
Abstract
Key message
A new strategy that integrated multiple public data resources was established to construct root gene co-expression network and mine genes regulating root system architecture in maize. A root gene co-expression network, containing 13,874 genes, was constructed. A total of 53 root hub genes and 16 priority root candidate genes were identified. One priority root candidate was further functionally verified using overexpression transgenic maize lines.
Abstract
Root system architecture (RSA) is crucial for crops productivity and stress tolerance. In maize, few RSA genes are functionally cloned, and effective discovery of RSA genes remains a great of challenge. In this work, we established a strategy to mine maize RSA genes by integrating functionally characterized root genes, root transcriptome, weighted gene co-expression network analysis (WGCNA) and genome-wide association analysis (GWAS) of RSA traits based on public data resources. A total of 589 maize root genes were collected by searching well-characterized root genes in maize or homologous genes of other species. We performed WGCNA to construct a maize root gene co-expression network containing 13874 genes based on public available root transcriptome data, and further discovered the 53 hub genes related to root traits. In addition, by the prediction function of obtained root gene co-expression network, a total of 1082 new root candidate genes were explored. By further overlapping the obtained new root candidate gene with the root-related GWAS of RSA candidate genes, 16 priority root candidate genes were identified. Finally, a priority root candidate gene, Zm00001d023379 (encodes pyruvate kinase 2), was validated to modulate root open angle and shoot-borne roots number using its overexpression transgenic lines. Our results develop an integration analysis method for effectively exploring regulatory genes of RSA in maize and open a new avenue to mine the candidate genes underlying complex traits.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maize (Zea mays L.) is one of the most important cereal crops in the world. The continuous increase in maize yield benefits from improvement of germplasm and hybrid breeding (Duvick 2001). Since the past century, the above-ground traits such as plant architecture, ear size, biotic and abiotic stress tolerance, etc., are considered as the main target traits of maize during genetic improvement and breeding process (Lopez-Reynoso and Hallauer 1998; Mock and Pearce 1975; Nepolean et al. 2018; Santiago et al. 2010). Recently, maize breeders are turning their attention to improvement of root traits for enhancing yield and stress tolerance (Gu et al. 2016; Li et al. 2015; Mu et al. 2015). However, root traits are difficult to be evaluated because plant roots are hidden into the soil, and therefore, genetic improvement of root traits seems to be neglected.
The root is an important plant organ that absorbs water, mineral nutrients and providing anchorage (Hodge et al. 2009; Lynch 1995, 2007). In maize, an ideal root system architecture (RSA) can enhance the ability of plants to capture water and nutrients from soils, and thus improve plant growth and productivity, particularly under stress conditions (Mi et al. 2016). The deep root system is proposed to be optimal for water and nitrogen acquisition (Lynch 2013). The RSA with less number of crown root is usually steep (Gao and Lynch 2016; Saengwilai et al. 2014). Meanwhile, the larger root diameter allows the root to penetrate deep soil (Lynch et al. 2014; Schneider et al. 2020). In addition, the RSA traits are closely associated with maize planting density tolerance and lodging resistance that is crucial for yield formation (Liu et al. 2012; Stamp and Kiel 1992). A simulation study in the USA Corn Belt showed that historical maize yield trends can be explained by the improvements of root systems (Hammer et al. 2009). Although many physiological studies demonstrate the importance of maize RSA, the genetic basis of RSA remains largely unknown, which severely limits RSA-based genetic improvement in maize breeding.
RSA is the complex traits, depending on genetic and environmental factors and their interactions (Lynch 1995; de Dorlodot et al. 2007). Quantitative trait loci (QTL) mapping and genome-wide association study (GWAS) are powerful tools to dissect the genetic basis of complex traits (Wang and Xu 2019). A large number of genetic loci for RSA traits are identified in maize, including root number, root length, root diameter, root angle, etc. (Guo et al. 2018; Hund et al. 2011; Tuberosa et al. 2003). Due to the strong plasticity of RSA traits, root growth and development are easily affected by the environment factors, especially under filed conditions, and therefore, identification of reliable genetic loci and underlying genes are difficult (Cai et al. 2012; Li et al. 2020). Using maize root mutants, such as rtcs1, rum1, rth3 and rth6, a small amount of maize root-related genes have been cloned (Bray and Topp 2018; Hochholdinger et al. 2018; Hostetler et al. 2021). However, how to quickly and efficiently clone the new root genes in maize remains a great of challenge.
Recently, the development of bioinformatics provides a variety of tools to identify target genes in maize. For example, GWAS approach is widely used to mine candidate genes of complex traits, owing to high diversity and rapid linkage disequilibrium (LD) decay (Yan et al. 2011). Using the plant comparative genomics database, the homologs of a functional gene in different species can be quickly explored (Conte et al. 2008; Gupta et al. 2016; Irish and Benfey 2004). By the weighted gene co-expression network analysis (WGCNA), thousands of genes can be divided into several modules, and hub genes in modules associated with traits can be further identified (Langfelder and Horvath 2008; Lv et al. 2019; Zhu et al. 2019). The prediction function of gene co-expression network further guides to identify the new candidate genes for target traits (Voineagu et al. 2011). Thus, an approach integrating these methods can be more advantageous to mine candidate genes underlying complex traits.
In this work, we first collected root genes of Arabidopsis, rice and barley from database and literatures, and then identified their homologs in maize. Second, based on obtained maize root genes and public available root transcriptome data, we constructed maize root gene co-expression network by WGCNA and further discovered the hub genes related to root traits. Additionally, by the prediction function of obtained root gene co-expression network, a number of new root candidate genes were explored. Third, by overlapping the obtained new root candidate gene with the root-related GWAS candidate genes, we found the priority root candidate genes, which were explored by root gene co-expression network and also detected by root-related GWAS. Finally, one of the priority root candidate genes was verified by overexpression transgenic maize lines. Taken together, our research highlighted an integration analysis for systemic identification of root genes in maize, and provided a valuable example how to mine the new genes underlying complex traits.
Materials and methods
Collection and identification maize root genes
Two approaches were used to collect root genes in maize, including previously functionally characterized root genes described in three review articles (Bray and Topp 2018; Hochholdinger et al. 2018; Hostetler et al 2021), and the homolog genes from well-known root genes of other plant species via sequence alignment by using EnsemblPlants online tool (Bolser et al. 2017). To identify maize root homolog genes, we first collected the root genes of rice and Arabidopsis from the funRiceGenes database (Yao et al. 2018) and the TAIR database (Reiser et al. 2017), respectively. In addition, a barley root gene collected from Kirschner et al. (2021) was used to identify maize homolog genes.
