
Abstract
Key message
Applying an integrated meta-analysis approach led to identification of meta-QTLs/ candidate genes associated with rice root system architecture, which can be used in MQTL-assisted breeding/ genetic engineering of root traits.
Abstract
Root system architecture (RSA) is an important factor for facilitating water and nutrient uptake from deep soils and adaptation to drought stress conditions. In the present research, an integrated meta-analysis approach was employed to find candidate genes and genomic regions involved in rice RSA traits. A whole-genome meta-analysis was performed for 425 initial QTLs reported in 34 independent experiments controlling RSA traits under control and drought stress conditions in the previous twenty years. Sixty-four consensus meta-QTLs (MQTLs) were detected, unevenly distributed on twelve rice chromosomes. The confidence interval (CI) of the identified MQTLs was obtained as 0.11–14.23 cM with an average of 3.79 cM, which was 3.88 times narrower than the mean CI of the original QTLs. Interestingly, 52 MQTLs were co-located with SNP peak positions reported in rice genome-wide association studies (GWAS) for root morphological traits. The genes located in these RSA-related MQTLs were detected and explored to find the drought-responsive genes in the rice root based on the RNA-seq and microarray data. Multiple RSA and drought tolerance-associated genes were found in the MQTLs including the genes involved in auxin biosynthesis or signaling (e.g. YUCCA, WOX, AUX/IAA, ARF), root angle (DRO1-related genes), lateral root development (e.g. DSR, WRKY), root diameter (e.g. OsNAC5), plant cell wall (e.g. EXPA), and lignification (e.g. C4H, PAL, PRX and CAD). The genes located within both the SNP peak positions and the QTL-overview peaks for RSA are suggested as novel candidate genes for further functional analysis. The promising candidate genes and MQTLs can be used as basis for genetic engineering and MQTL-assisted breeding of root phenotypes to improve yield potential, stability and performance in a water-stressed environment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is one of the most important staple crops, which is consumed by more than one-third of the world’s population. Among the abiotic factors, drought is the major limiting factor affecting rice growth and productivity. It is estimated that by the year 2050, more than 50% of the world’s arable land will be affected by drought (Singhal et al. 2016). Therefore, improving rice for drought-prone environments is a priority.
Dynamic responses of root system architecture (RSA) play a key role in efficiently using water and nutrients in crops (De Smet et al. 2012; Paez-Garcia et al. 2015), and also in drought avoidance mechanism and drought tolerance performance (Blum 2005). Improving the architecture and function of roots will be the key to the second green revolution in the future (Gewin 2010). However, breeding for root morphological traits has rarely been addressed, mostly because of the costs and time constraints as well as lack of reliable and efficient phenotyping techniques for root-related traits (Carvalho et al. 2014; Toyofuku et al. 2015). The RSA indicates the organization of the primary roots, crown roots, and lateral roots in the soil environment (De Smet et al. 2012). The genetic factors and interactions with environmental conditions mainly affect the RSA adaptation (Malamy 2005). Genotypic background determines the intrinsic morphological characters, whereas environmental factors modify the root morphologies on the basis of requirements for dynamically adapting to undesirable environmental conditions (Gruber et al. 2013; Soriano and Alvaro 2019). Mapping of quantitative trait loci (QTL) has been used as a powerful statistical method to identify genomic regions associated with traits of interest for breeding (Wang et al. 2019b). To date, many QTLs related to RSA traits were identified by linkage analysis from different population types and sizes across diverse moisture conditions (Courtois et al. 2003; de Dorlodot et al. 2007; Uga et al. 2011, 2013). In many QTL mapping studies, the overlap of QTLs for root and agronomic traits has been revealed, suggesting the profound implications that breeding for RSA will have on improving crop genotypes through enhancing crop productivity and high water/nutrients use efficiency under water-limited conditions (Jia et al. 2019; Ju et al. 2018; Maccaferri et al. 2016; Tuberosa et al. 2002). The growth angle and length of roots mainly determine the root system distribution of rice throughout the soil profile (Araki et al. 2002).
Various positions of a QTL in different mapping populations lead to a large confidence interval and uncertain position of the QTL. This can be further exacerbated by several additional factors such as different sizes of mapping populations and sampling errors (Darvasi and Soller 1997; Darvasi et al. 1993), differences in experimental replicates, marker density and QTL mapping models. Various approaches have been used so far to confirm the QTL results, such as QTL mapping using first-generation populations, and getting validated in advanced generation breeding populations of the same cross (Gelli et al. 2017). In other cases, QTL confirmation is performed using the candidate gene approach or positional cloning, followed by integrating functional and genetic data within a breeding process (de Dorlodot et al. 2007). This challenging process, however, requires high-density linkage maps, extensive genomic resources and logical informatics data (de Dorlodot et al. 2007).
To identify consensus QTL regions across multiple studies, QTL meta-analysis method was initially developed (Goffinet and Gerber 2000) using maximum likelihood estimation and was then improved (Veyrieras et al. 2007). Meta-analysis clearly estimates the numbers, positions and CI of meta-QTL regions in each chromosome. This method has been used to identify consensus regions of the genome across multiple QTL studies for their effect and consistency across different genetic backgrounds and environments, also to refine and confirm QTL positions on a consensus map via mathematical models. The QTL meta-analysis has been performed on root morphological traits in different species, including maize (Guo et al. 2018), bread wheat (Darzi-Ramandi et al. 2017; Soriano and Alvaro 2019; Bilgrami et al. 2020), durum wheat (Iannucci et al. 2017), and oilseed rape (Zhang et al. 2018).
In the current study, we integrated 425 QTLs from the 34 published QTL mapping studies for root traits in rice and identified consensus genomic regions through QTL meta-analysis method into a novel integrated consensus genetic map with 5447 loci. Thus far, only one QTL meta-analysis on rice root morphological traits has been reported based on the QTLs collected from 24 studies from 1995 to 2007, mostly using AFLP and RFLP markers, detecting MQTLs with 1.98 Mb average CI and a reduction of 61% in the number of QTLs (Courtois et al. 2009). However, out of 34 QTL mapping studies used in the current research, 27 studies, including 268 QTLs, were reported after 2007, using mostly SSR and some SNP markers, which had not been included in the QTL meta-analysis so far. Moreover, the supporting intervals of the identified MQTLs reduced to an average of 1.57 Mb corresponded to a reduction of 85% in the number of initial QTLs and then validated by the related GWAS studies. The genes located in the MQTLs were detected and functionally classified. In addition, the differentially expressed genes (DEGs) in the rice root under drought conditions were detected through the analysis of RNA-seq and microarray datasets, and the MQTL regions associated with RSA were explored to find the drought-responsive genes in the rice root. Conclusively, integrating QTLs, GWAS, and transcriptome data resulted in identification of the promising MQTLs and candidate genes, which can be used in MQTL-assisted breeding and genetic engineering to improve RSA-related traits in rice.
Materials and methods
Collecting RSA trait-associated QTLs from independent studies
A comprehensive review of the literature was conducted by searching the articles published from 2001 to 2020 on rice QTLs associated with RSA traits in normal conditions and drought stress (Table 1). The data collected for each QTL region included (1) root architecture traits, (2) parents of the population, (3) types of mapping population: F2, backcross (BC), doubled haploids (DH), recombinant inbred lines (RILs) and near-isogenic lines (NILs), (4) population size (N), (5) logarithm of odds ratio (LOD score), (6) proportion of phenotypic variance explained by the QTL (R2), (7) the flanking or single marker(s) for interval mapping and single-marker analysis (SMA), respectively. The present study assumed a LOD score of 3 in a few cases in which the published article reported a p-value statistic or stated that a minimum LOD score of 3 was adopted as the threshold for QTL analyses. Equation (1) proposed by (Nagelkerke 1991) was used to estimate the LOD score (where N represents the population size), which is the explanatory power of QTLs if the LOD score was not reported.
Ten traits associated with root architecture analyzed in the present study under normal and drought stress conditions included deep root ratio, root number (RN), root length (RL), root thickness (RT), root volume (RV), root to shoot ratio (RSR), root fresh weight (RFW), root dry weight (RDW), deep root ratio (DRR), root growth rate (RGR) and root surface area (RSA) were analyzed. The analysis encompassed the QTLs whose map positions, LOD scores and R2 values were available. All the QTL studies related to RSA conducted on these traits using markers including AFLP, SSR, SNP and RFLP were applied in the QTL meta-analysis, but the articles lacking proper genetic maps or QTL-associated data were excluded. The QTL mapping studies with any missing parameters were also discarded.
Developing the consensus map and projecting the QTLs
A novel integrated consensus genetic map was developed from four previous studies with high-density markers by BioMercator V4.2 (https://urgi.versailles.inra.fr/Tools/BioMercator-V4) with default parameters. The selected maps included (i) the map of the International Rice Microsatellite Initiative (IRMI) comprising 1684 SSR markers developed by (McCouch et al. 2002) and available at https://archive.gramene.org (IRMI_2003), (ii) the high-density rice genetic map comprising 3267 RFLP markers available at https://rgp.dna.affrc.go.jp (RGP_2001), (iii) the Cornel_SSR_2006 map comprising 706 markers, including 455 SSRs, 133 RFLPs and 116 specific gene markers, available at https://archive.gramene.org (Cornel_SSR_2001) and (iv) the CIAT_SSR_2006 map comprising 408 SSR markers available at https://archive.gramene.org (CIAT_SSR_2006). All the markers from the aforementioned maps were used to develop the consensus marker map. The meta-analysis was performed after eliminating the markers showing inversion on the consensus map. The projection of the QTLs position was performed on the basis of a simple scaling method between the interval of the QTL flanking markers on their original map and the interval of these markers on the consensus map (Supplementary Table S1).
After estimating the new confidence interval of the initial QTL on their original genetic map using the Gaussian distribution, the QTLs were projected on the consensus map. The 95% Cl for a QTL position was calculated according to the type of population used in Eqs. (2)–(4), in which N represents the population size, R2 the percentage of the phenotypic variance of the QTLs and CI the supporting or confidence interval for each initial QTL.
Equation (2) is presented by modeling to calculate the 95% CI of F2 and BC populations by (Darvasi and Soller 1997). Equation (3) was used for the population of DH lines (Visscher and Goddard 2004), and Eq. (4) for both RIL and NIL populations (Guo et al. 2006).
