
Abstract
Key message
Simulations highlight the potential of genomic selection to substantially increase genetic gain for complex traits in sugarcane. The success rate depends on the trait genetic architecture and the implementation strategy.
Abstract
Genomic selection (GS) has the potential to increase the rate of genetic gain in sugarcane beyond the levels achieved by conventional phenotypic selection (PS). To assess different implementation strategies, we simulated two different GS-based breeding strategies and compared genetic gain and genetic variance over five breeding cycles to standard PS. GS scheme 1 followed similar routines like conventional PS but included three rapid recurrent genomic selection (RRGS) steps. GS scheme 2 also included three RRGS steps but did not include a progeny assessment stage and therefore differed more fundamentally from PS. Under an additive trait model, both simulated GS schemes achieved annual genetic gains of 2.6–2.7% which were 1.9 times higher compared to standard phenotypic selection (1.4%). For a complex non-additive trait model, the expected annual rates of genetic gain were lower for all breeding schemes; however, the rates for the GS schemes (1.5–1.6%) were still greater than PS (1.1%). Investigating cost–benefit ratios with regard to numbers of genotyped clones showed that substantial benefits could be achieved when only 1500 clones were genotyped per 10-year breeding cycle for the additive genetic model. Our results show that under a complex non-additive genetic model, the success rate of GS depends on the implementation strategy, the number of genotyped clones and the stage of the breeding program, likely reflecting how changes in QTL allele frequencies change additive genetic variance and therefore the efficiency of selection. These results are encouraging and motivate further work to facilitate the adoption of GS in sugarcane breeding.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sugarcane is a major industrial crop predominantly grown in tropical and subtropical regions, and it accounts for more than 70% sugar produced worldwide (Ming et al. 2005). Sugarcane is also increasingly used as a bioenergy crop, e.g. for ethanol production and its total production has increased by threefold over the past 50 years, mainly due to a significant expansion in cultivation area. For instance, China and Brazil increased their acreage between 1973 and 2013 by 237% and 500%, respectively (Zhao and Li 2015). This, and developments in management practices like fertilisation and irrigation techniques, has substantially contributed to an increased total cane production worldwide (Morais et al. 2015). While breeding has considerably contributed to these trends, the current rates of improvement appear to have plateaued in many major production regions over the past few decades. For traits like resistance against smut, significant progress has been made through breeding (Bhuiyan et al. 2013). However, for more complex traits such as tonnes of cane per hectare (TCH), it has been more challenging to increase the rate of genetic gain (Wei and Jackson 2016; Yadav et al. 2020). From a sugarcane breeding perspective, substantial barriers to increased gains are (i) the very long breeding cycle in conventional breeding schemes and (ii) potentially a large proportion of non-additive genetic variance for key commercial traits (Wei and Jackson 2016). Genomic selection (GS), a modern breeding tool that has contributed to increased genetic gains in numerous crop and livestock genetic improvement programs (Gaffney et al. 2015; Cooper et al. 2014; Hickey et al. 2017; García-Ruiz et al. 2016), holds to the potential to accelerate the rate of genetic gain in sugarcane (Yadav et al. 2020). GS is particularly promising because it could help to reduce the breeding cycle length and contribute to a more accurate prediction of clonal performance and breeding values of parents.