The EnsemblPlants database was used to search for the homologous root genes in maize. The orthologs ID, percentage of the orthologs sequence matching the Arabidopsis, rice or barley sequence (Target %id), percentage of the Arabidopsis, rice or barley sequence matching the sequence of orthologs (Query %id) and evaluation results of homology with high %identity (high confidence, yes or no) were obtained. The high-confidence orthologs were considered as maize root genes. The gene ID and physical positions of all collected root genes were uniformly converted into B73 RefGen_v4. The detailed information is summarized as Supplementary Table S1.
During domestication, genes in certain regions of the maize genome undergo significant directional changes in allele frequency, methylation level or transcription level. These genes were defined as domesticated genes as described by Hufford et al. (2012), Swanson-Wagner et al. (2012) and Xu et al. (2020). Some identified root genes in this work displayed in the list of maize domestication genes, which was considered to be involved in the domestication process. The detailed information is summarized as Supplementary Table S1.
All maize root genes were assigned functionally into different groups using the AgriGO 2.0 online tools through the singular enrichment analysis (Tian et al. 2017). KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analysis was conducted using the KOBAS 3.0 online tools (Bu et al. 2021).
Construction of root gene co-expression network and identification of root hub genes
The raw reads of maize inbred B73 root RNA-sequencing were downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/sra/) via accession number PRJNA171684, including 50 samples of 18 root tissues from different tissues across different development stages (Stelpflug et al. 2016). The quality control, adapter trimming and quality filtering of raw sequencing data were performed by fastp (Chen et al. 2018). The mapping-based index was build based on a transcriptome annotation from B73_RefGen_v4. Transcripts Per Million (TPM) were used to estimate gene expression levels by Salmon v0.14.1 (Patro et al. 2017). Root gene co-expression networks were constructed using the weighted gene co-expression network analysis (WGCNA) package in R 4.0.0 (Langfelder and Horvath 2008). Using the blockwiseModules function with the parameters (power = 8; TOMType = unsigned; minModuleSize = 30; mergeCutHeight = 0.25; corType = Pearson), the gene co-expression modules were obtained. Using signed kME (module membership values) function, the kME of each gene was obtained. In each module, the top 10% genes with highest kME were defined as the hub genes. Root genes and their co-expression genes with weight > 0.15 were screened to construct co-expression network, named as maize root gene co-expression network.
Prediction of new root candidate genes and priority root candidate genes
According to the suggestion of Voineagu et al. (2011), the top ten connections (weight value) of root genes in the maize root gene co-expression network were predicted as new root candidate genes. Additionally, the candidate genes for GWAS of RSA were collected from the literature (Li et al. 2021; Moussa et al. 2021; Schneider et al. 2020; Sun et al. 2021; Wang et al. 2021; Zheng et al. 2020). The details of these GWAS are summarized in Supplementary Table S7. Then, an overlap analysis is performed between the set of new root candidate gene and GWAS candidate gene. Those genes that appeared in both gene sets were considered as priority root candidate genes.
Gene expression analysis of maize transgenic lines
Using transgenic technology, the maize overexpression lines (OE) were generated by overexpressing the target gene in maize inbred line ND101 driven by the ubiquitin promoter. Two independent lines were obtained from the Center for Crop Functional Genomics and Molecular Breeding, China Agricultural University, with the accession number CAUB0480. Maize seeds were sterilized for 30 min in a 2% solution of NaClO, washed 3 times with distilled water, soaked in saturated CaSO4 for 12 h and then germinated in the dark on moist filter paper at room temperature. After 3 days, the germinated seeds were wrapped in a moist filter paper roll and grown to two visible leaves. Nine seedlings were then randomly selected from 20 healthy seedlings and transferred into a plastic tub (22 × 15 × 28 cm, length × width × height) containing 10-L nutrient solution. The nutrient solution consisted of 2.0-mM Ca(NO3)2, 0.75-mM K2SO4, 0.65-mM MgSO4, 0.1-mM KCl, 0.25-mM KH2PO4, 0.13-mM EDTA-Fe, 1.0-μM H3BO3, 1.0-μM MnSO4, 1.0-μM ZnSO4, 0.1-μM CuSO4 and 0.005-μM (NH4)6Mo7O24, with pH 6.0. The nutrient solution was renewed every 3 days. Plants were grown in a growth chamber with controlled conditions: 28/22 °C during a 14/10-h light/dark cycle, with a light density of 250–300 μmol m−2 s−1.
After 14 days, the maize seeding has grown two layers of crown root. Then, whole roots were harvested, and total RNA was extracted using RNAiso Plus (Takara Bio, Shiga, Japan). cDNA was reverse transcribed using PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara Bio, Shiga, Japan). The qRT-PCR reaction was performed using IQ5 Real-Time PCR System (Bio-Rad) for three biological replicates. The gene Zm00001d013367 (ZmTUB4, F: GCTATCCTGTGATCTGCCCTGA; R: CGCCAAACTTAATAACCCAGTA) was used for normalizing gene expression (Gong et al. 2020). The target gene specific primers used in the qPCR analysis were as follows: F: TTGCTAAGTACAGGCCCAGC and R: GTGGATCAGCAAGCATAGGGA.
Root phenotypic analysis of maize transgenic lines grown in fields
The OE together with wild-type (WT) plants were field-grown in 2021 summer at the Shangzhuang Experimental Station, China Agricultural University, Beijing (116°11'N, 40°8'E), and 2021 winter at the Sanya Experimental Station, China Agricultural University, Hainan (18°23′N, 109°44′E). Maize plants were planted in single-row plots without repetitions, the two OE lines were adjacent to the WT, and each row was 2-m long, 0.5-m wide and contained eight plants. At the silking stage, the 6–8 healthy plants were selected within a row, and the root systems were excavated from soils in a depth of 25 cm. The remaining soil attached to roots was removed by water washing. Roots were transferred to a studio with stable LED light to collect two-dimensional images of the root systems using a camera (ILCE-5100, Sony, Tokyo, Japan). The REST program was used to analysis root images to obtain the root open angle (Colombi et al. 2015). According to the evaluation criteria for the number of crown root whorls described by Vanhees et al. (2020), the sixth whorl crown root was selected to measure root diameter (mm) with a vernier caliper. Number of shoot-borne roots in each layer was counted as described by Li et al. (2019a, b). Student’s t-tests were used to compare the differences of RSA traits between WT and OE transgenic lines (*P ≤ 0.05; **P ≤ 0.01 and ***P ≤ 0.001).