Meta-analysis and QTL-overview index
After projecting the QTLs on the consensus map, meta-analysis was conducted according to the QTL clusters on each chromosome using BioMercator V4.2 (Arcade et al. 2004) which contains algorithms from the MetaQTL software (Sosnowski et al. 2012; Veyrieras et al. 2007). Two different approaches were used depending on the number of initial QTLs in a chromosome, which was below 10 when the meta-analysis proposed by Gerber and Goffinet was employed. Based on this method, the most likely assumption is estimated in BioMercator among five MQTL models (1, 2, 3, 4, or N) with different AIC values. Moreover, the model with the minimum AIC was selected for QTL integration and identification of consensus MQTL positions. The method proposed by Veyrieras et al. was employed in case the number of QTLs in a chromosome equaled at least 10. According to this approach, two stages are taken in QTL meta-analyses. In the first stage, the collected QTLs are clustered on individual chromosomes with default parameters. The number of potential MQTLs is then estimated on the basis of the model choice criteria from AIC, the modified AIC (AICc and AIC3), Bayesian information criterion (BIC), and Approximate Weight of Evidence (AWE). A model with the minimum values of the selection criteria in at least 3 out of the 5 models was selected as the optimal MQTL model. In the next stage, the 95% CIs and positions of each MQTL were obtained as per the optimal model selected in the first stage. The QTLs were integrated in a way that the peak position of the initial QTLs lay in the MQTL confidence interval. The QTLs whose probability of membership in an MQTL exceeded 60% were assigned to the same MQTL.
Moreover, the QTL-overview statistic proposed by Chardon et al. (2004) was calculated to measure the contribution of a chromosome region to trait variations. The "pnorm" function in R software (R Core Team 2018) was used to obtain the QTL-overview statistic through a step-by-step calculation of the uniform probability that a 0.5-cM-long segment (x and x + 0.5) included a QTL in a test as per Eq. (5).
in which nbE represents the total number of experiments, Si2 the variance position of the individual QTLs on a chromosome, and nbQTL the number of QTLs. The value of the QTL-overview was obtained in a given chromosome interval based on (i) the number of QTLs lying in the vicinity of the chromosome interval and (ii) the power and accuracy in mapping for these QTLs (high R2 value or small CI). To observe genomic zones with a significant QTL peak, the mean statistic [U(x)] and a threshold for high values [H(x)] were empirically calculated as 5 times of the mean value (Chardon et al. 2004). Equations (6)–(7) were, respectively, used to calculate U(x) and H(x).
Identifying the genes located in the MQTL regions
To find the genes located in the identified MQTLs, the related flanking markers were detected on the Oryza sativa genetic map (Temnykh et al. 2001) (http://archive.gramene.org/markers/microsat/) (Supplementary Table S2). The physical positions were then determined after mapping the flanking markers onto the Oryza sativa Japonica group (IRGSP-1.0) reference genome (Kawahara et al. 2013). The genes located in the MQTL regions were ultimately detected in biomart on the ensemble website (https://plants.ensembl.org/biomart/martview/) (Supplementary Table S3).
Graphical representation
The distribution and position of the MQTLs on the individual chromosomes were presented as a heat-map based on the rice genome by the ggplot2 R package (Wickham et al. 2019). Moreover, a graphical summary of initial QTLs, MQTLs and QTL-overview statistics was drawn on all the 12 rice chromosomes by SOFIA package (Diaz-Garcia et al. 2017) in R environment.
Gene ontology enrichment analysis
The genes located in the MQTL regions were functionally classified using the web-based AgriGO 2.0 (systemsbiology.cau.edu.cn/agriGOv2/) by the singular enrichment analysis (SEA) tool based on the setting of parameters as follows: (i) p value < 0.05 as the level of statistical significance and (ii) the Fisher’s exact test using the adjustment method proposed by Benjamini–Yekutieli for controlling the false discovery rate in multiple tests under dependency. Moreover, the singular enrichment analysis was performed to detect gene ontology terms, i.e. molecular functions, biological processes and cellular components, which were significantly enriched by genes for the individual traits. Gene ontology (GO) enrichment analyses were performed for the genes located in the 64 identified RSA associated MQTL regions, 36 MQTL regions with an interval of less than 1 Mb, the differentially expressed candidate genes (DECGs), and constitutively expressed candidate genes (CECGs), independently.
Collection of datasets and analysis of gene expression
Several DEGs were collected from different microarray (5 published articles) and RNA-seq (15 published articles) experiments at https://www.ncbi.nlm.nih.gov. The genes with a cutoff of log2-fold of over 1 (two-fold absolute value) and a P-value of at most 0.05 were considered differentially expressed between the genotypes (Supplementary Table S4 and S5). Venn diagram was used to compare the rice root drought-responsive genes detected by RNA-seq or microarray data analysis, and the genes located in MQTL regions. Candidate genes shared between MQTL, microarray and RNA-seq data were analyzed in MapMan (Jung and An 2012). Four overviews, i.e. regulation, metabolism, proteasome and transcription, were used to describe any genes up-regulated in response to drought stress.
Collecting RSA-related genome-wide association studies (GWAS) and comparing them with MQTLs
GWAS studies for root morphological traits in rice were reviewed (Bettembourg et al. 2017; Biscarini et al. 2016; Courtois et al. 2013; Kadam et al. 2017; Li et al. 2017; Mai et al. 2020; Pariasca-Tanaka et al. 2020; Phung et al. 2016; Wang et al. 2018; Xu et al. 2020), and the reported SNP peak positions were collected to find the overlaps between their positions with MQTLs. The genes located in the SNP peak positions (± 25 kb) were extracted from Oryza sativa Japonica group (IRGSP-1.0) reference genome according to their physical positions.
Results
Genetic consensus map construction
A novel integrated consensus genetic map comprising 5447 markers was constructed in BioMercator V4.2 using 1923 SSRs, 3223 RFLPs, 25 ESTs, and 152 other genomic loci based on 5 previously published genetic maps (Supplementary Table S1). The total length of this consensus map was 1528.3 cM, with an average chromosome length of 127.4 cM and a range of 83.0 cM (Chromosome 10) to 181.8 cM (Chromosome 1). The mean number of markers in individual chromosomes was obtained as 454, with the lowest and highest numbers of markers being, respectively, associated with chromosome 10 (n = 256) and chromosome 1 (n = 833). The marker density was obtained as 2.6 to 4.6 markers per cM on chromosomes 12 and 1, respectively, with an average of 3.6 per cM.
Distribution of initial QTLs associated with RSA traits in rice
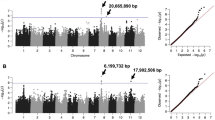
The reports in the literature and rice database (http://qtaro.abr.affrc.go.jp) were reviewed to collect QTLs data on the RSA of rice in normal and drought conditions (Table 1). The studies covered 34 different experimental crosses involving 56 parental lines and 9461 progeny lines, while population size ranged from 75 (Anis et al. 2019) to 480 individual genotypes (Catolos et al. 2017). The present QTL meta-analysis employed 425 out of the 568 initial QTLs associated with RSA traits (Supplementary Table S6). One hundred and forty-three QTLs were not used in the meta-analysis because either there was no common marker between individual genetic maps and the consensus genetic map, or some of QTLs showed a large confidence interval. Among the 425 initial QTLs, 233 (54.82%) and 192 (49%) were found under normal and water deficit conditions, respectively (Fig. 1a). The present study investigated different populations of QTL mapping studies on root traits, which included F2 (2 populations), BC (11 populations), RILs (17 populations), DH (5 populations) and NILs (8 populations) (Table 1). The number of markers used in the previous reports of QTL mapping studies ranged from 215 (Courtois et al. 2003) to 584 (Catolos et al. 2017). The distribution pattern of the initial QTLs on all the twelve rice chromosomes showed that the highest frequency of initial QTLs was related to chromosomes 1 (n = 60), 4 (n = 59) and 7 (n = 58) and the lowest to chromosomes 10 (n = 5), 12 (n = 6) and 5 (n = 16) (Table 2 and Fig. 1b). The QTLs were unevenly distributed on rice chromosomes, with different combinations of QTLs for different root traits. The average number of initial QTLs for each individual RSA trait was 39, ranging from 13 for the root-to-shoot ratio to 97 for the root length, followed by 65 (15.3%) initial QTLs for root weight traits (i.e. root fresh weight and root dry weight) (Fig. 1b and Table 3). With an average value of 14.83 cM, the 95% confidence intervals (CI) varied between 1.18 and 58.40 cM, of which approximately 42% of the collected initial QTLs had a CI lower than 10 cM, and 75% had a CI lower than 20 cM (Fig. 2a). The proportion of phenotypic variance explained (PVE) by the single QTLs ranged from 2.0 to 66.6% with an average of 13.4% (Fig. 2b). The proportion of phenotypic variance explained by each of the 425 initial QTLs was used to rank them in terms of all the RSA traits (Fig. 2c). A total of 271(59.3%) of the 425 QTLs showed a PVE of more than 10%, whereas 186 QTLs (40.7%) explained less than 10% of the phenotypic variance. For the root length, among the 97 represented QTLs, 87 QTLs explained more than 10% of the phenotypic variance (Fig. 2c).

Distribution of QTLs for root architecture traits on all the rice chromosomes; a The number of initial QTLs distributed on the individual rice chromosomes under normal and water shortage conditions, b The number of initial QTLs for root traits on each rice chromosome used in meta-analysis of QTL

Summary of 425 initial QTLs related to RSA traits used in the QTL meta-analysis. a The frequency distribution of initial QTLs density based on different levels of the 95% confidence interval, b The phenotypic variance explained (PVE), c The number of initial QTLs with different phenotypic variance explained (R2 < 10 and R2 ≥ 10) for each root morphology trait, d The distribution of initial QTLs and MQTLs on the twelve chromosomes in rice. DRR: deep root length ratio, RDR: ratio of deep rooting, RDW: root dry weight, RFW: root fresh weight, RGR: root growth rate, RL: root length, RN: root number, RSA: root surface area, RSR: root to shoot ratio, RT: root thickness, RV: root volume
Map projection and meta-analysis of QTLs controlling RSA traits
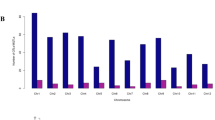
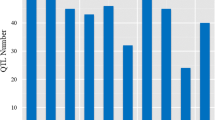
The collected initial QTLs were projected on the integrated consensus map with its high-density SSR and RFLP markers. A total of 64 MQTLs were developed from the 425 initial QTLs for RSA traits with at least 2 MQTLs on each of the twelve rice chromosomes. The number of MQTLs per chromosome ranged from 9 MQTL on chromosome 2, to 2 MQTL on chromosomes 10 and 12 (Figs. 2d, 3 and 4; Table 3). Integrated QTL data showed that all rice chromosomes seemed to be involved in genetic control of root traits. The frequency of clustered initial QTLs per MQTL lay between 2 QTLs in 8 MQTLs (MQTL1-8, MQTL2-1, MQTL6-2, MQTL7-4, MQTL9-2, MQTL10-1, MQTL11-2 and MQTL12-1) and 22 QTLs in MQTL3-1 on chromosome 3 (Table 3). Forty-four (67.8%) MQTLs were obtained by clustering QTLs from at least 3 different experiments, which involved diverse types of the mapping population. The stability of these MQTLs was more likely to be preserved in a variety of environments. The mean of phenotypic variance explained (PVE) by each MQTL ranged from 6.8 to 30.7% with an average of 13.6%, while the 95% CIs reported for the MQTLs ranged from 0.11 cM (0.16 Mb) for the intervals C847–RM473A on chromosome 7 to 14.23 cM (7.98 Mb) for the intervals RM6179–RM4455 on chromosome 10 (Table 3).