There are numerous studies in crops which demonstrate that quantitative traits can be accurately predicted using DNA markers, for example in maize (Riedelsheimer et al. 2012; Bernardo and Yu 2007; Zhao et al. 2012), rice (Spindel et al. 2015), wheat (Heffner et al. 2011; Poland et al. 2012; Rutkoski et al. 2012), sorghum (Hunt et al. 2018; Fernandes et al. 2018), barley (Zhong et al. 2009; Lorenz et al. 2012), rapeseed (Werner et al. 2018) or cassava (Oliveira et al. 2012; Ly et al. 2013). However, for sugarcane, there are only a few studies that investigated the potential of GS. It is important to note that modern sugarcane cultivars are typically interspecific hybrids between Saccharum officinarum and Saccharum spontaneum with an extreme level of ploidy. Recent next-generation DNA sequencing efforts have made significant progress towards improving our understanding of how the highly complex sugarcane genome has evolved over time (Garsmeur et al. 2018; Zhang et al. 2018). With regard to implementing GS, a main potential challenge that could arise from this high genome ploidy is allele dosage, which is known to affect phenotypic trait variation in crops, e.g. in maize or tomato (Osborn et al. 2003). The few studies in sugarcane that have tested the accuracy of genomic prediction of complex traits all assumed a biallelic genetic model. For example, Gouy et al. (2013) investigated genomic prediction accuracies for ten commercially important traits in two sugarcane panels from a commercial breeding program (167 clones each) that were genotyped with 1499 DArT markers. They reported prediction accuracies between 0.11 and 0.62 within panels and 0.13–0.55 between panels, depending on the trait and the prediction method. In a recent study, Deomano et al. (2020) reported average genomic prediction accuracies for sugar content and cane yield between 0.25 and 0.45 in three larger commercial sugarcane panels consisting of almost 2,400 clones using cross-validation of prediction accuracies. Hayes et al. (2021) investigated different genomic prediction approaches in forward validations, i.e. predicting the performance of new germplasm tested in new years and regions, and found similar prediction accuracies for TCH, CCS and fibre content. These levels of prediction accuracy seem encouraging for implementing GS, especially given that a simplified diploid model was assumed in these studies.
The explicit consideration of allele dosage in genomic prediction has been shown to provide a more realistic representation of genotypic class effects and therefore improve the accuracy of genomic prediction. This was demonstrated in several autotetraploid species, including potato (Endelman et al. 2018; Slater et al. 2016), Guinea grass (C Lara et al. 2019) and blueberry (Bem Oliveira et al. 2019). In sugarcane, the polyploidy level and hence the number of chromosome copies varies within and across individuals. Garcia et al. (2013) have quantitatively assessed the level of allele dosage using 1034 genome-wide SNP markers and found that the largest portion of SNP showed multi-allelic calls with estimated dosages ranging from 6 to 20. Using cytogenetic analyses, Piperidis and D'hont (2020) showed that the number of copies in modern sugarcane ranges from 10 to 13. Aitken et al. (2016) developed an Axiom SNP array with over 400,000 SNPs from which about 40,000 were single-dose and covered the whole genome. Single-dosage means that there is only a single copy of an allele for a locus. Studies have demonstrated that single-dose markers (i.e. markers heterozygous and present on only one copy of the homologous chromosomes) are prevalent throughout the genome (Aitken et al. 2005). This shows that the implementation of allele dosage in GS in sugarcane is much more complicated than in other species with precisely defined levels of polyploidy.
Today, all sugarcane breeding programs rely on conventional phenotypic selection which involves (i) the creation of a large number of seedlings through targeted crossing, (ii) the assessment of seedlings in unreplicated progeny assessment trials (PAT), (iii) the testing of the best individuals from the best families that were clonally propagated and planted in partially replicated single-row 10 m plots in clonal assessment trials (CAT) and iv) very intensive final assessment trials (FAT) in which the best clones from the CAT trial are tested in 4-row 10 m plots across 4–5 environments and 3 crops (years). One breeding cycle of a phenotypic selection scheme takes around 9–10 years, which imposes significant challenges for generating rapid genetic gains. Seedlings that were generated through targeted crossing are propagated as clones throughout the whole breeding cycle. This means that recombination through crossing, and hence reshuffling of alleles, is only carried out once every 10 years per individual breeding cycle. Furthermore, key traits such as tonnes of cane per hectare (TCH) were reported to show very low narrow-sense heritabilities, and therefore selection of parents based on their predicted breeding value was reported to be inefficient for such highly complex traits (Wei and Jackson 2016). GS holds the potential to overcome problems associated with the long generation intervals and inaccurate selection, but the question of how to best incorporate the technology in a breeding program needs to be addressed.
Therefore, we adopted a simulation-based evaluation approach in our study. Because of the notorious complexity of the sugarcane genome, we made simplified assumptions of a biallelic genetic model with single-dose markers and QTL that affect a quantitative trait (e.g. TCH) in the base population. Albeit being likely overly simplistic, this was inspired by recent empirical work that showed that of 400,000 genome-wide SNP identified in a panel of sugarcane clones about 40,000 were single-dose (Aitken et al. 2016), and that single-dose markers can represent over 75% of polymorphic markers in an individual cross (George and Aitken 2010; Baker et al. 2010). This simplification also facilitated the simulation of non-additive genetic effects, in particular QTL x QTL epistasis, which is challenging to model in a high ploidy context (i.e. interaction effects between multi-dose QTL). Therefore, we consider our study as a first step towards addressing important features and considerations for the implementation of GS in sugarcane, while undoubtedly further work is required, e.g. to specifically address the effect of allele dosage.
