993) indicated that annexins may play a role in the regulation of callose (1,3-β-glucan) synthesis. Those authors suggest that annexins may serve as an anchor for other proteins that interact with the glucan synthase complex. They draw these conclusions, in part, from the observation that ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, a chelating agent used for the determination of calcium in the presence of magnesium, eluted a set of proteins from the plasma membrane including annexins, protein kinases, and sucrose synthase. A subsequent study by Shin and Brown (1999) also confirm the essential role of Ca2+ in the entrapment of β-glucan synthase with annexins; nevertheless, their results did not confirm a role for annexins in the activity of β-glucan synthase.
S-adenosylmethionine decarboxylase (SAMDC) is a key enzyme in the biosynthesis of the polyamines spermidine and spermine, and it is also known to influence the rate of the biosynthesis of ethylene (Kumar et al. 1996). In addition, a microarray analysis of cell wall biosynthesis in loblolly pine revealed a SAMDC as being abundantly expressed in juvenile wood when compared to mature wood (Whetten et al. 2001). The role this enzyme plays in the changes observed in M292 is unknown.
The protein r40g2 was identified in a proteomic characterization of wheat amyloplasts and was isolated in the soluble fraction (Andon et al. 2002) and in both the soluble and membrane-bound fractions by Balmer et al. (2006). The differential expression of the r40g2 gene observed in our study, and the association of the protein with the amyloplast, is interesting in the context of a starch synthesis mutant influencing grain-fill in barley and warrants further study.
A cDNA clone (GenBank BE444846) fits within the TIGR (http://www.tigr.org) tentative consensus (TC) sequence TC233204 which has 49% ID to Pfam05755, a rubber elongation factor protein (REF). Other related proteins to REF are a small rubber particle protein and stress-related protein (SRP). The function of these proteins is unknown, but EST sequences representing these proteins have been isolated from a range of cereal EST libraries, including root, seed, and ovary. Given the relationship to the SRP, the up-regulation of this gene in M292 is possibly related to a stress response in the mutant seed.
In the M292 seed, there was a decrease in expression of the hordoindoline a-gene. This decrease in expression is associated with a 27% increase in grain hardness of the M292 grain. In a previous microarray analysis, Clarke and Rahman (2005) found that the puroindoline a-gene was consistently differentially expressed in a comparison between hard and soft wheat lines. It has also been noted that changes in cell wall polysaccharides also affect the hardness of the mature barley grain (Tsuchiya et al. 2005; Fincher and Stone 1986).
The cDNA clone CV60392 forms part of TC147907. Other EST sequences representing this TC were isolated from a range of EST libraries according to the “expression summary” in the TIGR db. The consensus sequence has no BLAST hit with an E score below E0.57 against either the protein or nucleotide database. This gene is, however, strongly down-regulated in M292 (Table 2).
Plant serpins (serine proteinase inhibitors) can contribute up to 4% of total protein in the cereal grain, but their physiological function remains unknown (Østergaard et al. 2000). However, Guerin et al. (1992) identified a protein Z type serpin coupled with β-amylase in a bound inactive complex associated with the barley starch granule. In this study, the transcript levels of both serpin (protein Z type) and β-amylase are reduced in the mutant line, indicating that these proteins may be coordinately regulated to ensure inactive β-amylase in the developing grain.
The expression of the α-amylase inhibitor (BDAI-I type) gene is also reduced in M292. The α-amylase inhibitors have been linked to the inhibition of α-amylase activity in the gut of insect that utilize starch as an energy source (Franco et al. 2000). It has been suggested that the production of α-amylase inhibitors is primarily a defense mechanism that has evolved in plants to control insect pests that attack seeds (Konarev 1996). The reduction in expression of the α-amylase inhibitor in M292 could be related to the reduced starch content.
Conclusion
This work provides an overview of the transcriptional changes that occur in the endosperm of the mutant barley M292 where the activity of SSIIa has been abolished. Northern blot analysis, used to confirm these changes, verified the general trends observed on the microarray. A striking feature of this comparison is the small number of transcriptional changes observed between the mutant and wild type seed. Considering the changes in grain composition, detailed above, we expected to see a greater influence on gene regulation.