Concentric circles show different features drawn in SOFIA package (Diaz-Garcia et al. 2017) in the R environment. The twelve rice chromosomes portrayed along the perimeter of every circle; a Colored bars showing the twelve rice chromosomes and chromosomal positions (cM) presented along the chromosomes. b Flanking markers of each MQTL on the consensus genetic map. c Distribution of the initial QTLs on the twelve chromosomes of rice (blue lines) and position of MQTLs with the 95% confidence intervals (red). d QTL-overview index (probability density) for the root traits QTLs on consensus genetic map. e Proportion of phenotypic variance explained (R2) for each initial QTLs. f Density of initial QTL for each MQTL

Circos diagram for initial QTLs and MQTLs associated with root traits on the physical map of the rice japonica variety Nipponbare. The twelve rice chromosomes are portrayed along the perimeter of each circle. a Colored bars showing the twelve rice chromosomes and physical positions (100 Kb) are presented along the chromosomes. b Flanking markers of each MQTL on the rice physical map. c Distribution of initial QTLs on twelve chromosomes of rice (black lines) and position of MQTLs with confidence intervals (red areas). d Coefficient of reduction in physical CI from mean original QTLs to MQTL. (e) Number of genes involved in each MQTL interval
The confidence interval was narrower in the individual MQTL regions than the mean CI of the original QTLs in that position. At 17 MQTL regions, the CI was reduced to less than 2 cM, with a reduction in length by 12.6 times of the mean CI initial QTLs. Moreover, the maximum decrease was observed in the CI of the initial QTLs on chromosome 7, whereas the MQTL7-7 CI was by 136.9 times lower than the CI mean in the clustered QTLs and corresponded to a confidence interval of merely 0.11 cM (0.16 Mb). A total of 14 MQTLs had the physical length of around 500 kb and 10 of which also had less than 2 cM genetic distance (Table 3). These ten MQTL regions also had a mean phenotypic variance of 14.8%. The flanking markers of these ten MQTL regions were appropriate for molecular breeding and marker-assisted selection in future genetic improvement programs of root morphological traits in rice. The physical intervals of 36 (56.2%) MQTLs were below 1 Mb. The physical length of MQTL ranged from 0.08 Mb (MQTL3-5) to 7.98 Mb (MQTL10-1). The number of RSA traits in each MQTL ranged from one (root length) in 1 MQTL (MQTL1-8) to eight in 4 MQTLs (MQTL1-6, MQTL4-3, MQTL4-4 and MQTL7-7). The individual QTLs for the root length and root thickness were present in 49 MQTLs out of the 64 detected MQTL regions, the most for any root morphological traits, while the individual QTLs for the root surface area was only present in 11 MQTLs, the least among the RSA traits (Table 2).
Estimation of QTL-overview index for RSA QTLs in rice
After projecting the 425 QTLs onto the consensus genetic map, the density of the presence of QTLs described as “QTL-overview index” was calculated for the considered interval of 0.5 cM on each chromosome to identify genomic regions significantly associated with RSA traits (Fig. 3 and Supplementary Fig. S1). Sixty-four out of 73 overview index peaks obtained were higher than 0.0122 as the mean value of the statistic over the genome and showed ‘real QTLs’ that affect RSA traits in rice. According to Supplementary Fig. S1, fifteen out of the 64 peaks also exceeded 0.061 as the high-value threshold. The number of significant peaks ranged from one in chromosomes 1, 2 and 5 to three in chromosomes 4 and 9. In the latter case, all the 5 peaks exceeded the high-value threshold. The results revealed that the estimated CIs identified through the QTL-overview analysis were similar to the meta-analysis method. The QTL-overview index is a statistical approach that has been widely used for traits such as flowering time in maize (Chardon et al. 2004), the response of leaf growth to water deficit in maize (Welcker et al. 2007), yield and related traits in maize (Martinez et al. 2016), the fatty acid content in soybean (Qin et al. 2018) and root-related traits in bread wheat (Soriano and Alvaro 2019).
Detecting differentially expressed genes (DEGs) in the rice root under drought conditions
The drought-responsive genes in the rice root were collected from RNA-seq and microarray datasets (Supplementary Table S5). Based on the RNA-seq data analysis, 10,239 up-regulated and 7214 down-regulated rice root DEGs were observed at drought compared to normal conditions. Microarray meta-analysis suggested 22,307 DEGs, among which, 16,290 and 6017 were up- and down-regulated, respectively (Supplementary Table S7 and Fig. S2). Finally, MQTL regions associated with RSA were explored to find the drought-responsive genes in the rice root. A total of 5448 and 1338 common genes were revealed using the Venn diagram between the DEGs derived from RNA-seq and microarray data and the genes located in MQTL regions (Supplementary Table S8 and Fig. S2) for all the 64 MQTLs (Supplementary Fig. S2b) and the 36 MQTLs with CI of less than 1 Mb (Supplementary Fig. S2a), respectively. These genes that are both differentially expressed at drought conditions in rice roots and located in MQTL regions can be considered as differentially expressed candidate genes (DECGs).
Gene ontology analysis of the genes located in the RSA-associated MQTL regions
Enrichment analysis of the genes located in the 64 MQTLs, 36 MQTLs, DECGs and CECGs, independently revealed many shared and a few diverged enriched GO terms (Supplementary Table S9 and Fig. S3). It confirms the significance of the results and provides evidence that genes with associated functions might be clustered and contributed to the QTL traits. The common terms included gene expression, protein modification process, RNA metabolic process, phosphotransferase activity, protein serine/threonine kinase activity, calcium ion binding transporter activity, nucleus, membrane part and ribosome. However, as expected, some terms were enriched in the drought-responsive candidate genes, but not in the constitutively expressed candidate genes, such as ARF protein signal transduction, response to stress, ion transport, response to oxidative stress, and plant-type cell wall organization. Under the gene expression term, many RSA-related genes were observed such as members from WRKY, NAC, ARF, WOX, AUX/IAA, RR, RAA, and HSFA2 families, which were also detected by MapMan as drought-responsive TFs. Likewise, other significant GO terms such as the protein modification processes were significant through both MapMan and GO analysis and contained RSA-related genes (e.g. ACA8, SAPK 6,10,7 and CCR3).
Analyzing the drought-responsive candidate genes in MapMan
The fold-change data and Locus IDs for 5448 and 1338 drought-responsive genes of the 64 and 36 MQTLs (with an interval of less than 1 Mb), respectively, were uploaded to MapMan toolkit (Supplementary Table S10) and the various overviews were obtained (Supplementary Fig. S4a to S4f). It is worth noting that almost similar results were obtained from MapMan analysis of the DEGs located in the 64 and 36 MQTL regions. Investigating the regulation overview of the 5448 genes showed the up-regulation of 417 transcription factors (TFs), 276 genes related to protein degradation, and 192 genes associated with protein modification in the rice root under drought stress (Supplementary Fig. S4b). The highest frequency of TFs and their importance in the regulation of the root response and structure under drought stress, warrant further investigations (Supplementary Fig. S4c).
The most significantly enriched genes in the signaling pathway included those for G-protein and calcium regulation. Thioredoxin as the reduction–oxidation response element dominated the cellular processes of drought-responsive genes in rice. A G protein-mediating calcium-signaling cascade, therefore, functions upstream of drought-responsive TFs in the hierarchy of signaling pathways to regulate the reduction–oxidation reaction through the thioredoxin activity and trigger the drought tolerance. This evidence suggests the cooperative role of thioredoxin, G-proteins and calcium regulation in responding to drought in the rice root. It is worth noting that MapMan analysis results of the DEGs located in the 64 and 36 MQTL regions were somehow similar (Supplementary Fig. S4a to S4f).
Validation of MQTLs with the related GWAS studies
Significant overlaps existed between the MQTLs detected using meta-analysis and the SNPs detected using the GWAS method that are linked to RSA traits in rice genome. Interestingly, 52 MQTLs out of the 64 identified MQTLs were co-located with 171 SNP peak positions reported in rice GWAS for root morphological traits (Figs. 5 and 6). A total of 752 rice genes were located in the SNP peak positions (± 25 kb) overlapping with MQTLs, among them 49 SNPs were occurred exactly within a gene (Supplementary Table S11).

Genomic collinearity of the MQTLs with the significant loci in the recent GWAS results for RSA traits. The MQTLs are shown on the right side of each chromosome, with black segments indicating their physical intervals. The genomic positions of the MQTL regions correspond to Table 3. The significant SNPs identified by GWAS for RSA traits are displayed on the left side of each chromosome, along with the related traits and the genomic position of the significant loci with base pairs (bp) positions (Supplementary Table S11)

Genetic loci controlling phenotypic variations of RSA traits in rice. The red rectangles represents the MQTLs identified on the 12 chromosomes of rice through meta-analysis method, while rectangle width shows their confidence interval. The mean of phenotypic variance explained (PVE) by each MQTL was ruled on the left side of plot. Points with different colors represents significant SNPs identified by previous GWAS studies for different root traits. RA: root angle, RDW: root dry weight, RL: root length, RN: root number, RSR: root to shoot ratio, RT: root thickness, RV: root volume
Discussion
MQTLs for RSA traits and the perspective of their implications in MQTL-assisted breeding
The RSA plays a key role in absorbing nutrients and water by a plant, its anchoring to the soil and its yield (Subira et al. 2016). Linkage analysis and linkage disequilibrium mapping indicate that root morphology in rice is a complex trait that involves multiple loci with small effects. Meta-analysis is a powerful statistical technique that can be performed on QTL data collected from different genetic backgrounds and environments to determine consistent QTLs, enhance the accuracy of their chromosomal locations, and integrate the QTL data collected from numerous independent experiments (Goffinet and Gerber 2000). In the present QTL meta-analysis, we collected the data of 425 QTLs from 34 articles published between 2001 and 2020 (Supplementary Table S6) to identify genomic regions linked to RSA and the drought stress tolerance in rice. We could detect 64 MQTLs by performing a meta-analysis based on the modified Akaike information criterion (AIC) (Table 3). The supporting intervals of the identified MQTLs with an average of 3.79 cM (1.57 Mb) reduced 3.88 times as compared to the mean of corresponding original QTLs. The only similar previous work (Courtois et al. 2009) just covered 306 QTLs reported until 2007, detecting 119 MQTLs with 4.14 Mb average CI for the initial QTLs and 1.98 Mb for MQTLs. In our study, 376 out of the 425 initial QTLs were SSR flanking markers, of which 268 QTLs belonged to the reports after 2007. In other QTL meta-analyses on diverse quantitative traits in various crops, including bread wheat, barley, maize and soybean, has been reported a reduction of 10%-21% in the total number of initial QTLs compared to the number of MQTLs, and the average reduction in the CI of the MQTLs varied from two to four times of the initial QTLs (Ballini et al. 2008; Courtois et al. 2009; Darzi-Ramandi et al. 2017; Hao et al. 2010; Khahani et al. 2020; Lanaud et al. 2009; Rong et al. 2007; Soriano and Alvaro 2019). In the current research, the genetic CI of 71.8% and physical CI of 75% MQTL’s were narrower than 5 cM and 2 Mb, respectively. The present study found several MQTLs in recombination hot spot regions with a narrow CI such as MQTL7-7 (0.16 Mb, 0.11 cM), MQTL3-5 (0.08 Mb, 0.22 cM), MQTL6-6 (0.27 Mb, 0.47 cM), MQTL9-6 (0.27 Mb, 0.48 cM), MQTL5-3 (0.21 Mb, 0.71 cM) and MQTL7-2 (0.51 Mb, 0.82 cM) to be useful for the fine-mapping studies of quantitative trait loci (Table 3). The present findings suggested that analyzing MQTLs can help discover consensus genomic regions and accurately locate linked QTLs for RSA traits in rice.