Evaluation of root gravity bending
Maize seeds (WT and OE) were sterilized for 30 min in a 2% solution of NaClO, washed 3 times with distilled water and soaked in saturated CaSO4 for 12 h. Then, the seeds were wrapped in wet filter paper rolls and incubated for 3 days under 28 °C in the dark. When the primary roots grow to 4–5 cm, 40 seedlings with consistent growth be moved to 2% agar plate (13 × 13 cm, length × width). The seedlings were first cultured vertically for 12 h and then were rotated by 90° to continue culture. The whole process is carried out under light conditions (the root is in the dark) and maintained at a constant temperature of 28 °C. After 24 h, the camera (ILCE-5100, Sony, Tokyo, Japan) was used to take pictures, and the angle of root bending was measured with a protractor. Student’s t-tests were used to compare the differences between WT and OE transgenic lines (*P ≤ 0.05; **P ≤ 0.01 and ***P ≤ 0.001).
Results
Genomic distribution and functional annotation of collected maize root genes
By the literature review, we first found thirty-four previously well-characterized maize root genes, such as rtcs1, rum1, rth3 and rth6 (Fig. 1A and Supplementary Table S1). We also explored the maize homolog genes based on homology analysis with well-known root functional genes of other plant species. A large number of maize homolog genes (555) were then identified, including those highly homology to genes of Arabidopsis (296 genes), rice (257 genes) and barley (2 genes). In total, 589 maize root genes were collected, and their distribution across the 10 maize chromosomes is illustrated in Fig. 1A. Among them, 78 genes are likely domestication genes, including 56 genes domesticated on genomics, 21 genes domesticated on DNA methylation and one gene domesticated on transcriptome.

Genomic distribution and functional annotation of 589 maize root genes collected. A The root genes distributed across ten maize chromosomes. B The fifteen most significant GO terms obtained by GO enrichment analysis of maize root genes. The GO terms related to root development and hormone were highlighted in red and blue, respectively. C The twenty most significant pathways obtained by KEGG pathway analysis of root genes. The vertical red line represents the threshold (FDR ≤ 0.05) (color figure online)
To explore the biological functions of obtained the 589 maize root genes, GO (Gene Ontology) enrichment and KEGG pathway analysis were performed. A total of 968 significant GO terms were identified (FDR ≤ 0.05), involved in the biological process (815 GO terms), cellular component (52 GO terms) and molecular function (101 GO terms). The top 15 GO terms with high significances were mainly associated with the biological process, including root development (183 genes, FDR = 4.4E-109), root morphogenesis (122 genes, FDR = 1.1E-69) and hormone responses (210 genes, FDR = 3.7E-58) (Fig. 1B and Supplementary Table S2). A total of 12 significantly enriched pathways (FDR ≤ 0.05) were identified, including plant hormone signal transduction (32 genes, FDR = 2.01E-16), MAPK signaling pathway (17 genes, FDR = 1.67E-08), inositol phosphate metabolism (7 genes, FDR = 1.52E-03) and fatty acid metabolism (5 genes, FDR = 4.22E-03) (Fig. 1C and Supplementary Table S3).
Construction of maize root gene co-expression network
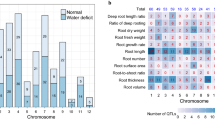
We performed WGCNA analysis to construct maize root genes co-expression network, and on the basis, we identified the root hub genes in maize. After excluding those genes with low expression levels, a total of 26,336 highly expressed genes (including 522 maize root genes) were obtained and further classified into 20 gene co-expression modules (Fig. 2A). The turquoise and blue modules contained 140 and 116, respectively, accounted for the large proportion of maize root genes. The followed yellow, green and brown modules contained 43, 43 and 41 root genes, respectively, and a few root genes remained in other modules (Fig. 2B and Supplementary Table S4). The genes in different modules showed specific expression patterns. For example, those in yellow module revealed high expression levels only in brace_roots_node6_v13, and those in green module showed high expression levels in root_stele_3 days. By contrast, root genes in blue module were constitutively expressed among multiple root tissues (Fig. 2B and Supplementary Table S4). Based on root genes and their co-expression genes with weight > 0.15, a total of 16 root gene co-expression subnetwork, containing 13,874 genes, were constructed (Fig. 3 and Supplementary Table S5).

Weighted gene co-expression network analysis (WGCNA) of root transcriptome. A The twenty gene co-expression modules revealed by hierarchical cluster tree and heatmap. B The heatmap for the expression patterns of root genes in different co-expression modules. 1. Primary_Root_3DAS, primary root 3 d after sowing; 2. Root_MZ_and_Ez_3DAS, primary root meristematic and elongation zones 3 d after sowing; 3. Root_DZ_3DAS, primary root differentiation zone of 3 d after sowing; 4. Root_CP_3DAS, root cortical parenchyma of primary root differentiation zone 3 d after sowing; 5. Root_Stele_3DAS, root stele of primary root differentiation zone 3 d after sowing; 6. Primary_Root_6DAS, primary root 6 d after sowing; 7. Root_System_7DAS, primary and seminal roots 7 d after sowing; 8. Primary_Root_7DAS, primary root 7 d after sowing; 9. Seminal_Roots_7DAS, seminal roots 7 d after sowing; 10. Primary_Root_Z1_7DAS, first cm of primary root tip 7 d after sowing; 11. Primary_Root_Z2_7DAS, primary root from end of first cm of root tip to the point of root hair or lateral root initiation 7 d after sowing; 12. Primary_Root_Z3_7DAS, primary root lower half of differentiation zone 7 d after sowing; 13. Primary_Root_Z4_7DAS, primary root upper half of differentiation zone 7 d after sowing; 14. Crown_Roots_Nodes1–3_V7, crown roots of nodes 1–3 at developmental stage V7; 15. Crown_Roots_Nodes4_V7, crown roots of node 4 at developmental stage V7; 16. Crown_Roots_Nodes5_V7, crown roots of node 5 at developmental stage V7; 17. Crown_Roots_Nodes5_V13, crown roots of node 5 at developmental stage V13; 18. Brace_Roots_Node6_V13, brace roots of node 6 at developmental stage V13

Root gene co-expression network and root hub genes. Different network colors indicate different root gene co-expression modules. The orange font in the box indicates root genes with known function, green font indicates root genes domesticated on genomics and purple font indicates root genes domesticated on DNA methylation (color figure online)
Identification of root hub genes in maize
Based on their high kME values (top 10% kME value in gene co-expression modules), a total of 53 root hub genes were identified (Fig. 3 and Supplementary Table S1). These root hub genes were mainly assigned into the turquoise (19 genes) and blue modules (17 genes). Among them, three maize genes that have been functionally characterized were determined. The Zm00001d012883 (bige1) encodes a putative membrane-localized protein regulating maize brace root whorl number (Suzuki et al. 2015). The Zm00001d046492 (elm2) encodes a heme-oxygenase that significantly increased the lateral root length, number and density when it is overexpressed in Arabidopsis (Han et al. 2012). The Zm00001d036031 (prh17) encodes a serine/threonine protein phosphatase 2A isoform. This gene can control multiple root traits simultaneously, thus improves low phosphorus tolerance in maize (Wang et al. 2017).