Our simulations were designed to compare a conventional phenotypic selection scheme with two proposed GS-based breeding schemes with regard to genetic gain and changes in genetic variance over time. The objectives were to (i) investigate the effect of GS-based breeding schemes on the rate of genetic gain, (ii) investigate the effect of trait architecture on the rate of genetic gain and genetic variance over several selection cycles through comparisons of an additive with a non-additive genetic model and (iii) assess the cost–benefit ratio associated for the proposed GS schemes.
Materials and methods
Simulation of the base population and additive QTL effects
A base population of 1000 clones was simulated with 100 chromosomes of 110 cM each that were evenly distributed across ten subgenomes. Inspired by empirical work which identified 40,000 out of 400,000 genome-wide SNP as single-dose in a panel of sugarcane (Aitken et al. 2016), and studies which show that single-dose markers can represent over 75% of polymorphic markers in an individual cross (George and Aitken 2010; Baker et al. 2010), we simulated a total of 10,000 biallelic single-dose SNP markers with allele frequencies between 0.3 and 0.6 which were evenly distributed across the 100 chromosomes. The simplification of assuming single-dose loci, which have been shown to be very informative in a high polyploidy (Wu et al. 1992; George and Aitken 2010; Baker et al. 2010) and bivalent pairing of homologous chromosomes during meiosis was made to approximate the extreme genomic complexity of sugarcane. This was also inspired by work that shows that there is evidence of some preferential pairing probably involving chromosomes from the same species (Jannoo et al. 2004; Aitken et al. 2005). These simplified assumptions are unlikely to reflect the full genome complexity of sugarcane, but they facilitated the simulation of non-additive gene action which is challenging under the assumption of multi-dosage and random pairing (see below). Further investigations are needed for more detailed considerations of topics related to genome complexity, in particular allele dosage and random chromosome pairing. A total of 1000 biallelic QTL with intermediate allele frequencies contributing to the simulated quantitative trait were randomly assigned to the 100 chromosomes which resulted in 6–16 QTL per chromosome. We used the random number generator implemented in R (R Core Team 2018) to generate the base population. For each of the 10,000 SNP, we randomly sampled a value for the alternate allele frequency from a uniform distribution ranging from 0.3 to 0.6. Based on that, the total number of alternate alleles per SNP were randomly distributed over the 1000 simulated clones (e.g. for a sampled allele frequency of 0.3, 600 alleles were randomly distributed over 2000 potential copies). For each of the 1000 QTL, we assumed intermediate allele frequencies based on which alternate QTL alleles (i.e. ‘active copies’) were randomly distributed over the 1000 clones. To introduce linkage disequilibrium (LD) among SNP and QTL, we used a burn-in phase of phenotypic selection which resulted in LD levels that were comparable to those observed in a real population of elite sugarcane clones (see below).
QTL effects \(Q\) for the 1000 QTL were randomly sampled from a normal distribution with mean = 0 and variance = 1. The QTL effects ranged from − 2.94 to 2.97. Because the accuracy of genomic selection is critically dependent on the level of linkage disequilibrium (LD) among markers and QTL, we simulated five phenotypic selection cycles as a burn-in phase to allow LD to build up (see below). We calibrated this genome simulation to the actual observed levels of LD observed in 1041 elite sugarcane clones from the Sugar Research Australia breeding program that were genotyped with the Affymetrix sugarcane SNP array, which yielded 23,957 high-quality SNP markers. LD was calculated as r2 among all possible pairs of the 23,957 SNP that were segregating in that sample population. LD was also calculated among all possible SNP pairs in the simulated data after five cycles of burn-in phenotypic selection in 100 simulated clones, and the distribution of LD in the real and simulated data was compared. Figure 1 shows that the distribution of LD in the simulated data was comparable to that observed for the empirical SNP data from a real sugarcane population.