Although there is still a high diversity for the quantitative inheritance nature of RSA traits, rice breeders can take advantage of the current genetic diversity to select the most appropriate MQTLs for improving the root characteristics and drought tolerance in their elite breeding materials. According to Löffler et al. (2009), the MQTLs selected for breeding purposes should have (i) a narrow CI, (ii) comprise a large number of initial QTLs, and (iii) a high proportion of its mean phenotypic variance is explained by initial QTLs. Using some of the identified MQTLs for MQTL-assisted breeding to improve the RSA traits to enhance drought tolerance appears promising. Interestingly, eleven of the identified MQTLs contained at least 10 initial QTLs with a 95% CI of below 2 cM (Table 3). Among them, the MQTL1-7, MQTL3-5, MQTL4-3, MQTL4-5, MQTL6-6, MQTL7-7, and MQTL9-6 span less than 500 kb and the original QTLs constitute six to nine independent studies. These MQTLs could be used as multi-effect hotspot regions affecting RSA traits, which is worth applying in breeding programs in the near future.
Potential candidate genes involved in RSA and drought tolerance
Multiple genes associated with root-related traits in rice were found to be located in MQTL intervals (Supplementary Table S3) which were used to reconstruct the related molecular network, and will be discussed in the following (Fig. 7 and Supplementary Table S4).

The molecular network associated with rice root development using potential candidate genes located in MQTL regions. An arrow shows a positive regulatory action, and lines with a flat head in their end show negative regulatory actions. Text color code: biological processes: purple, hormones: blue, genes/proteins located in MQTLs: black, genes/proteins located in both MQTL regions and SNP peak positions reported in rice GWAS for root morphological traits: dark blue, Ca+2: red, root traits: pink, root parts: green
Plant hormone-mediated root growth and development
We observed several genes involved in auxin biosynthesis or signaling located in the MQTL regions (Fig. 7 and Supplementary Table S4). Auxins regulate many growth and development dimensions of root through elongating primary roots and root hairs and raising the number of lateral root primordia (Overvoorde et al. 2010). Some members of the rice YUCCA gene family involved in the biosynthesis of indole-3-acetic acid (IAA), including OsYUCCA1 located in MQTL1-5, OsCOW1, OsYUCCA8 and OsNAL7 located in MQTL3-1, play a key role in the root growth (Fujino et al. 2008; Woo et al. 2007). It is reported that overexpression of a YUCCA gene, which encodes the rate limiting enzyme of auxin biosynthesis, significantly increases the proliferation of crown roots depending on the presence of WOX TFs (Zhang et al. 2018). The WOX6 transcription factor lays in MQTL3-3. The WOX transcription is activated by auxin, which then initiates the crown root development in rice by establishing the YUC-Auxin-WOX module (Zhang et al. 2018). WOX was also found to interact with ERF and bind to the OsRR2 (MQTL2-3) promoter as a response regulator of cytokinin signaling to regulate crown root morphological traits, root number, root to shoot ratio and root volume (Zhao et al. 2015). As a RING-H2 membrane-anchor E3 ubiquitin ligase, EL5 (MQTL2-5) preserves cell viability after initiating root primordia through cytokinin-mediated signaling (Koiwai et al. 2007).
A rice zinc finger protein (OsZFP) in MQTL1-2 interacts with a cyclophilin (LRT2/OsCYP2) and influences root development through the IAA pathway (Cui et al. 2017). LRT2 (LATERAL ROOTLESS2), the cyclophilin protein (OsCYP2) located in MQTL2.1 regulates auxin signaling and lateral root initiation in rice (Zheng et al. 2013). LRT2/OsCYP2 was found to contribute to degrading AUX/IAA proteins (Kang et al. 2013). Several OsAUX/IAA15 and OsAUX/IAA22 members are found in MQTL5-1 and MQTL6-3 regions, which act as the negative regulators of ARFs (Mockaitis and Estelle 2008). Based on our GO analysis results, regulation of ARF GTPase activity and ARF protein signal transduction were enriched in the biological processes. The activity of ARF GTPase activator was also enriched among the molecular functions. Two major protein families that regulate auxin signaling include ARF proteins as the DNA-binding transcriptional regulators of auxin responses and, Aux/IAA proteins as the negative regulators of ARF (Mockaitis and Estelle 2008). The rice lateral root formation requires Auxin/IAA protein and ARF-mediated signaling (Yamauchi et al. 2019). OsARF1 was found to lie in MQTL5-2, OsARF7 in MQTL2-5 and OsARF16 in MQTL6-2. It is reported that OsARF7 and OsARF16 regulate RSA through AUX/LAX in auxin signaling (Lee et al. 2019; Mockaitis and Estelle 2008; Yamauchi et al. 2019). As functional auxin influx carriers, the AUX/LAX family of proteins mediate auxin-related developmental programs. Responses to abiotic stress, root gravitropism and root hair development are controlled by AUX1. Lateral root development is affected by LAX3 and AUX1 (Swarup and Bhosale 2019; Swarup and Péret 2012). OsAUX3 is expressed in root hairs, primary and lateral roots. OsAUX3 mutations were found to decrease the length of primary roots and lateral root density and increase the length of root hairs (Wang et al. 2019a). OsAUX3/LAX2 was located in MQTL3-2. Adventitious root formation in Arabidopsis to be affected by LBD18 and LBD16 functioning downstream of ARF19 and ARF7 (Lee et al. 2019). We found LBD38 gene in the MQTL3-4 region that is a class II type LBD protein, acting as a transcriptional activator (Pan et al. 2017).
Oryza sativa root architecture associated 1 (OsRAA1) located in MQTL1-2 play a role in the auxin-mediated development of root in rice (Ge et al. 2004). The transition from metaphase to anaphase during cell division is inhibited by OsRAA1, and its degradation by the ubiquitin–proteasome system is essential for initiating anaphase in mitosis during root development (Xu et al. 2010). OsMADS57, a MADS-box transcription factor, was found in MQTL2-8. The inhibition of seminal root elongation in OsMADS57 mutants can be explained by elevated auxin and polar auxin transport to the root tips of the mutant plants (Huang et al. 2019). MAIF1, a rice F-box domain gene located in MQTL2-5, was found to contribute to several signaling pathways in the regulation of root growth. Auxin, abscisic acid, cytokinin and abiotic stress can induce the expression of MAIF1. The root growth in rice can be stimulated through the over-expression of MAIF1 (Yan et al. 2011).
OsEIL1, located in the MQTL3-3 interval, activates the OsYUCCA8 transcription and, therefore, auxin biosynthesis, regulating ethylene-inhibited root elongation in rice seedlings (Qin et al. 2017). ACC (1-aminocyclopropane-1-carboxylic acid) oxidase gene of OsACO1 (MQTL9-4) was enhanced in transgenic plants overexpressing OsEIL1 (Mao et al. 2006). Ethylene signaling is regulated in rice through activating OsEIL1 by RAVL1 (MQTL4-4), as an upstream component of brassinosteroid biosynthesis and signaling (Zhu et al. 2018). Mediating the degradation of OsEIL1 through the ubiquitination pathway by OsEBF1 (MQTL6-4) as an E3 ligase suggested the negative regulation of the ET signaling pathway in response to the infestation of BPH (Ma et al. 2020).
Abscisic acid (ABA) plays a key role in raising the length of root hairs in plants. It can accumulate auxin along the root hair by promoting the biosynthesis and polar transport of auxin in the root tip (Qin et al. 2017). Expression of some ABA biosynthesis genes could regulate early signaling genes, e.g. OsPP2C68 (MQTL9-2), SAPK6 (MQTL2-5) and late responsive gene OsLEA3 (MQTL6-3) during droughts (Xiong et al. 2014). SAPK6 is effective in root elongation during drought conditions (Xiong et al. 2014), and longer root hairs were reported in transgenic rice overexpressing SAPK10 (MQTL3-4) (Qin et al. 2017). Jasmonate (JA) signaling components such as OsJAZ1 (MQTL4-6) was also found, of which OsJAZ1 is reported to play a negative role in regulating drought tolerance through ABA and JA pathways (Fu et al. 2017). The degradation of jasmonate ZIM-domain (JAZ) proteins releases TFs known to interact with JAZ proteins, which can generate responses to growth and stress (Tian et al. 2019). OsJAZ1 was found to be associated with the root weight, and potentially regulate rice root development at different developmental steps (Pan et al. 2017). OsCKI1, a rice casein kinase I detected in MQTL2-6, was found to contribute to different hormone-signaling pathways and regulate the development of rice lateral roots (Liu et al. 2003).