A total of eight root hub genes were determined to be domestication genes (Fig. 3 and Supplementary Table S1), including four genomic domestication genes (Zm00001d015788, Zm00001d003598, Zm00001d015289 and Zm00001d011890) and four DNA methylation domestication genes (Zm00001d041407, Zm00001d010974, Zm00001d054088 and Zm00001d016601; Fig. 3). The Zm00001d011890 is a homolog gene of rice OsCKX4 that regulates crown root development by integrating cytokinin and auxin pathway (Gao et al. 2014). The Zm00001d003598 is a homolog gene of OsRR1 that encodes a rice type-A response regulator of cytokinin signaling, and involved in crown root initiation (Kitomi et al. 2011). The Zm00001d041407 presents a homolog gene of Arabidopsis SOS4 that encodes a pyridoxal kinase essential for root hair development (Shi and Zhu 2002). The Zm00001d054088 is a homolog gene of Arabidopsis XLG3, a member of extra-large G proteins family involved in root morphogenesis (Ding et al. 2008).
Prediction of new root candidate genes and priority root candidate genes in maize
According to Voineagu et al. (2011), we considered the top 10 connections (weight value) of root genes in root gene co-expression network as maize new root candidate genes. As shown in Fig. 4A and Supplementary Table S6, after filtering out the root gene co-expression network gene set, 1082 genes were predicted as new root candidate gene due to their highly connected with root genes.

Prediction of priority root candidate genes based on root gene co-expression network and RSA-GWAS. A Co-expression network of 377 root genes and 1082 new root candidate genes. The red, green and blue circles represent root hub genes, root genes and new root candidate genes, respectively. B A total of 16 priority root candidate genes identified based on set of new root candidate gene and RSA-GWAS candidate gene. The red, green, blue and yellow circles represent root hub genes, root genes, new root candidate genes and priority root candidate genes, respectively. C List of 16 priority root candidate genes. Genes colored in red are co-expressed with the root hub gene (color figure online)
To clarify those new root candidate genes with natural variation controlling maize RSA (root number, root diameter and root angle), we integrated GWAS candidate genes of RSA in analysis. A gene set containing 500 non-redundant genes was obtained from the predecessor GWAS of RSA (Li et al. 2021; Moussa et al. 2021; Schneider et al. 2020; Sun et al. 2021; Wang et al. 2021; Zheng et al. 2020). Comparing this set of GWAS gene with the new root candidate genes, a total of 16 genes that were overlapped among these two genes sets were determined as the priority root candidate gene (Fig. 4B). These priority root candidate genes were significantly associated with root diameter (10 genes), root number (4 genes) and root angle (2 genes). These genes were mainly involved in the regulatory pathway that might be functional in root growth and development, such as WRKY transcription factor (Khan et al. 2022; Liu et al. 2022), protein kinase (Gamuyao et al. 2012; Sukumar et al. 2009), etc. In addition, the two genes Zm00001d007960 and Zm00001d023379 were found to be directly co-expressed with the root hub gene (Fig. 4B and C).
Verification of a priority root candidate gene function by overexpression transgenic lines
The function of one of the candidate genes in regulating RSA in maize was verified. To this end, transgenic maize lines overexpressing Zm00001d023379 were generated (Fig. 5A), and their RSA phenotypes were evaluated under field conditions. The Zm00001d023379 gene encodes a pyruvate kinase 2 (pyk2), which is co-expressed with the root hub genes Zm00001d020810 (Fig. 4B). Two independent OE transgenic lines OE#3 and OE#5 were identified, and their expression levels of transgenes were increased up to 8.7- and 17.1-fold, respectively, compared with that of wild type (Fig. 5B). In the field, the two OE lines showed steeper crown root than that of wild-type plants (Fig. 5C). The root opening angle of OE lines decreased significantly by more than 10 degrees in both locations of Beijing and Sanya, while the root diameter did not differ among them. The number of shoot-borne roots decreased significantly by 4–13 in Beijing while they did not show any significantly change in Sanya (Fig. 5D, E and F). This result clarified that pyk2 was a functional gene regulating RSA in maize. In addition, the root tips of the two OE lines exhibited greater bending angles under gravity (Fig. 6, P < 0.001), indicating that they respond more strongly to gravity, which may be an important reason why their root opening angles are smaller than those of wild type.

Functional validation of priority root candidate genes. A Gene model of Zm00001d023379 (pyk2) and construct for generation of pyk2-overexpression lines. B Relative expression of in pyk2-overexpression transgenic lines (OE). C RSA phenotype of WT and OE transgenic lines under field conditions. D, E and F Evaluation of root open angle, crown root diameter and shoot-borne roots number for WT and OE transgenic lines

Evaluation of root gravity bending in transgenic maize lines overexpressing pyk2. A Root bending phenotype of WT and OE transgenic lines in the agar plate. B Frequency distribution of root bending angle in WT and OE transgenic lines. C Average of root bending angles for WT and OE transgenic lines
Discussion
Given the importance of RSA in absorbing nutrients and water, providing anchorage against lodging in crops (Lynch 1995, 2007; Hodge et al. 2009; Stamp and Kiel 1992), genetic improvement of RSA is an effective way to achieve efficient utilization of resources and increase yield (Steele et al. 2006; Landi et al. 2010; Chin et al. 2011). In the past few decades, a large number of genetic loci controlling maize RSA have been identified by QTL mapping and GWAS, but underlying genes remain to be identified. Although few root genes for maize RSA have been map-based cloned via mutants so far, the natural variation with breeding value needs to be further explored. In this study, we proposed a new bioinformatics strategy for mining root candidate genes and identified a number of new maize root candidate genes that may be valuable for breeding. This work then provides new insights for the genetic study of maize RSA.