Distribution of linkage disequilibrium (r2) in real and simulated sugarcane populations. The X-axes represent the level of linkage disequilibrium (LD) between pairwise markers, and the Y-axes represent the fraction of marker pairs with this level of LD relative to the total number of marker pairs. In both cases, the proportion of markers with moderate LD (r2 > 0.2) was 0.26. The real data consisted of 1041 elite sugarcane clones that were genotyped for 23,957 single-nucleotide-polymorphism (SNP) markers. The simulated data consisted of 100 clones after five burn-in cycles of phenotypic selection under the additive A_model scenario genotyped for 10,000 SNPs (see materials and methods for details)
Genetic models and quantitative traits
An additive trait genetic model (A_model) was simulated as follows. Firstly, an additive genotypic effect (total genotypic value) for each clone was calculated as the sum of the allele effects \( Q\) across all 1000 QTL. The phenotype was simulated by adding to the additive total genotypic value a random environmental error term that was randomly sampled from a normal distribution with a mean = 0 and a variance = Ve. Ve was calculated as \(\frac{{V_{g} }}{{H^{2} }} - V_{g}\) at each selection stage, following the general formula for broad-sense heritability \(H^{2} = \frac{{V_{g} }}{{V_{g} + V_{e} }}\) where \(V_{g}\) is the total genetic variance (variance of the total genotypic values among genotypes in the population) and \(V_{e}\) is the variance for the residual term. We assumed different estimates of broad-sense heritability for the three phenotypic selection stages, i.e. H2 = 0.15 for the progeny assessment trial (PAT, stage 1), H2 = 0.4 for the clonal assessment trial (CAT, stage 2) and H2 = 0.7 in the final assessment trial (FAT, stage 3), as suggested by Australian sugarcane breeders. Throughout the simulated program, H2 calculations were based on the current genetic variance at the respective stages of the breeding cycle.
The influence of trait genetic architecture was further investigated by introducing non-additive genetic effects to the trait genetic model. The total genotypic value of the quantitative trait that was also affected by dominance and additive-by-additive epistasis (i.e. interactions between two or more QTL) were simulated as the sum of the additive genotypic value and a dominance and epistasis term (ADE_model). Supplementary Figure S1 summarises how the non-additive genetic effects were simulated. This was inspired by earlier work that investigated the relative importance of gene networks on breeding selection (Podlich and Cooper 1998; Cooper et al. 2002). Dominance was simulated by assigning an effect of \(d*Q\) to heterozygous QTL, where \(Q\) designates the additive QTL effect that was defined as described above. The parameter \(d\) was defined as the absolute value from a random draw from a normal distribution with mean = 0 and variance = 0.25. Values of \(d\) that were > 1 were set to 1. Because the dominance term was added to the additive component, in the most extreme case there was full dominance at a heterozygous QTL, i.e. the effect of the heterozygous QTL was equal to the effect of the QTL homozygous for the allele with the sampled additive effect. To define the additive-by-additive epistatic component, a total of 441 interactions involving 674 genome-wide QTL were randomly assigned (Supplementary Figure S1). Epistatic effects were sampled from continuous numbers between − 5.87 and 5.94 (twice the range of the additive QTL effects), with a corresponding U-shaped beta density distribution with alpha = beta = 0.1 (Supplementary Figure S1c). Pairwise epistatic effects were only assumed to be present if both interacting QTL were homozygous for the alternate allele (i.e. A1/A1 and A2/A2 for QTL1 and QTL2 where A is the alternate allele with a sampled additive effect). The amount of non-additivity (the sum of dominance and epistasis effects) in the trait genetic model was defined in preliminary adjustment runs in which the true narrow-sense heritability was calculated in 10 cycles of phenotypic selection. Using the above settings for dominance and epistasis, the true narrow-sense heritability (i.e. the offspring’s total genotypic value regressed on the parents’ average total genotypic value) ranged between h2 = 0.0—0.6 across the six phenotypic selection cycles and across 30 replications, with an average of around h2 = 0.3 (Supplementary Figure S2). After obtaining the total genotypic values for each clone, which were calculated as the sum of the additive, dominance and epistasis component, the phenotypes were simulated following the same procedure as described above.