A possible explanation for the relatively few expression changes observed in the ssIIa mutant could be that only a limited number of alternative pathways are available for carbon storage in the developing grain. In wild-type Himalaya, sucrose transported into the grain can be efficiently converted to starch for storage. However, in M292, this pathway is disrupted by the ssIIa mutation, causing a 25% reduction in amylose content and an 84% reduction in amylopectin. Previous work (Schulman et al. 1995) suggests that there may be a limit to the total proportion of starch which may be synthesized as amylose. Therefore, the failure of the mutant to utilize the available sugars in starch production would lead to their accumulation in the grain (Doehlert and Kuo 1994). This accumulation of sugars would alter the osmotic conditions resulting in the up-regulation of a number of stress-related genes, as observed. The only other major form of carbohydrate in the grain is the NSP, 16.7% of the total carbohydrate in wild-type, and 32.5% in the mutant grain. The small increase in expression of the carbohydrate-synthesizing genes, observed here, may be related to the increase in NSP.
References
Andon NL, Hollingworth S, Koller A, Greenland AJ, Yates JR, Haynes PA (2002) Proteomic characterization of wheat amyloplasts using identification of proteins by tandem mass spectrometry. Proteomics 2:1156–1168
Andrawis A, Solomon M, Delmer DP (1993) Cotton fiber annexins: a potential role in the regulation of callose synthase. Plant J 3:763–772
Appels R, Dvorak J (1982) The wheat ribosomal DNA spacer region: its structure and variation in populations and among species. Theor Appl Genet 63:337–348
Ball SG, Morell MK (2003) From bacterial glycogen to starch: understanding the biogenesis of the plant starch granule. Annu Rev Plant Biol 54:207–233
Balmer Y, Vensel WH, DuPont FM, Buchanan BB, Hurkman WJ (2006) Proteome of amyloplasts isolated from developing wheat endosperm presents evidence of broad metabolic capability. J Exp Bot 57:1591–1602
Bernfeld P (1955) Amylases alpha and beta. In: Colowick S, Kaplan N (eds) Methods in enzymology. Academic, NY, p 149
Bird AR, Flory C, Davies DA, Usher S, Topping DL (2004a) A novel barley cultivar (Himalaya 292) with a specific gene mutation in starch synthase IIa raises large bowel starch and short chain fatty acids in rats. J Nutr 134:831–835
Bird AR, Jackson M, King RA, Davies DA, Usher S, Topping DL (2004b) A novel high amylose barley cultivar (Hordeum vulgare var. Himalaya 292) lowers plasma cholesterol and alters indices of large-bowel fermentation in pigs. Br J Nutr 92:607–615
Boston RS, Viitanen PV, Vierling E (1996) Molecular chaperones and protein folding in plants. Plant Mol Biol l32:191–222
Campbell JA, Hansen RW, Wilson JR (1999) Cost-effective colorimetric microtitre plate enzymatic assays for sucrose, glucose and fructose in sugarcane tissue extracts. J Sci Food Agric 79:232–236
Clarke B, Rahman S (2005) A microarray analysis of wheat grain hardness. Theor Appl Genet 110:1259–1267
Clarke BC, Hobbs M, Skylas D, Appels R (2000) Genes active in developing wheat endosperm. Funct Integr Genomics 1:44–55
da Silva PMFR, Eastmond PJ, Hill LM, Smith AM, Rawsthorne S (1997) Starch metabolism in developing embryos of oilseed rape. Planta 203:480–487
Davies C, Robinson SP (2000) Differential screening indicates a dramatic change in mRNA profiles during grape berry ripening. Cloning and characterization of cDNAs encoding putative cell wall and stress response proteins. Plant Physiol 122:803–812