Integration analysis provides an effective approach to mine genes controlling root system architecture in maize
In the previous studies, QTL meta-analysis (MQTL) was often used to mine reliable QTLs underlying root traits in maize and even to further narrow the QTL position intervals (Guo et al. 2018; Hund et al. 2011). However, owing to dozens to hundreds of genes underlying the MQTL intervals, it is difficult to determine the proper candidate genes. Recently, GWAS has been widely explored in the studies on maize root genetics (Li et al. 2021; Moussa et al. 2021; Schneider et al. 2020; Sun et al. 2021; Wang et al. 2021; Zheng et al. 2020). Its accuracy becomes relatively higher, and only a few candidate genes are found to be associated with significant SNPs. Due to the lack of gene annotation information based on mutant analyses, identification of the target gene from a large number of genes remains challengeable (Andorf et al. 2016). Therefore, reliable new methods are demanded for rapid and efficient identification of genes underlying complex traits.
Schaefer et al. (2018) developed a computational approach, Camoco, which integrates genetic loci identified by GWAS with the gene functional information derived from gene co-expression networks. Using this Camoco, a total of 610 prioritized candidate genes, regulating maize grain elements accumulation, have been selected from thousands of candidate genes based on a large-scale GWAS. Li et al. (2021) used WGCNA analysis to identify gene co-expression modules associated with root plasticity in response to salt stress. By integrating WGCNA approach with differentially expressed genes (DEGs) and GWAS, the number of candidate genes was subsequently reduced from 587 to 130. We suppose that the integration of GWAS and gene co-expression network analysis may be more effective to mining the candidate genes underlying complex traits.
In the present study, we developed a new method to predict new root candidate genes (Fig. 7). Unlike methods provided by Li et al. (2019a, b) and Moisseyev et al. (2020), who used a similar approach to identify maize root-associated genes mainly through gene tissue-specific expression information. In our present work, however, an integrated approach was used including the homologous genes, transcriptome and GWAS genetic loci. Thus, our work is novel and open a new avenue to mine the candidate genes for complex root traits. Compared to the previous studies by Li et al. (2021) and Schaefer et al. (2018), there are many advantages to our integration approach. First, multiple databases related to maize roots have been integrated, including homologous genes, transcriptome and GWAS genetic loci, providing more abundant information. This is obviously better than a simple analysis based on single dataset (Li et al. 2021). Second, our established maize root gene co-expression network with a large number of root genes (522) is more comprehensive and reliable, and powerful to discover new root candidate genes as well root hub genes. By contrast, Schaefer et al. (2018) simply used the co-expression between pairs of unknown functional genes to mine candidate genes. Third, unlike other previous work (Gong et al. 2020; Wang et al. 2021), this approach allows to identify the priority root candidate genes that have yet to be functionally characterized. Thus, we establish an integration analysis method for effectively exploring regulatory genes of RSA in maize and open a new avenue to mine the candidate genes underlying complex traits.

Data analysis process in the present study. Each analysis step and the corresponding output are shown in the different colored boxes. The key parameters of this analysis are also indicated (color figure online)
The 53 root hub genes and 16 priority root candidate genes deserve further studied
Hub genes are mostly located in the upstream of gene pathway and play regulatory roles in plants (Langfelder and Horvath 2008). The identification of hub gene is crucial for understanding of gene regulatory network of target traits. In maize, WGCNA analysis has been recently widely used to analyze hub genes in biological processes such as salt tolerance, drought resistance, ion transport and root elongation (Liang et al. 2022; Liu et al. 2021; Ma et al. 2021; Wang et al. 2021).
In this study, 53 root hub genes were identified from 589 maize root genes by WGCNA analysis. The functions of these genes are, e.g., related to transcription factors, actin, protein kinases, hormone and nitrogen metabolism. They are considered as the upstream of other root genes and play important regulatory roles in roots (Supplementary Tables S2 and S3). In addition, these root hub genes including three well-known genes in maize (prh17, elm2 and bige1), four domesticated on genomics (prc4, cko3, Zm00001d003598 and Zm00001d015289) and four were domesticated on DNA methylation (phd17, xlg2, Zm00001d016601 and Zm00001d045599; Fig. 3 and Supplementary Table S1). These evidence support that the identified hub genes may play key roles in regulating development of maize roots, and present valuable gene resources for further genetic improvement of maize roots.
Furthermore, the 16 priority root candidate genes were identified in this work, including transcription factors, protein kinases and so on (Fig. 4B and C). These genes were significantly associated with RSA traits and may have functional allelic variation being valuable for breeding. These genes provide new genetic resources for root genetic improvement in maize. It is worthy to note that a root priority candidate gene pyk2, encoding a pyruvate kinase involved in in the glycolysis process (GO: 0,006,096), was functionally verified by the overexpression lines. In the field, the two OE lines of pyk2 significantly reduced the root open angles more than 10 degrees (Fig. 5C), but slight and no changes in crown roots number and diameter, respectively, compared to those of WT (Fig. 5D and E). The reduced root open angle in the OE lines could be further explained by the increased root gravitropism (Fig. 6). It is assumed that the pyk2 regulates root gravity response and subsequently determine RSA in maize.
The glycolytic process plays a vital role in plant growth and development of plants, and the pyruvate kinase is one of the main rate-limiting enzymes in this process (Fernie et al. 2004). In rice, the loss function of pyruvate kinase OsPK1 leads to plant dwarfing, abnormal inflorescence development and reduced fruiting rate, and the loss function of another homolog gene OsPK2 leads to low grain weight, decreased starch content and alteration of starch physicochemical properties (Cai et al. 2018; Zhang et al. 2012). As a metabolic substrate for the glycolysis process, glucose plays an important role in regulating plant root growth and development (Kircher and Schopfer 2012; Singh et al. 2014). Glucose not only acts as a nutrient and signaling molecule to regulate root apical meristem activity, primary root elongation, lateral root and root hair formation, but also interacts with hormones such as auxin, cytokinin, abscisic acid and brassinosteroid, indirectly regulating root growth and gravitropism (Gupta et al. 2015; Kushwah and Laxmi 2017; Mishra et al. 2009; Xiong et al. 2013; Yuan et al. 2014). We then assumed that the pyk2 gene regulates sugar metabolism by influencing the glycolysis process, thereby regulating RSA. The extensive interaction between glucose and plant hormones is also determined (Gupta et al. 2015; Kushwah and Laxmi 2017; Mishra et al. 2009; Xiong et al. 2013; Yuan et al. 2014). Therefore, pyk2 may also control the RSA by indirectly regulating the hormone pathway. For further research, we still need more genetic materials (mutant or gene editing) and experimental evidence (molecular or biochemical) to verify the function of other root candidate genes.