The crossing and selection steps in the simulations of the breeding schemes were run with routines developed in the C programming language, using an interface to the statistical software R version 3.4.4 (R Core Team 2018) which is implemented in the R-package SelectionTools 18.1 (population-genetics.uni-giessen.de/ ~ software/). Each breeding scheme simulation was run with 100 replications. Recombination during meiosis was modelled using a count-location algorithm (Karlin and Liberman 1978), assuming that the number of crossovers on a chromosome of length λ in Morgan (M) is a Poisson distributed random variable with parameter λ, which was a constant of 1.1 in our study (i.e. 110 cM per chromosome). The location of a crossover on the chromosome was modelled as a random variable with uniform distribution. These assumptions are mathematically equivalent to those of Haldane’s map function (Haldane 1919), assuming no interference in crossover formation.
The simulated breeding schemes
Phenotypic selection
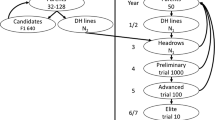
Based on discussions with breeders from Sugar Research Australia and CSIRO, and based on the information provided in Park et al. (2007), we developed a classical phenotypic selection (PS) breeding scheme (Fig. 2) and compared expected rates of genetic gain with two alternative breeding schemes that incorporate GS (Figs. 3, 4). All three schemes were assumed to have the same cycle length of approximately 9–10 years. It is important to note that under PS, crossing was only carried out at the start of the breeding cycle while in the GS schemes three additional rounds of intercrossing were simulated. Therefore, the term ‘breeding cycle’ in our study rather reflects a temporal classification, i.e. the amount of increase in gain that can be achieved through varying intercrossing and selection steps in a timeframe of 9–10 years. For each of the three breeding programs, we simulated phenotypic selection as outlined in Fig. 2 for the first five cycles and considered this the burn-in phase. Comparisons of genetic gain and genetic variance between the three different breeding schemes were made over five breeding cycles after the first five burn-in PS cycles.

Simulated phenotypic selection scheme

Simulated genomic selection scheme 1

Simulated genomic selection scheme 2
All three breeding program simulations were initiated by randomly sampling 83 clones from the base population as parents. Typically, about 40% of the clones selected as parents cannot be used in the mating design due to complications with flowering initiation and synchronisation. Therefore, we randomly dropped out 33 of the 83 parents to account for this practical constraint. The 50 remaining clones were each randomly mated to five other clones to generate a total of 250 F1 families with 100 offspring each. The resulting 25,000 seedlings were tested in a simulated stage 1 field trial in which H2 = 0.15 was assumed. We performed between and within family selection based on the simulated phenotypes. At first, the best 50% of the families were selected based on their mean trait phenotype, resulting in identification and advancement of 125 families. Then, the seedlings with the highest trait phenotypic value within each family were selected. Breeders typically prioritise better performing families which means that more seedlings are selected from the top-performing families. This process was simulated by imposing a linear gradual reduction of selected seedlings so that the best families of the 125 selected contributed 30 clones to the next selection stage whereas the worst families of the 125 contributed only 10 individuals.
The selected 2500 clones were tested in a simulated stage 2 trial assuming H2 = 0.4. From this stage on, clones were bulked and selected on an individual performance-basis. The best 150 clones were selected in stage 2 and went into the final stage 3 trial in which H2 = 0.7 was assumed. The best 83 clones from stage 3 were selected as parents to initiate the next breeding cycle. In each of the five breeding cycles, the total genotypic value and genetic variance of the 83 selected best clones were recorded. It should be noted that in a conventional phenotypic selection program, breeders typically perform across-generation selection of parents to initiate a new breeding cycle. This means that the best parents of the current breeding population are typically evaluated with the newly selected parents to form a new breeding population, i.e. only a fraction of the old parents is replaced by new clones. This ensures that clones that produce crosses of high genetic merit are retained until better cross-combinations are identified. This practice reflects the very high complexity around trait genetic architectures and improvement via selection. In our simulations, we made the assumption that all parents were replaced after a breeding cycle. The numbers of parents and population sizes were adjusted to represent one of four regional Australian breeding programs, which are typically interconnected through germplasm exchange (Park et al. 2007).