Doehlert DC, Kuo TM (1994) Gene expression in developing kernels of some endosperm mutants of maize. Plant Cell Physiol 35:411–418
Fincher GB, Stone BA (1986) Cell walls and their components in cereal grain technology. Adv Cereal Sci Technol 8:207–295
Franco OL, Rigden DJ, Melo FR, Bloch JC, Silva CP, Grossi de Sa MF (2000) Activity of wheat alpha-amylase inhibitors towards bruchid alpha-amylases and structural explanation of observed specificities. Eur J Biochem 267:2166–2173
Giroux MJ, Boyer C, Feix G, Hannah LC (1994) Coordinated transcriptional regulation of storage product genes in the maize endosperm. Plant Physiol 106:713–722
Guerin JR, Lance RCM, Wallace W (1992) Release and activation of barley beta-amylase by malt endopeptidases. J Cereal Sci 15:5–14
Harvey AJ, Hrmova M, Fincher GB (2001) Regulation of genes encoding β-D-gucan glucohydrolases in barley (Hordeum vulgare). Physiol Plant 113:108–120
Hrmova M, Fincher GB (2001) Structure–function relationships and β-d-glucan endo- and exohydrolases from higher plants. Plant Mol Biol 47:73–91
James MG, Denyer K, Myers AM (2003) Starch synthesis in the cereal endosperm. Curr Opin Plant Biol 6:215–222
Konarev AV (1996) Interaction of insect digestive enzymes with plant protein inhibitors and host-parasite co-evolution. Euphytica 92:89–94
Kossmann J, Lloyd J (2000) Understanding and influencing starch biochemistry. Crit Rev Plant Sci 19:171–226
Kumar A, Taylor MA, Mad Arif SA, Davies HV (1996) Potato plants expressing antisense and sense S-adenosylmethionine decarboxylase (SAMDC) transgenes show altered levels of polyamines and ethylene: antisense plants display abnormal phenotypes. Plant J 9:147–158
Laudencia-Chingcuanco DL, Stamova BS, You FM, Lazo GR, Beckles DM, Anderson OD (2007) Transcriptional profiling of wheat caryopsis development using cDNA microarrays. Plant Mol Biol 63:651–668
Lunn JE, Hatch MD (1995) Primary partitioning and storage of photosynthate in sucrose and starch in leaves of C4 plants. Planta 197:385–391
Merendino N, D’Aquino M, Molinari R, De Gara L, D’Egidio MG, Paradiso A, Cecchini C, Corradini C, Tomassi G (2006) Chemical characterization and biological effects of immature durum wheat in rats. J Cereal Sci 43:129–136
Millar HA, Sweetlove LJ, Giege P, Leaver CJ (2001) Analysis of the Arabidopsis mitochondrial proteome. Plant Physiol 127:1711–1727
Morell MK, Rahman S, Regina A, Appels R, Li Z (2001) Wheat starch biosynthesis. Euphytica 119:55–58
Morell MK, Kosar-Hashemi B, Cmiel M, Samuel MS, Chandler P, Rahman S, Buleon A, Batey IL, Li Z (2003) Barley sex6 mutants lack starch synthase IIa activity and contain a starch with novel properties. Plant J 34:173–185
Morell MK, Li Z, Regina A, Rahman S, d’Hulst C, Ball SG (2006) Control of starch biosynthesis in vascular plants and algae. In: Plaxton WC, McManus MT (eds) Control of primary metabolism in plants. Annual Plant Reviews 22:258–289. Blackwell Publishing, Oxford, UK
Myers AM, Morell MK, James MG, Ball SG (2000) Recent progress towards understanding the amylopectin crystal. Plant Physiol 122:989–997
Nijtmans LGJ, de Jong L, Sanz MA, Coates PJ, Berden JA, Back JW, Muijsers AO, van der Spek H, Grivell LA (2000) Prohibitins act as a membrane-bound chaperone for the stabilization of the mitochondrial proteins. EMBO J 19:2444–2451
Ohdan T, Francisco PB Jr, Sawada T, Hirose T, Terao T, Satoh H, Nakamura Y (2005) Expression profiling of genes involved in starch synthesis in sink and source organs of rice. J Exp Bot 56:3229–3244