Data availability
Data supporting the findings of this work are available within the paper and its Supplementary Information files.
References
Andorf CM, Cannon EK, Portwood JL 2nd, Gardiner JM, Harper LC, Schaeffer ML, Braun BL, Campbell DA, Vinnakota AG, Sribalusu VV, Huerta M, Cho KT, Wimalanathan K, Richter JD, Mauch ED, Rao BS, Birkett SM, Sen TZ, Lawrence-Dill CJ (2016) MaizeGDB update: new tools, data and interface for the maize model organism database. Nucleic Acids Res 44:D1195–D1201
Bolser DM, Staines DM, Perry E, Kersey PJ (2017) Ensembl plants: integrating tools for visualizing, mining, and analyzing plant genomic data. Methods Mol Biol 1533:1–31
Bray AL, Topp CN (2018) The quantitative genetic control of root architecture in maize. Plant Cell Physiol 59:1919–1930
Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, Wu Y, Zhao L, Liu J, Guo J, Fang S, Cao W, Yi L, Zhao Y, Kong L (2021) KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res 49:W317–W325
Cai H, Chen F, Mi G, Zhang F, Maurer HP, Liu W, Reif JC, Yuan L (2012) Mapping QTLs for root system architecture of maize (Zea mays L.) in the field at different developmental stages. Theor Appl Genet 125:1313–1324
Cai Y, Li S, Jiao G, Sheng Z, Wu Y, Shao G, Xie L, Peng C, Xu J, Tang S, Wei X, Hu P (2018) OsPK2 encodes a plastidic pyruvate kinase involved in rice endosperm starch synthesis, compound granule formation and grain filling. Plant Biotechnol J 16:1878–1891
Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884–i890
Chin JH, Gamuyao R, Dalid C, Bustamam M, Prasetiyono J, Moeljopawiro S, Wissuwa M, Heuer S (2011) Developing rice with high yield under phosphorus deficiency: pup1 sequence to application. Plant Physiol 156:1202–1216
Colombi T, Kirchgessner N, Le Marié CA, York LM, Lynch JP, Hund A (2015) Next generation shovelomics: set up a tent and rest. Plant Soil 388:1–20
Conte MG, Gaillard S, Lanau N, Rouard M, Périn C (2008) GreenPhylDB: a database for plant comparative genomics. Nucleic Acids Res 36:D991–D998
de Dorlodot S, Forster B, Pagès L, Price A, Tuberosa R, Draye X (2007) Root system architecture: opportunities and constraints for genetic improvement of crops. Trends Plant Sci 12:474–481
Ding L, Pandey S, Assmann SM (2008) Arabidopsis extra-large G proteins (XLGs) regulate root morphogenesis. Plant J 53:248–263
Duvick DN (2001) Biotechnology in the 1930s: the development of hybrid maize. Nat Rev Genet 2:69–74
Fernie AR, Carrari F, Sweetlove LJ (2004) Respiratory metabolism: glycolysis, the TCA cycle and mitochondrial electron transport. Curr Opin Plant Biol 7:254–261
Gamuyao R, Chin JH, Pariasca-Tanaka J, Pesaresi P, Catausan S, Dalid C, Slamet-Loedin I, Tecson-Mendoza EM, Wissuwa M, Heuer S (2012) The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 488:535–539
Gao Y, Lynch JP (2016) Reduced crown root number improves water acquisition under water deficit stress in maize (Zea mays L.). J Exp Bot 67:4545–4557
Gao S, Fang J, Xu F, Wang W, Sun X, Chu J, Cai B, Feng Y, Chu C (2014) CYTOKININ OXIDASE/DEHYDROGENASE4 integrates cytokinin and auxin signaling to control rice crown root formation. Plant Physiol 165:1035–1046
Gong X, Liu X, Pan Q, Mi G, Chen F, Yuan L (2020) Combined physiological, transcriptome, and genetic analysis reveals a molecular network of nitrogen remobilization in maize. J Exp Bot 71:5061–5073
Gu R, Chen F, Long L, Cai H, Liu Z, Yang J, Wang L, Li H, Li J, Liu W, Mi G, Zhang F, Yuan L (2016) Enhancing phosphorus uptake efficiency through QTL-based selection for root system architecture in maize. J Genet Genom 43:663–672
Guo J, Chen L, Li Y, Shi Y, Song Y, Zhang D, Li Y, Wang T, Yang D, Li C (2018) Meta-QTL analysis and identification of candidate genes related to root traits in maize. Euphytica 214:223
Gupta A, Singh M, Laxmi A (2015) Interaction between glucose and brassinosteroid during the regulation of lateral root development in arabidopsis. Plant Physiol 168:307–320
Gupta P, Naithani S, Tello-Ruiz MK, Chougule K, D’Eustachio P, Fabregat A, Jiao Y, Keays M, Lee YK, Kumari S, Mulvaney J, Olson A, Preece J, Stein J, Wei S, Weiser J, Huerta L, Petryszak R, Kersey P, Stein LD, Ware D, Jaiswal P (2016) Gramene database: navigating plant comparative genomics resources. Curr Plant Biol 7–8:10–15
Hammer GL, Dong ZS, McLean G, Doherty A, Messina C, Schussler J, Zinselmeier C, Paszkiewicz S, Cooper M (2009) Can changes in canopy and/or root system architecture explain historical maize yield trends in the U.S. corn belt? Crop Sci 49:299–312
Han B, Xu S, Xie YJ, Huang JJ, Wang LJ, Yang Z, Zhang CH, Sun Y, Shen WB, Xie GS (2012) ZmHO-1, a maize haem oxygenase-1 gene, plays a role in determining lateral root development. Plant Sci 184:63–74
Hochholdinger F, Yu P, Marcon C (2018) Genetic control of root system development in maize. Trends Plant Sci 23:79–88
Hodge A, Berta G, Doussan C, Merchan F, Crespi M (2009) Plant root growth, architecture and function. Plant Soil 321:153–187
Hostetler AN, Khangura RS, Dilkes BP, Sparks EE (2021) Bracing for sustainable agriculture: the development and function of brace roots in members of poaceae. Curr Opin Plant Biol 59:101985
Hufford MB, Xu X, van Heerwaarden J, Pyhäjärvi T, Chia JM, Cartwright RA, Elshire RJ, Glaubitz JC, Guill KE, Kaeppler SM, Lai J, Morrell PL, Shannon LM, Song C, Springer NM, Swanson-Wagner RA, Tiffin P, Wang J, Zhang G, Doebley J, McMullen MD, Ware D, Buckler ES, Yang S, Ross-Ibarra J (2012) Comparative population genomics of maize domestication and improvement. Nat Genet 44:808–811