Root growth angle
Deeper rooting 1 (DRO1) (MQTL9-4), regulated through ARF by auxin, appears to contribute to elongating root tip cells causing the asymmetric growth of root and its downward bending in response to gravity. Induction of the DRO1 expression raises the angle of root growth, increasing the downward growth of the root (Uga et al. 2013). Controlling RSA by DRO1 raises the rice yield during drought stress (Uga et al. 2013). Moreover, DRO1 was found to be expressed in the root tip in the vicinity of the root apical meristem as well as in basal shoots in the crown root primordia (Uga et al. 2013). Interestingly, we further found some other candidate genes which might regulate branching and angle in lateral roots; for example, OsLAZY1 (MQTL6-1), OsSOL1 (suppressor of lazy1) (MQTL6-1) and OsTAC3 (MQTL3-5). DRO1-related genes, including TAC1 and LAZY1, belong to the IGT gene family, contain a C-terminal EAR-like motif IVLEI and are observed in different plant phyla (Ashraf et al. 2019). The gravitropism of the root and shoots is affected by LAZY1 and its orthologues in rice, maize, Medicago and Arabidopsis (Ashraf et al. 2019). HSFA2D located in MQTL3.1 functions as a positive regulator of the LAZY1-dependent asymmetric distribution of auxin in the upstream. Auxin causes the asymmetrical expression of WOX6 (MQTL3.3) to connect gravitropism to controlling the tiller angle in rice (Zhang et al. 2018). PROG1 (PROSTRATE GROWTH1) (MQTL7.1), and LPA1 (LOOSE PLANT ARCHITECTURE1) (MQTL3.2) were reported as the main genetic regulators of the tiller angle in rice (Li et al. 2007).
Lateral root development
The enhancement of lateral root formation causes a potentially useful adaptation to drought in lowland rice under drought stress (Hazman and Brown 2018). A panel of lateral root development-related potential candidate genes was located in MQTL regions (Fig. 7 and Supplementary Table S4). O. sativa drought stress response-1 (OsDSR-1), a calmodulin-like gene, was found in MQTL10-1. The binding of its protein to Ca2+ leads to conformational changes, and increases drought tolerance by scavenging the reactive oxygen species in rice (Yin et al. 2017). The expression of OsDSR-1 in Arabidopsis was found to enhance developing lateral root in high K+ concentrations and improve sensitivity to ABA (Yin et al. 2011). OsWRKY31 found in MQTL6-3 can act in the auxin signal transduction pathway, and its ectopic expression reduced the length and formation of lateral roots (Zhang et al. 2008). OsWRKY76 lying in MQTL9-3 was found to regulate cellular responses to abiotic and biotic types of stress (Yokotani et al. 2013) partly by regulating the expression of RSOsPR10 in the lateral roots of rice, improving the mass and growth of root and increasing tolerance to high salt environments and soil desiccation (Yamamoto et al. 2018).
Root diameter
As a transcriptional activator and stress-responsive protein lying in MQTL11-2, OsNAC5 improves stress tolerance through the up-regulation of stress-inducible genes of rice, e.g. DjC10 (MQTL1-4) and OsLEA3 (MQTL6-3). The overexpression of OsNAC5 increases the root diameter in rice and improves grain yield and drought tolerance (Jeong et al. 2013). The root-specific overexpression of OsNAC10 (MQTL7-6) significantly improves the rice yield, especially during drought stress (Redillas et al. 2012). OsNAC10 was found to induce 34 root-specific target genes such as those encoding cytochrome P450, mitogen-activated protein kinase kinases (MAPKK), LEA and TFs such as WRKYs and NACs (Jeong et al. 2010). As a key factor in preserving calcium homoeostasis, calcium‐transporting ATPase serves as an early response to drought, salinity stress and low temperature in plant cells (Knight 1999). A member of this family, ACA8 (Ca2+ P-Type ATPase 8) (Knight 1999), was located in MQTL10-2. ACA8 was up-regulated in OsNAC5 overexpressed plants (Jeong et al. 2013).
Cell wall organization
Plant-type cell wall organization was significantly enriched based on the GO analysis of DECGs. Several genes of expansins were found in MQTL positions including OsEXPA8, EXPA9 (MQTL1-2), ExpA10 (MQTL4-4), ExpA4 (MQTL5-2), EXPA14, EXPA13, EXPA22 (MQTL2-3) and EXPA6 (MQTL3-3) (Fig. 7 and Supplementary Table S4). With key roles in the cell growth of plants and cell elongation of root tips, expansins serve as a family of closely related nonenzymatic proteins in the cell wall of plants (Shin et al. 2005). The cell wall relaxation required for the cell expansion can be achieved through the actions of expansin genes (EXPs) and endoglucanases (GLUs) (Hayashi et al. 1984; McQueen-Mason et al. 1992; Okamoto and Okamoto 1995). Some members of the endoglucanases such as OsGLU1 (MQTL3-3), OsGLU14 (MQTL2-1) and OsGLU30 (MQTL9-5) were found in MQTL regions. OsGLU1 mutation leads to a decrease in the cellulose content and cell elongation (Zhou et al. 2006). OsARD4 lying in MQTL10-2 encodes an acireductone dioxygenase, which improves RSA in rice (Ramanathan et al. 2018). The OsARD4 overexpression contributed to the root growth by increasing the rates of radical emergence, elongation of primary roots and initiation of lateral/crown roots and raising the root biomass compared to that of nontransgenic plants (Ramanathan et al. 2018). Plant root lignification plays an effective role in tolerating abiotic stresses such as drought (Seo et al. 2020). As an important enzyme of the lignin biosynthetic pathway, Cinnamoyl-CoA reductase (CCR) controls the quality and quantity of lignin (Jones et al. 2001). The other important enzymes in the lignin biosynthesis include C4H, PAL, PRX and CAD (Lee et al. 2016), whose members including CAD3 (MQTL10-2), CAD1 (MQTL10-1), CCR3 (MQTL5-1), CCR19 (MQTL9-3), PAL1 (MQTL2-6), PAL, PAL8 (MQTL2-6), PAL5, PAL3 (MQTL2-6), PAL1, PAL6, PAL06, ZB8, pal/zb8 (MQTL2-6) and Prx15 (MQTL1-3) were found in MQTL regions.
Multi-level data integration to discover novel candidate genes
A large number of studies have shown that QTL mapping and genome-wide association study (GWAS) are two complementary methods for mapping causal genes and dissecting the genetic basis of the traits of interest (Mahuku et al. 2016; Tao et al. 2013). Meta-analysis is an effective approach to integrate QTLs from several independent experiments, and GWAS is a robust method for detecting significant effects in diverse germplasms, each has its own merits and demerits, which can complement each other (Sonah et al. 2015). A major challenge of QTL analysis is the reduction of the CI to discover the most relevant genomic regions, and the integration of GWAS results with meta-analysis of QTLs could help to do so very effectively. In our study, significant overlaps existed between the MQTLs detected using meta-analysis and the SNPs detected using the GWAS method linked to RSA traits in rice genome. Out of the 64 identified MQTLs, 52 MQTLs were overlapped with 171 SNP peak positions reported for root morphological traits (Figs. 5 and 6). Among them, all MQTLs on chromosomes 1, 2, 8, 10 and 12 were in co-linear region with GWAS signals. This integrative approach leads to the detection of 49 rice genes located in both the MQTL regions and SNP peak positions reported in rice GWAS for root morphological traits including genes encoding EXPA6, MADS59 and RR3. In addition, looking at a 25 kb region on either side of the SNP peak positions, 755 genes were found comprising many novel genes and some genes with known RSA-related functions such as DRO1, WRKY62, CKI1, ARF8, SAUR30 and HOX11 (Supplementary Table S11).
To find the chromosome regions with the most contribution to a desired phenotype, the QTL-overview index is calculated based on the number of original QTLs, accuracy in mapping for these QTLs and the high phenotypic variance (Chardon et al. 2004). In the current research, the genes located in the significant peaks of both SNPs and the QTL-overview for root architecture traits were considered as novel candidate genes (Table 4). These 10 novel genes are interesting candidates for functional analysis, while the transcript level of rNBS41, GELP58, EME1 and bZIP64 was also drought-responsive in the rice roots.
Conclusion
Root system architecture plays critical roles in plant growth and productivity as well as plant tolerance to abiotic stresses such as drought. Therefore, improving RSA through molecular breeding or genetic engineering is a promising strategy to develop the yield and stress tolerance in rice. In the present research, a genome-wide meta-analysis of RSA in rice led to the identification of 64 MQTLs. Interestingly, seven of these MQTLs have a physical length of around 500 kb and genetic distance of less than two cM, contained at least ten initial QTLs while the original QTLs were derived from six to nine independent studies, and cover a mean phenotypic variance of 14.8%. Two of them (MQTL1-7 and MQTL6-6) were further confirmed with SNP peak positions reported in rice GWAS for root morphological traits. Conclusively, these MQTL regions can be appropriate for MQTL-assisted breeding in future genetic improvement programs of RSA-related traits in rice. On the other hand, the DEGs in the rice root under drought conditions were detected through the analysis of RNA-seq and microarray datasets, and the MQTL regions associated with RSA were explored to find the drought-responsive genes in the rice root. Gene ontology enrichment analysis of the genes located in these MQTLs provides evidence that the genes with RSA associated functions are clustered and contributed to the QTL traits. Multiple genes with functions associated with RSA and drought tolerance were found in the MQTLs comprising some genes from YUCCA, WOX, RR, LRT2/CYP2, AUX/IAA, ARF, RAA, MAIF1, EIL, PP2C, SAPK, DRO, DSR, WRKY, RSOsPR, NAC, C4H, PAL, PRX and CAD, EXPA, GLU and ARD families. The genes located within both the SNP positions and in the QTL-overview peaks for root architecture traits are suggested as interesting candidate genes for further functional analysis. Conclusively, we integrated meta-analysis of QTLs associated with RSA traits in normal and drought stress conditions, GWAS studies results for root morphological traits, and the transcriptome analysis data of the root under drought conditions leading to find 189 candidate genes (Supplementary Fig. S5), which might play an important role in rice RSA under drought stress. After functional analysis, the promising candidate genes would be applicable for root genetic engineering aimed at improving yield potential, stability and performance in drought conditions.
Availability of data and materials
All data supporting the conclusions of this article are provided within the article and its supplementary.