Østergaard H, Rasmussen SK, Roberts TH, Hejgaard J (2000) Inhibitory serpins from wheat with reactive centers resembling glutamine-rich repeats of prolamin storage proteins. J Biol Chem 275:33272–33279
Poole R, Barker G, Wilson ID, Coghill JA, Edwards KJ (2007) Measuring global gene expression in polyploidy; a cautionary note from allohexaploid wheat. Funct Integr Genomics 7:207–219
Prosky L, Asp N-G, Furda I, DeVries JW, Schweizer TF, Harland BF (1985) Determination of total dietary fibre in foods and food product: collaborative study. J Assoc Off Agric Chem 68:677
RACI (2003) Official testing methods of the Cereal Chemistry Division of the Royal Australian Chemical Institute, Method 02-03. Dumas (combustion) total nitrogen determination
Rahman S, Li Z, Batey I, Cochrane MP, Appels R, Morell MK (2000) Genetic alteration of starch functionality in wheat. J Cereal Sci 31:91–110
Regina A, Bird A, Topping D, Bowden S, Freeman J, Barsby T, Kosar-Hashemi B, Li Z, Rahman S, Morell M (2006) High-amylose wheat generated by RNA interference improves indices of large-bowel health in rats. Proc Natl Acad Sci USA 103:3546–3551
Ruuska S, Rebetzke G, van Herwaarden A, Richards RA, Fettell N, Tabe L, Jenkins CLD (2006) Genotypic variation in water-soluble carbohydrate accumulation in wheat. Funct Plant Biol 33:799–809
Schulman AH, Tomooka S, Suzuki A, Myllarinen P, Hizukuri S (1995) Structural analysis of starch from normal and shx (shrunken endosperm) barley (Hordeum vulgare L.). Carbohydr Res 275:361–369
Shin H, Brown RM Jr (1999) GTPase Activity and biochemical characterization of a recombinant cotton fiber annexin. Plant Physiol 119:925–934
Smith AM (2001) The biosynthesis of starch granules. Biomacromolecules 2:335–341
Tetlow IJ, Morell MK, Emes MJ (2004) Recent developments in understanding the regulation of starch metabolism in higher plants. J Exp Bot 55:2131–2145
Theander O, Aman P, Westerlund E, Andersson R, Pettersson D (1995) Total dietary fibre determined as neutral sugar residues, uronic acid residues and Klason lignin (the Uppsala method). J AOAC Int 78:1030–1044
Topping DL, Morell KM, King RA, Li Z, Bird AR, Noakes M (2003) Resistant starch and health—Himalaya 292, a novel barley cultivar to deliver benefits to consumers. Starke 55:539–545
Tsuchiya K, Urahara T, Konishi T, Kotake T, Tohno-oka T, Komae K, Kawada N, Tsumuraya Y (2005) Biosynthesis of (1-3),(1-4)-β-glucan in developing endosperms of barley (Hordeum vulgare). Physiol Plant 125:181–191
Whetten R, Sun Y-H, Zhang E, Sederoff R (2001) Functional genomics and cell wall biosynthesis in loblolly pine. Plant Mol Biol 47:275–291
Wilson DL, Buckley MJ, Helliwell CA, Wilson IW (2003) New normalisation methods for cDNA microarray data. Bioinformatics 19:1325–1332
Winter H, Huber SC (2000) Regulation of sucrose metabolism in higher plants: localization and regulation of activity of key enzymes. Crit Rev Plant Sci 19:31–67
Wobus U, Sreenivasulu N, Borisjuk L, Rolletschek H, Panitz R, Gubatz S, Weschke W (2004) Molecular physiology and genomics of developing barley grains. Recent Res Dev Plant Mol Biol 2:1–29
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Clarke, B., Liang, R., Morell, M.K. et al. Gene expression in a starch synthase IIa mutant of barley: changes in the level of gene transcription and grain composition. Funct Integr Genomics 8, 211–221 (2008). https://doi.org/10.1007/s10142-007-0070-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10142-007-0070-7