Hund A, Reimer R, Messmer R (2011) A consensus map of QTLs controlling the root length of maize. Plant Soil 344:143–158
Irish VF, Benfey PN (2004) Beyond arabidopsis. Translational biology meets evolutionary developmental biology. Plant Physiol 135:611–614
Khan N, Zhang Y, Wang J, Li Y, Chen X, Yang L, Zhang J, Li C, Li L, Ur Rehman S, Reynolds MP, Zhang L, Zhang X, Mao X, Jing R (2022) TaGSNE, a WRKY transcription factor, overcomes the trade-off between grain size and grain number in common wheat and is associated with root development. J Exp Bot 73:6678–6696
Kircher S, Schopfer P (2012) Photosynthetic sucrose acts as cotyledon-derived long-distance signal to control root growth during early seedling development in arabidopsis. Proc Natl Acad Sci USA 109:11217–11221
Kitomi Y, Ito H, Hobo T, Aya K, Kitano H, Inukai Y (2011) The auxin responsive AP2/ERF transcription factor crown rootless5 is involved in crown root initiation in rice through the induction of OsRR1, a type-A response regulator of cytokinin signaling. Plant J 67:472–484
Kushwah S, Laxmi A (2017) The interaction between glucose and cytokinin signaling in controlling arabidopsis thaliana seedling root growth and development. Plant Signal Behav 12:e1312241
Landi P, Giuliani S, Salvi S, Ferri M, Tuberosa R, Sanguineti MC (2010) Characterization of root-yield-1.06, a major constitutive QTL for root and agronomic traits in maize across water regimes. J Exp Bot 61:3553–3562
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinform 9:559
Li P, Chen F, Cai H, Liu J, Pan Q, Liu Z, Gu R, Mi G, Zhang F, Yuan L (2015) A genetic relationship between nitrogen use efficiency and seedling root traits in maize as revealed by QTL analysis. J Exp Bot 66:3175–3188
Li J, Chen F, Li Y, Li P, Wang Y, Mi G, Yuan L (2019a) ZmRAP2.7, an AP2 transcription factor, is involved in maize brace roots development. Front Plant Sci 10:820. https://doi.org/10.3389/fpls.2019.00820
Li Y, Liu X, Chen R, Tian J, Fan Y, Zhou X (2019b) Genome-scale mining of root-preferential genes from maize and characterization of their promoter activity. BMC Plant Biol 19:584
Li P, Fan Y, Yin S, Wang Y, Wang H, Xu Y, Yang Z, Xu C (2020) Multi-environment QTL mapping of crown root traits in a maize RIL population. Crop J 8:645–654
Li P, Yang X, Wang H, Pan T, Wang Y, Xu Y, Xu C, Yang Z (2021) Genetic control of root plasticity in response to salt stress in maize. Theor Appl Genet 134:1475–1492
Liang T, Qing C, Liu P, Zou C, Yuan G, Pan G, Shen Y, Ma L (2022) Joint GWAS and WGCNA uncover the genetic control of calcium accumulation under salt treatment in maize seedlings. Physiol Plant 174(1):e13606
Liu S, Song F, Liu F, Zhu X, Xu H (2012) Effect of planting density on root lodging resistance and its relationship to nodal root growth characteristics in maize (Zea mays L.). J Agric Sci 4:182–189
Liu S, Zenda T, Dong A, Yang Y, Wang N, Duan H (2021) Global transcriptome and weighted gene co-expression network analyses of growth-stage-specific drought stress responses in maize. Front Genet 12:645443
Liu X, Yang Y, Wang R, Cui R, Xu H, Sun C, Wang J, Zhang H, Chen H, Zhang D (2022) GmWRKY46, a WRKY transcription factor, negatively regulates phosphorus tolerance primarily through modifying root morphology in soybean. Plant Sci 315:111148
Lopez-Reynoso JJ, Hallauer AR (1998) Twenty-seven cycles of divergent mass selection for ear length in maize. Crop Sci 38:1099–1107
Lv Y, Xu L, Dossa K, Zhou K, Zhu M, Xie H, Tang S, Yu Y, Guo X, Zhou B (2019) Identification of putative drought-responsive genes in rice using gene co-expression analysis. Bioinformation 15:480–489
Lynch J (1995) Root architecture and plant productivity. Plant Physiol 109:7–13
Lynch JP (2007) Roots of the second green revolution. Aust J Bot 55:493–512
Lynch JP (2013) Steep, cheap and deep: an ideotype to optimize water and N acquisition by maize root systems. Ann Bot 112:347–357
Lynch JP, Chimungu JG, Brown KM (2014) Root anatomical phenes associated with water acquisition from drying soil: targets for crop improvement. J Exp Bot 65:6155–6166
Ma L, Zhang M, Chen J, Qing C, He S, Zou C, Yuan G, Yang C, Peng H, Pan G, Lübberstedt T, Shen Y (2021) GWAS and WGCNA uncover hub genes controlling salt tolerance in maize (Zea mays L.) seedlings. Theor Appl Genet 134:3305–3318
Mi G, Chen F, Yuan L, Zhang F (2016) Ideotype root system architecture for maize to achieve high yield and resource use efficiency in intensive cropping systems. Adv Agron 139:73–97
Mishra BS, Singh M, Aggrawal P, Laxmi A (2009) Glucose and auxin signaling interaction in controlling arabidopsis thaliana seedlings root growth and development. PLoS ONE 4:e4502
Mock JJ, Pearce RB (1975) An ideotype of maize. Euphytica 24:613–623
Moisseyev G, Park K, Cui A, Freitas D, Rajagopal D, Konda AR, Martin-Olenski M, Mcham M, Liu K, Du Q, Schnable JC, Moriyama EN, Cahoon EB, Zhang C (2020) RGPDB: database of root-associated genes and promoters in maize, soybean, and sorghum. Database (Oxford) 2020:baaa038
Mu X, Chen F, Wu Q, Chen Q, Wang J, Yuan L, Mi G (2015) Genetic improvement of root growth increases maize yield via enhanced post-silking nitrogen uptake. Eur J Agron 63:55–61
Nepolean T, Kaul J, Mukri G, Mittal S (2018) Genomics-enabled next-generation breeding approaches for developing system-specific drought tolerant hybrids in maize. Front Plant Sci 9:361
Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14:417–419
Reiser L, Subramaniam S, Li D, Huala E (2017) Using the Arabidopsis information resource (TAIR) to find information about arabidopsis genes. Curr Protoc Bioinformatics 60:1.11.1-1.11.45