References
Anis GB, Zhang Y, Islam A, Zhang Y, Cao Y, Wu W, Cao L, Cheng S (2019) RDWN6XB, a major quantitative trait locus positively enhances root system architecture under nitrogen deficiency in rice. BMC Plant Biol 19:12
Araki H, Morita S, Tatsumi J, Iijima M (2002) Physiol-morphological analysis on axile root growth in upland rice. Plant Prod Sci 5:286–293
Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, Joets J (2004) BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics 20:2324–2326
Ashraf A, Rehman OU, Muzammil S, Leon J, Naz AA, Rasool F, Ali GM, Zafar Y, Khan MR (2019) Evolution of Deeper Rooting 1-like homoeologs in wheat entails the C-terminus mutations as well as gain and loss of auxin response elements. PLoS One 14:e0214145
Ballini E, Morel JB, Droc G, Price A, Courtois B, Notteghem JL, Tharreau D (2008) A genome-wide meta-analysis of rice blast resistance genes and quantitative trait loci provides new insights into partial and complete resistance. MPMI 21:859–868
Bettembourg M, Dardou A, Audebert A, Thomas E, Frouin J, Guiderdoni E, Ahmadi N, Perin C, Dievart A, Courtois B (2017) Genome-wide association mapping for root cone angle in rice. Rice 10:1–17
Bilgrami SS, Ramandi HD, Shariati V, Razavi K, Tavakol E, Fakheri BA, Mahdi Nezhad N, Ghaderian M (2020) Detection of genomic regions associated with tiller number in Iranian bread wheat under different water regimes using genome-wide association study. Sci Rep 10:14034
Biscarini F, Cozzi P, Casella L, Riccardi P, Vattari A, Orasen G, Perrini R, Tacconi G, Tondelli A, Biselli C (2016) Genome-wide association study for traits related to plant and grain morphology, and root architecture in temperate rice accessions. PLoS One 11:e0155425
Blum A (2005) Drought resistance, water-use efficiency, and yield potential—are they compatible, dissonant, or mutually exclusive? Aust J Agric Res 56:1159–1168
Carvalho P, Azam-Ali S, Foulkes MJ (2014) Quantifying relationships between rooting traits and water uptake under drought in Mediterranean barley and durum wheat. J Integr Plant Biol 56:455–469
Catolos M, Sandhu N, Dixit S, Shamsudin NAA, Naredo MEB, McNally KL, Henry A, Diaz MG, Kumar A (2017) Genetic loci governing grain yield and root development under variable rice cultivation conditions. Front Plant Sci 8:1–17
Chardon F, Virlon B, Moreau L, Falque M, Joets J, Decousset L, Murigneux A, Charcosset A (2004) Genetic architecture of flowering time in maize as inferred from quantitative trait loci meta-analysis and synteny conservation with the rice genome. Genetics 168:2169–2185
Courtois B, Ahmadi N, Khowaja F, Price AH, Rami J-F, Frouin J, Hamelin C, Ruiz M (2009) Rice root genetic architecture: meta-analysis from a drought QTL database. Rice 2:115–128
Courtois B, Audebert A, Dardou A, Roques S, Ghneim-Herrera T, Droc G, Frouin J, Rouan L, Gozé E, Kilian A (2013) Genome-wide association mapping of root traits in a japonica rice panel. PLoS One 8:e78037
Courtois B, Shen L, Petalcorin W, Carandang S, Mauleon R, Li Z (2003) Locating QTLs controlling constitutive root traits in the rice population IAC 165× Co39. Euphytica 134:335–345
Cui P, Liu H, Ruan S, Ali B, Gill RA, Ma H, Zheng Z, Zhou W (2017) A zinc finger protein, interacted with cyclophilin, affects root development via IAA pathway in rice. J Integr Plant Biol 59:496–505
Darvasi A, Soller M (1997) A simple method to calculate resolving power and confidence interval of QTL map location. Behav Genet 27:125–132
Darvasi A, Weinreb A, Minke V, Weller J, Soller M (1993) Detecting marker-QTL linkage and estimating QTL gene effect and map location using a saturated genetic map. Genetics 134:943–951
Darzi-Ramandi H, Shariati JV, Tavakol E, Najafi-Zarini H, Bilgrami SS, Razavi K (2017) Detection of consensus genomic regions associated with root architecture of bread wheat on groups 2 and 3 chromosomes using QTL meta-analysis. Aust J Crop Sci 11:777–785
de Dorlodot S, Forster B, Pagès L, Price A, Tuberosa R, Draye X (2007) Root system architecture: opportunities and constraints for genetic improvement of crops. Trends Plant Sci 12:474–481
De Smet I, White PJ, Bengough AG, Dupuy L, Parizot B, Casimiro I, Heidstra R, Laskowski M, Lepetit M, Hochholdinger F (2012) Analyzing lateral root development: how to move forward. Plant Cell 24:15–20
Diaz-Garcia L, Covarrubias-Pazaran G, Schlautman B, Zalapa J (2017) SOFIA: an R package for enhancing genetic visualization with Circos. J Hered 108:443–448
Fu J, Wu H, Ma S, Xiang D, Liu R, Xiong L (2017) OsJAZ1 attenuates drought resistance by regulating JA and ABA signaling in rice. Front Plant Cci 8:2108
Fujino K, Matsuda Y, Ozawa K, Nishimura T, Koshiba T, Fraaije MW, Sekiguchi H (2008) NARROW LEAF 7 controls leaf shape mediated by auxin in rice. Mol Genet Genom 279:499–507
Ge L, Chen H, Jiang JF, Zhao Y, Xu ML, Xu YY, Tan KH, Xu ZH, Chong K (2004) Overexpression of OsRAA1 causes pleiotropic phenotypes in transgenic rice plants, including altered leaf, flower, and root development and root response to gravity. Plant Physiol 135:1502–1513
Gelli M, Konda AR, Liu K, Zhang C, Clemente TE, Holding DR, Dweikat IM (2017) Validation of QTL mapping and transcriptome profiling for identification of candidate genes associated with nitrogen stress tolerance in sorghum. BMC Plant Biol 17:123
Gewin V (2010) Food: an underground revolution. Nature 466:552–553
Goffinet B, Gerber S (2000) Quantitative trait loci: a meta-analysis. Genetics 155:463–473
Gruber BD, Giehl RF, Friedel S, von Wirén N (2013) Plasticity of the Arabidopsis root system under nutrient deficiencies. Plant Physiol 163:161–179
Guo B, Sleper D, Lu P, Shannon J, Nguyen H, Arelli P (2006) QTLs associated with resistance to soybean cyst nematode in soybean: meta-analysis of QTL locations. Crop Sci 46:595–602
Guo J, Chen L, Li Y, Shi Y, Song Y, Zhang D, Li Y, Wang T, Yang D, Li C (2018) Meta-QTL analysis and identification of candidate genes related to root traits in maize. Euphytica 214:223
Hao Z, Li X, Liu X, Xie C, Li M, Zhang D, Zhang S (2010) Meta-analysis of constitutive and adaptive QTL for drought tolerance in maize. Euphytica 174:165–177
Hayashi T, Wong Y-S, Maclachlan G (1984) Pea xyloglucan and cellulose. II. Hydrolysis by pea endo-1,4-glucanase. Plant Physiol 75:605–610
Hazman M, Brown KM (2018) Progressive drought alters architectural and anatomical traits of rice roots. Rice 11:62
Horii H, Nemoto K, Miyamoto N, Harada J (2006) Quantitative trait loci for adventitious and lateral roots in rice. Plant Breed 125:198–200
Huang S, Liang Z, Chen S, Sun H, Fan X, Wang C, Xu G, Zhang Y (2019) A transcription factor, OsMADS57, regulates long-distance nitrate transport and root elongation. Plant Physiol 180:882–895
Iannucci A, Marone D, Russo MA, De Vita P, Miullo V, Ferragonio P, Blanco A, Gadaleta A, Mastrangelo AM (2017) Mapping QTL for root and shoot morphological traits in a durum wheat× T. dicoccum segregating population at seedling stage. Int J Genomics 2017:6876393
Ikeda H, Kamoshita A, Manabe T (2007) Genetic analysis of rooting ability of transplanted rice (Oryza sativa L.) under different water conditions. J Exp Bot 58:309–318
Jeong JS, Kim YS, Baek KH, Jung H, Ha S-H, Do Choi Y, Kim M, Reuzeau C, Kim J-K (2010) Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol 153:185–197
Jeong JS, Kim YS, Redillas MC, Jang G, Jung H, Bang SW, Choi YD, Ha SH, Reuzeau C, Kim JK (2013) OsNAC5 overexpression enlarges root diameter in rice plants leading to enhanced drought tolerance and increased grain yield in the field. Plant Biotechnol J 11:101–114
Jia Z, Liu Y, Gruber BD, Neumann K, Kilian B, Graner A, Von Wirén N (2019) Genetic dissection of root system architectural traits in spring barley. Front Plant Sci 10:400
Jones L, Ennos AR, Turner SR (2001) Cloning and characterization of irregular xylem4 (irx4): a severely lignin-deficient mutant of Arabidopsis. Plant J 26:205–216
Ju C, Zhang W, Liu Y, Gao Y, Wang X, Yan J, Yang X, Li J (2018) Genetic analysis of seedling root traits reveals the association of root trait with other agronomic traits in maize. BMC Plant Biol 18:171
Jung K-H, An G (2012) Application of MapMan and RiceNet drives systematic analyses of the early heat stress transcriptome in rice seedlings. J Plant Biol 55:436–449
Kadam NN, Tamilselvan A, Lawas LM, Quinones C, Bahuguna RN, Thomson MJ, Dingkuhn M, Muthurajan R, Struik PC, Yin X (2017) Genetic control of plasticity in root morphology and anatomy of rice in response to water deficit. Plant Physiol 174:2302–2315
Kamoshita A, Zhang J, Siopongco J, Sarkarung S, Nguyen H, Wade L (2002) Effects of phenotyping environment on identification of quantitative trait loci for rice root morphology under anaerobic conditions. Crop Sci 42:255–265
Kang B, Zhang Z, Wang L, Zheng L, Mao W, Li M, Wu Y, Wu P, Mo X (2013) OsCYP2, a chaperone involved in degradation of auxin-responsive proteins, plays crucial roles in rice lateral root initiation. Plant J 74:86–97
Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6:1–10
Khahani B, Tavakol E, Shariati V, Fornara F (2020) Genome wide screening and comparative genome analysis for Meta-QTLs, ortho-MQTLs and candidate genes controlling yield and yield-related traits in rice. BMC Genomics 21:1–24
Kim PS, Kim DM, Kang JW, Lee HS, Ahn SN (2015) QTL mapping of Rice root traits at different NH4+ levels in hydroponic condition. Plant Breed Biotechnol 3:244–252
Kitomi Y, Kanno N, Kawai S, Mizubayashi T, Fukuoka S, Uga Y (2015) QTLs underlying natural variation of root growth angle among rice cultivars with the same functional allele of DEEPER ROOTING 1. Rice 8:16
Kitomi Y, Nakao E, Kawai S, Kanno N, Ando T, Fukuoka S, Irie K, Uga Y (2018) Fine mapping of QUICK ROOTING 1 and 2, quantitative trait loci increasing root length in rice. G3 8:727–735