Saengwilai P, Tian X, Lynch JP (2014) Low crown root number enhances nitrogen acquisition from low-nitrogen soils in maize. Plant Physiol 166:581–589
Santiago R, Reid LM, Zhu X, Butrón A, Malvar RA (2010) Gibberella stalk rot (Fusarium graminearum) resistance of maize inbreds and their F1 hybrids and their potential for use in resistance breeding programs. Plant Breed 129:454–456
Schaefer RJ, Michno JM, Jeffers J, Hoekenga O, Dilkes B, Baxter I, Myers CL (2018) Integrating coexpression networks with GWAS to prioritize causal genes in maize. Plant Cell 30:2922–2942
Schneider HM, Klein SP, Hanlon MT, Nord EA, Kaeppler S, Brown KM, Warry A, Bhosale R, Lynch JP (2020) Genetic control of root architectural plasticity in maize. J Exp Bot 71:3185–3197
Shi H, Zhu JK (2002) SOS4, a pyridoxal kinase gene, is required for root hair development in arabidopsis. Plant Physiol 129:585–593
Singh M, Gupta A, Laxmi A (2014) Glucose control of root growth direction in arabidopsis thaliana. J Exp Bot 65:2981–2993
Stamp P, Kiel C (1992) Root morphology of maize and its relationship to root lodging. J Agron Crop Sci 168:113–118
Steele KA, Price AH, Shashidhar HE, Witcombe JR (2006) Marker-assisted selection to introgress rice QTLs controlling root traits into an Indian upland rice variety. Theor Appl Genet 112:208–221
Stelpflug SC, Sekhon RS, Vaillancourt B, Hirsch CN, Buell CR, de Leon N, Kaeppler SM (2016) An expanded maize gene expression atlas based on RNA sequencing and its use to explore root development. Plant Genome. https://doi.org/10.3835/plantgenome2015.04.0025
Sukumar P, Edwards KS, Rahman A, Delong A, Muday GK (2009) PINOID kinase regulates root gravitropism through modulation of PIN2-dependent basipetal auxin transport in arabidopsis. Plant Physiol 150:722–735
Suzuki M, Sato Y, Wu S, Kang B-H, McCarty DR (2015) Conserved functions of the mate transporter big embryo1 in regulation of lateral organ size and initiation rate. Plant Cell 27:2288–2300
Swanson-Wagner R, Briskine R, Schaefer R, Hufford MB, Ross-Ibarra J, Myers CL, Tiffin P, Springer NM (2012) Reshaping of the maize transcriptome by domestication. Proc Natl Acad Sci USA 109:11878–21183
Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, Xu W, Su Z (2017) Agrigo v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 45:W122–W129
Tuberosa R, Salvi S, Sanguineti MC, Maccaferri M, Giuliani S, Maccaferri M, Sanguineti MC, Landi P, Tuberosa R, Giuliani S, Salvi S (2003) Searching for quantitative trait loci controlling root traits in maize: a critical appraisal. Plant Soil 255:35–54
Vanhees DJ, Loades KW, Bengough AG, Mooney SJ, Lynch JP (2020) Root anatomical traits contribute to deeper rooting of maize under compacted field conditions. J Exp Bot 71:4243–4257
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474:380–384
Wang M, Xu S (2019) Statistical power in genome-wide association studies and quantitative trait locus mapping. Heredity 123:287–306
Wang J, Pei L, Jin Z, Zhang K, Zhang J (2017) Overexpression of the protein phosphatase 2A regulatory subunit a gene ZmPP2AA1 improves low phosphate tolerance by remodeling the root system architecture of maize. PLoS ONE 12:e0176538
Wang Y, Sun H, Wang H, Yang X, Xu Y, Yang Z, Xu C, Li P (2021) Integrating transcriptome, co-expression and QTL-seq analysis reveals that primary root growth in maize is regulated via flavonoid biosynthesis and auxin signal transduction. J Exp Bot 72:4773–4795
Xiong Y, McCormack M, Li L, Hall Q, Xiang C, Sheen J (2013) Glucose-TOR signalling reprograms the transcriptome and activates meristems. Nature 496:181–186
Xu G, Lyu J, Li Q, Liu H, Wang D, Zhang M, Springer NM, Ross-Ibarra J, Yang J (2020) Evolutionary and functional genomics of DNA methylation in maize domestication and improvement. Nat Commun 11:5539
Yan J, Warburton M, Crouch J (2011) Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci 51:433–449
Yao W, Li G, Yu Y, Ouyang Y (2018) funRiceGenes dataset for comprehensive understanding and application of rice functional genes. Gigascience 7:1–9
Yuan TT, Xu HH, Zhang KX, Guo TT, Lu YT (2014) Glucose inhibits root meristem growth via ABA INSENSITIVE 5, which represses PIN1 accumulation and auxin activity in arabidopsis. Plant Cell Environ 37:1338–1350
Zhang Y, Xiao W, Luo L, Pang J, Rong W, He C (2012) Downregulation of OsPK1, a cytosolic pyruvate kinase, by T-DNA insertion causes dwarfism and panicle enclosure in rice. Planta 235:25–38
Zhu M, Xie H, Wei X, Dossa K, Yu Y, Hui S, Tang G, Zeng X, Yu Y, Hu P, Wang J (2019) WGCNA analysis of salt-responsive core transcriptome identifies novel hub genes in rice. Genes 10:719
Funding
The study was financially supported by the National Key Research and Development Program of China (grant no. 2021YFF1000500) and the National Natural Science Foundation of China (grant no. 31972485).
Author information
Authors and Affiliations
Contributions
LY, FC and GM designed the experiments; KH and ZZ performed the experiments; KH and WR performed data analysis; LC generated maize transgenic lines; ZC and QP provided the scientific advice; KH prepared and wrote the article and LY revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by Thomas Lubberstedt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
He, K., Zhao, Z., Ren, W. et al. Mining genes regulating root system architecture in maize based on data integration analysis. Theor Appl Genet 136, 127 (2023). https://doi.org/10.1007/s00122-023-04376-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00122-023-04376-0