Knight H (1999) Calcium signaling during abiotic stress in plants. Int Rev Cytol 195:269–324
Koiwai H, Tagiri A, Katoh S, Katoh E, Ichikawa H, Minami E, Nishizawa Y (2007) RING-H2 type ubiquitin ligase EL5 is involved in root development through the maintenance of cell viability in rice Plant J 51:92–104
Lanaud C, Fouet O, Clément D, Boccara M, Risterucci A-M, Surujdeo-Maharaj S, Legavre T, Argout X (2009) A meta–QTL analysis of disease resistance traits of Theobroma cacao L. Mol Breed 24:361–374
Lee D-K, Jung H, Jang G, Jeong JS, Kim YS, Ha S-H, Do Choi Y, Kim J-K (2016) Overexpression of the OsERF71 transcription factor alters rice root structure and drought resistance. Plant Physiol 172:575–588
Lee HW, Cho C, Pandey SK, Park Y, Kim M-J, Kim J (2019) LBD16 and LBD18 acting downstream of ARF7 and ARF19 are involved in adventitious root formation in Arabidopsis. BMC Plant Biol 19:46
Li J, Zhu S, Song X, Shen Y, Chen H, Yu J, Yi K, Liu Y, Karplus VJ, Wu P (2006) A rice glutamate receptor–like gene is critical for the division and survival of individual cells in the root apical meristem. Plant Cell 18:340-349
Li P, Wang Y, Qian Q, Fu Z, Wang M, Zeng D, Li B, Wang X, Li J (2007) LAZY1 controls rice shoot gravitropism through regulating polar auxin transport. Cell Res 17:402–410
Li J, Wang D, Xie Y, Zhang H, Hu G, Li J, Dai A, Liu L, Li Z (2011) Development of upland rice introgression lines and identification of QTLs for basal root thickness under different water regimes. J Genet Genomics 38:547–556
Li J, Han Y, Liu L, Chen Y, Du Y, Zhang J, Sun H, Zhao Q (2015) qRT9, a quantitative trait locus controlling root thickness and root length in upland rice. J Exp Bot 66:2723–2732
Li X, Guo Z, Lv Y, Cen X, Ding X, Wu H, Li X, Huang J, Xiong L (2017) Genetic control of the root system in rice under normal and drought stress conditions by genome-wide association study. PLoS Genet 13:e1006889
Liang YS, Zhan XD, Wang HM, Gao ZQ, chuan Lin Z, Chen DB, Shen XH, Cao LY, Cheng SH (2013) Locating QTLs controlling several adult root traits in an elite Chinese hybrid rice. Gene 526:331–335
Liu W, Xu ZH, Luo D, Xue HW (2003) Roles of OsCKI1, a rice casein kinase I, in root development and plant hormone sensitivity. Plant J 36:189–202
Löffler M, Schön CC, Miedaner T (2009) Revealing the genetic architecture of FHB resistance in hexaploid wheat (Triticum aestivum L.) by QTL meta-analysis. Mol Breed 23:473–488
Lou Q, Chen L, Mei H, Wei H, Feng F, Wang P, Xia H, Li T, Luo L (2015) Quantitative trait locus mapping of deep rooting by linkage and association analysis in rice. J Exp Bot 66:4749–4757
Ma F, Yang X, Shi Z, Miao X (2020) Novel crosstalk between ethylene-and jasmonic acid-pathway responses to a piercing–sucking insect in rice. New Phytol 225:474–487
Maccaferri M, El-Feki W, Nazemi G, Salvi S, Canè MA, Colalongo MC, Stefanelli S, Tuberosa R (2016) Prioritizing quantitative trait loci for root system architecture in tetraploid wheat. J Exp Bot 67:1161–1178
Mahuku G, Chen J, Shrestha R, Narro LA, Guerrero KVO, Arcos AL, Xu Y (2016) Combined linkage and association mapping identifies a major QTL (qRtsc8-1), conferring tar spot complex resistance in maize. Theor Appl Genet 129:1217–1229
Mai NTP, Mai CD, Nguyen HV, Le KQ, Duong LV, Tran TA, To HTM (2020) Discovery of new genetic determinants of morphological plasticity in rice roots and shoots under phosphate starvation using GWAS. J Plant Physiol 257:153340
Malamy J (2005) Intrinsic and environmental response pathways that regulate root system architecture. Plant Cell Environ 28:67–77
Mao C, Wang S, Jia Q, Wu P (2006) OsEIL1, a rice homolog of the Arabidopsis EIN3 regulates the ethylene response as a positive component. Plant Mol Biol 61:141
Martinez AK, Soriano JM, Tuberosa R, Koumproglou R, Jahrmann T, Salvi S (2016) Yield QTLome distribution correlates with gene density in maize. Plant Sci 242:300–309
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
McQueen-Mason S, Durachko DM, Cosgrove DJ (1992) Two endogenous proteins that induce cell wall extension in plants. Plant Cell 4:1425–1433
Mockaitis K, Estelle M (2008) Auxin receptors and plant development: a new signaling paradigm. Annu Rev Cell Dev Biol 24:55–80
Nagelkerke NJ (1991) A note on a general definition of the coefficient of determination. Biometrika 78:691–692
Niones JM, Inukai Y, Suralta RR, Yamauchi A (2015) QTL associated with lateral root plasticity in response to soil moisture fluctuation stress in rice. Plant Soil 391:63–75
Obara M, Takeda T, Hayakawa T, Yamaya T (2011) Mapping quantitative trait loci controlling root length in rice seedlings grown with low or sufficient supply using backcross recombinant lines derived from a cross between Oryza sativa L. and Oryza glaberrima Steud. Soil Sci Plant Nutr 57:80-92
Okamoto A, Okamoto H (1995) Two proteins regulate the cell wall extensibility and the yield threshold in glycerinated hollow cylinders of cowpea hypocotyl. Plant, Cell Environ 18:827–830
Overvoorde P, Fukaki H, Beeckman T (2010) Auxin control of root development. Cold Spring Harb Perspect Biol 2(6):a001537
Paez-Garcia A, Motes CM, Scheible W-R, Chen R, Blancaflor EB, Monteros MJ (2015) Root traits and phenotyping strategies for plant improvement. Plants 4:334–355
Pan Y, Hu X, Li C, Xu X, Su C, Li J, Song H, Zhang X, Pan Y (2017) SlbZIP38, a tomato bZIP family gene downregulated by abscisic acid, is a negative regulator of drought and salt stress tolerance. Genes 8:402
Pariasca-Tanaka J, Baertschi C, Wissuwa M (2020) Identification of loci through genome-wide association studies to improve tolerance to sulfur deficiency in rice. Front Plant Sci 10:1668
Phung NTP, Mai CD, Hoang GT, Truong HTM, Lavarenne J, Gonin M, Le Nguyen K, Ha TT, Do VN, Gantet P (2016) Genome-wide association mapping for root traits in a panel of rice accessions from Vietnam. BMC Plant Biol 16:1–19
Price AH, Steele K, Moore B, Jones R (2002) Upland rice grown in soil-filled chambers and exposed to contrasting water-deficit regimes: II. Mapping quantitative trait loci for root morphology and distribution. Field Crops Res 76:25–43
Qin H, Liu Z, Wang Y, Xu M, Mao X, Qi H, Yin Z, Li Y, Jiang H, Hu Z (2018) Meta-analysis and overview analysis of quantitative trait locis associated with fatty acid content in soybean for candidate gene mining. Plant Breed 137:181–193
Qin H, Zhang Z, Wang J, Chen X, Wei P, Huang R (2017) The activation of OsEIL1 on YUC8 transcription and auxin biosynthesis is required for ethylene-inhibited root elongation in rice early seedling development. PLoS Genet 13:e1006955
Qu Y, Mu P, Zhang H, Chen CY, Gao Y, Tian Y, Wen F, Li Z (2008) Mapping QTLs of root morphological traits at different growth stages in rice. Genetica 133:187–200
R Core Team (2018) R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. https://www.R-project.org
Ramanathan V, Rahman H, Subramanian S, Nallathambi J, Kaliyaperumal A, Manickam S, Ranganathan C, Muthurajan R (2018) OsARD4 encoding an acireductone dioxygenase improves root architecture in rice by promoting development of secondary roots. Sci Rep 8:1–15
Redillas MC, Jeong JS, Kim YS, Jung H, Bang SW, Choi YD, Ha SH, Reuzeau C, Kim JK (2012) The overexpression of OsNAC9 alters the root architecture of rice plants enhancing drought resistance and grain yield under field conditions. Plant Biotechnol J 10:792–805
Rong J, Feltus FA, Waghmare VN, Pierce GJ, Chee PW, Draye X, Saranga Y, Wright RJ, Wilkins TA, May OL (2007) Meta-analysis of polyploid cotton QTL shows unequal contributions of subgenomes to a complex network of genes and gene clusters implicated in lint fiber development. Genetics 176:2577–2588
Sabar M, Shabir G, Shah SM, Aslam K, Naveed SA, Arif M (2019) Identification and mapping of QTLs associated with drought tolerance traits in rice by a cross between Super Basmati and IR55419-04. Breed Sci 69:169-178
Sandhu N, Torres RO, Sta Cruz MT, Maturan PC, Jain R, Kumar A, Henry A (2015) Traits and QTLs for development of dry direct-seeded rainfed rice varieties. J Exp Bot 66:225–244
Seo DH, Seomun S, Choi YD, Jang G (2020) Root development and stress tolerance in rice: the key to improving stress tolerance without yield penalties. Int J Mol Sci 21:1807
Shin JH, Jeong DH, Park MC et al (2005) Characterization and transcriptional expression of the α-expansin gene family in rice. Mol Cells 20:210–218
Singhal P, Jan AT, Azam M, Haq QMR (2016) Plant abiotic stress: a prospective strategy of exploiting promoters as alternative to overcome the escalating burden. Front Life Sci 9:52–63
Sonah H, O’Donoughue L, Cober E, Rajcan I, Belzile F (2015) Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soybean. Plant Biotechnol Journal 3:211–221
Soriano JM, Alvaro F (2019) Discovering consensus genomic regions in wheat for root-related traits by QTL meta-analysis. Sci Rep 9:1–14
Sosnowski O, Charcosset A, Joets J (2012) BioMercator V3: an upgrade of genetic map compilation and quantitative trait loci meta-analysis algorithms. Bioinformatics 28:2082–2083
Srividhya A, Vemireddy LR, Ramanarao PV, Sridhar S, Jayaprada M, Anuradha G, Srilakshmi B, Reddy HK, Hariprasad AS, Siddiq EA (2011) Molecular mapping of QTLs for drought related traits at seedling stage under PEG induced stress conditions in rice. Am J Plant Sci 2:190–201
Steele K, Virk D, Kumar R, Prasad S, Witcombe J (2007) Field evaluation of upland rice lines selected for QTLs controlling root traits. Field Crops Res 101:180-186
Subira J, Ammar K, Álvaro F, Del Moral LFG, Dreisigacker S, Royo C (2016) Changes in durum wheat root and aerial biomass caused by the introduction of the Rht-B1b dwarfing allele and their effects on yield formation. Plant Soil 403:291–304
Suji K, Prince KSJ, Mankhar PS, Kanagaraj P, Poornima R, Amutha K, Kavitha S, Biji K, Gomez SM, Babu RC (2012) Evaluation of rice (Oryza sativa L.) near iso-genic lines with root QTLs for plant production and root traits in rainfed target populations of environment. Field Crops Res 137:89–96
Swarup R, Bhosale R (2019) Developmental roles of AUX1/LAX auxin influx carriers in plants. Front Plant Sci 10:1306
Swarup R, Péret B (2012) AUX/LAX family of auxin influx carriers-an overview. Front Plant Sci 3:225
Tao Y, Jiang L, Liu Q, Zhang Y, Zhang R, Ingvardsen CR, Frei UK, Wang B, Lai J, Lübberstedt T, Xu M (2013) Combined linkage and association mapping reveals candidates for Scmv1, a major locus involved in resistance to sugarcane mosaic virus (SCMV) in maize. BMC Plant Biol 13:162
Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Tian J, Cao L, Chen X, Chen M, Zhang P, Cao L, Persson S, Zhang D, Yuan Z (2019) The OsJAZ1 degron modulates jasmonate signaling sensitivity during rice development. Development 146:dev173419
Toyofuku K, Matsunami M, Ogawa A (2015) Genotypic variation in osmotic stress tolerance among rice cultivars and its association with L-type lateral root development. Plant Prod Sci 18:246–253
Tuberosa R, Sanguineti MC, Landi P, Giuliani MM, Salvi S, Conti S (2002) Identification of QTLs for root characteristics in maize grown in hydroponics and analysis of their overlap with QTLs for grain yield in the field at two water regimes. Plant Mol Biol 48:697–712
Uga Y, Okuno K, Yano M (2008) QTLs underlying natural variation in stele and xylem structures of rice root. Breed Sci 58:7-14
Uga Y, Okuno K, Yano M (2010) Fine mapping of Sta1, a quantitative trait locus determining stele transversal area, on rice chromosome 9. Mol Breed 26:533–538
Uga Y, Okuno K, Yano M (2011) Dro1, a major QTL involved in deep rooting of rice under upland field conditions. J Exp Bot 62:2485–2494
Uga Y, Hanzawa E, Nagai S, Sasaki K, Yano M, Sato T (2012) Identification of qSOR1, a major rice QTL involved in soil-surface rooting in paddy fields. Theor Appl Genet 124:75-86
Uga Y, Sugimoto K, Ogawa S, Rane J, Ishitani M, Hara N, Kitomi Y, Inukai Y, Ono K, Kanno N, Inoue H, Takehisa H, Motoyama R, Nagamura Y, Wu J, Matsumoto T, Takai T, Okuno K, Yano M (2013) Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat Genet 45:1097–1102
Uga Y, Kitomi Y, Ishikawa S, Yano M (2015) Genetic improvement for root growth angle to enhance crop production. Breed Sci 65:111-119
Veyrieras J-B, Goffinet B, Charcosset A (2007) MetaQTL: a package of new computational methods for the meta-analysis of QTL mapping experiments. BMC Bioinformatics 8:49
Visscher PM, Goddard ME (2004) Prediction of the confidence interval of quantitative trait loci location. Behav Genet 34:477–482
Wang H, Xu X, Zhan X, Zhai R, Wu W, Shen X, Dai G, Cao L, Cheng S (2013) Identification of qRL7, a major quantitative trait locus associated with rice root length in hydroponic conditions. Breed Sci 63:267–274
Wang F, Longkumer T, Catausan SC, Calumpang CLF, Tarun JA, Cattin-Ortola J, Ishizaki T, Pariasca Tanaka J, Rose T, Wissuwa M (2018) Genome-wide association and gene validation studies for early root vigour to improve direct seeding of rice. Plant Cell Environ 41:2731–2743
Wang M, Qiao J, Yu C, Chen H, Sun C, Huang L, Li C, Geisler M, Qian Q, Jiang DA (2019a) The auxin influx carrier, OsAUX3, regulates rice root development and responses to aluminium stress. Plant Cell Environ 42:1125–1138
Wang X, Liu H, Pang M, Fu B, Yu X, He S, Tong J (2019b) Construction of a high-density genetic linkage map and mapping of quantitative trait loci for growth-related traits in silver carp (Hypophthalmichthys molitrix). Sci Rep 9:1–12
Welcker C, Boussuge B, Bencivenni C, Ribaut J, Tardieu F (2007) Are source and sink strengths genetically linked in maize plants subjected to water deficit? A QTL study of the responses of leaf growth and of anthesis-silking interval to water deficit. J Exp Bot 58:339–349
Wickham H, Averick M, Bryan J, Chang W, McGowan LDA, François R, Grolemund G, Hayes A, Henry L, Hester J (2019) Welcome to the Tidyverse. Journal of Open Source Software 4:1686
Woo Y-M, Park H-J, Su’udi M, Yang J-I, Park J-J, Back K, Park Y-M, An G (2007) Constitutively wilted 1, a member of the rice YUCCA gene family, is required for maintaining water homeostasis and an appropriate root to shoot ratio. Plant Mol Biol 65:125–136
Xiong H, Li J, Liu P, Duan J, Zhao Y, Guo X, Li Y, Zhang H, Ali J, Li Z (2014) Overexpression of OsMYB48-1, a novel MYB-related transcription factor, enhances drought and salinity tolerance in rice. PLoS One 9:e92913
Xu X, Ye J, Yang Y, Zhang M, Xu Q, Feng Y, Yuan X, Yu H, Wang Y, Yang Y (2020) Genome-wide association study of rice rooting ability at the seedling stage. Rice 13:1–15
Xu Y, Cao H, Chong K (2010) APC-targeted RAA1 degradation mediates the cell cycle and root development in plants. Plant Signal Behav 5:218–223
Yamamoto T, Yoshida Y, Nakajima K, Tominaga M, Gyohda A, Suzuki H, Okamoto T, Nishimura T, Yokotani N, Minami E (2018) Expression of RSOsPR10 in rice roots is antagonistically regulated by jasmonate/ethylene and salicylic acid via the activator OsERF87 and the repressor OsWRKY76, respectively. Plant Direct 2:e00049
Yamauchi T, Tanaka A, Inahashi H, Nishizawa NK, Tsutsumi N, Inukai Y, Nakazono M (2019) Fine control of aerenchyma and lateral root development through AUX/IAA-and ARF-dependent auxin signaling. Proc Natl Acad Sci 116:20770–20775
Yan Y-S, Chen X-Y, Yang K, Sun Z-X, Fu Y-P, Zhang Y-M, Fang R-X (2011) Overexpression of an F-box protein gene reduces abiotic stress tolerance and promotes root growth in rice. Mol Plant 4:190–197
Yin X, Huang L, Wang M, Cui Y, Xia X (2017) OsDSR-1, a calmodulin-like gene, improves drought tolerance through scavenging of reactive oxygen species in rice (Oryza sativa L.). Mol Breed 37:75
Yin XM, Rocha PS, Wang ML, Zhu YX, Li LY, Song SF, Xia X (2011) Rice gene OsDSR-1 promotes lateral root development in Arabidopsis under high-potassium conditions. J Plant Biol 54:180–189
Yokotani N, Sato Y, Tanabe S, Chujo T, Shimizu T, Okada K, Yamane H, Shimono M, Sugano S, Takatsuji H, Kaku H, Minami E, Nishizawa Y (2013) WRKY76 is a rice transcriptional repressor playing opposite roles in blast disease resistance and cold stress tolerance. J Exp Bot 64:5085–5097
Yue B, Xiong L, Xue W, Xing Y, Luo L, Xu C (2005) Genetic analysis for drought resistance of rice at reproductive stage in field with different types of soil. Theor Appl Genet 111:1127-1136
Zhang J, Peng Y, Guo Z (2008) Constitutive expression of pathogen-inducible OsWRKY31 enhances disease resistance and affects root growth and auxin response in transgenic rice plants. Cell Res 18:508–521
Zhang N, Yu H, Yu H, Cai Y, Huang L, Xu C, Xiong G, Meng X, Wang J, Chen H, Liu G, Jing Y, Yuan Y, Liang Y, Li S, Smith SM, Li J, Wang Y (2018) A core regulatory pathway controlling rice tiller angle mediated by the LAZY1-dependent asymmetric distribution of auxin. Plant Cell 30:1461–1475
Zhao Y, Cheng S, Song Y, Huang Y, Zhou S, Liu X, Zhou D-X (2015) The interaction between rice ERF3 and WOX11 promotes crown root development by regulating gene expression involved in cytokinin signaling. Plant Cell 27:2469–2483
Zheng B, Yang L, Zhang W, Mao C, Wu Y, Yi K, Liu F, Wu P (2003) Mapping QTLs and candidate genes for rice root traits under different water-supply conditions and comparative analysis across three populations. Theor Appl Genet 107:1505-1515
Zheng B, Yang L, Mao C, Huang Y, Wu P (2008) Comparison of QTLs for rice seedling morphology under different water supply conditions. J Genet Genomics 35:473-484
Zheng H, Li S, Ren B, Zhang J, Ichii M, Taketa S, Tao Y, Zuo J, Wang H (2013) Lateral Rootless2, a cyclophilin protein, regulates lateral root initiation and auxin signaling pathway in rice. Mol Plant 6:1719–1721
Zhou H-L, He S-J, Cao Y-R, Chen T, Du B-X, Chu C-C, Zhang J-S, Chen S-Y (2006) OsGLU1, a putative membrane-bound endo-1, 4-ß-d-glucanase from rice, affects plant internode elongation. Plant Mol Biol 60:137–151
Zhou G, Ren N, Qi J, Lu J, Xiang C, Ju H, Cheng J, Lou Y (2014) The 9‐lipoxygenase Osr9‐LOX1 interacts with the 13‐lipoxygenase‐mediated pathway to regulate resistance to chewing and piercing‐sucking herbivores in rice. Physiol Plant 152:59-69
Zhu XF, De Peng Yuan CZ, Li TY, Xuan YH (2018) RAVL1, an upstream component of brassinosteroid signalling and biosynthesis, regulates ethylene signalling via activation of EIL1 in rice. Plant Biotechnol J 16:1399
Acknowledgements
The authors would like to express their gratitude to Mr. Mohammad Jedari to help in creating the artworks.
Funding
This study was financially supported by the Agricultural Biotechnology Research Institute of Iran (ABRII) (grant No: 013–15-1505–114-97027–971295) and the Biotechnology Development Council of the Islamic Republic of Iran (grant No: 970801).
Author information
Authors and Affiliations
Contributions
PD and HDR performed the meta-analysis, drafted the manuscript and drawing graphs. PD and RMM analyzed the microarray data. The project was conceived, coordinated and supervised by Z-SS, who also revised the manuscript. The final manuscript was checked by SD and GHS. The final version of the manuscript was reviewed and approved by all the authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interests/competing interests.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Communicated by Matthias Wissuwa.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Daryani, P., Darzi Ramandi, H., Dezhsetan, S. et al. Pinpointing genomic regions associated with root system architecture in rice through an integrative meta-analysis approach. Theor Appl Genet 135, 81–106 (2022). https://doi.org/10.1007/s00122-021-03953-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-03953-5