Genomic selection schemes 1
The first evaluated GS scheme (GS1, Fig. 3) followed the same routines as the phenotypic selection scheme until stage 2. After stage 2, three rapid recurrent GS (RRGS) cycles were implemented as follows. The 2500 clones at stage 2 were used as a training population and genome-wide marker effects for genomic selection were predicted using a ridge regression BLUP (rrBLUP) model (Meuwissen et al. 2001). The best 83 clones were selected based on their genomic estimated breeding values (GEBVs) and used to initiate the first of three RRGS cycles. Again, 40% of the 83 clones were dropped out to simulate complications associated with flowering synchronisation and the remaining 50 clones were used to generate 250 families with 40 offspring each, resulting in a total of 10,000 seedlings. These 10,000 seedlings were assumed to be genotyped for the 10,000 SNP markers described above. The previously predicted marker effects from stage 2 were used to obtain GEBVs for each of the 10,000 seedlings. The 83 clones with the highest GEBVs were selected to initiate the next RRGS cycle. After three RRGS cycles, the total genotypic values of the best 83 genotypes and the corresponding genetic variance were recorded.
Genomic Selection Scheme 2
The second GS scheme (GS2, Fig. 4) was initiated by using the best 83 clones from the last burn-in cycle to initiate a population of 2500 clones. This population of 2500 clones was used as a training population for the next three RRGS cycles involving 10,000 seedlings each, with the same routines as described above. After that, the best 2500 clones were taken forward to a stage 2-like trial with a broad-sense heritability of 0.4. From this trial, the best 83 clones were selected to initiate the next GS2 cycle and their total genotypic values and the corresponding genetic variance were recorded.
For both GS breeding schemes, it was assumed that the breeding cycle, and hence selection of new parents for the next cycle, was concluded before these clones were evaluated in FAT trials, which was assumed to be part of the product development process. Each of the three simulated breeding programs was run for a total of ten cycles (5 × burn-in cycles plus 5 × breeding cycles) with 100 replications.
Estimating relative cost–benefits of different GS breeding schemes with regard to genotyping costs
Typically, breeders work with limited financial resources that can be allocated to different steps in the breeding program. A key question that arises when considering the implementation of GS in a breeding program is how much genetic gain could be generated for the money that was spent for genotyping. To address that question for sugarcane, we compared the two breeding schemes GS1 and GS2 in terms of predicted genetic gains that could be generated when 500, 1000, 2500, 5000 or 10,000 clones were genotyped per RRGS step (i.e. 1500, 3000, 7500, 15,000 or 30,000 per breeding cycle). We took the most recent cost figure for genotyping using the Affymetrix Axiom SNP chip which is $95/sample as an estimate (CSIRO, personal communication). We ran 100 × replicates for each combination of genetic model (A_model or ADE_model) and varying numbers of clones genotyped per RRGS step (500, 1000, 2500, 5000 or 10,000). To compare the relative benefit of the two simulated GS schemes we compared the rate of genetic gain to the conventional PS scheme.
Results
Genomic prediction accuracies
For the A_model, prediction accuracies for the simulated trait ranged between 0.3 and 0.5 and were ~ 0.4 on average, with a decreasing trend over the three first RRGS steps, reflecting increased genetic distance and less shared haplotypes between the reference population and the prediction population. This is in accordance with average prediction accuracies for commercial cane sugar (CCS) (0.43–0.47) and fibre content (0.43–0.45), and slightly above total cane harvested (TCH) (0.28–0.30) as reported in the forward predictions by Hayes et al. (2021).
For the complex ADE_model, prediction accuracies for the simulated trait ranged from − 0.25–0.4, depending on the breeding cycle, with average accuracies of about 0.2. Given that the TCH predictions based on breeding trial data as reported by Hayes et al. (2021) were higher than those in our ADE_model simulations, the ADE_model can be considered to represent a scenario with potentially stronger non-additive effects than what is observed in an elite population of sugarcane clones. Taken together, the results for the A_model and ADE_model simulations could be considered as an informative upper and lower boundary for expected results for GS from an empirical breeding program.
Genetic gain and genetic variance for a quantitative trait with an additive genetic architecture
For all three breeding schemes, genetic improvement as defined by the increase in the average total genotypic value of the population was observed for the A_model (Fig. 5a). After the five burn-in PS cycles, the improvement rates followed an almost linear trend. A slightly asymptotic behaviour in the increase in the average total genotypic value was observed for both GS schemes, however without reaching a plateau by breeding cycle 5. Both GS schemes achieved rates of genetic gain that were almost twice as high as for the conventional PS scheme. The average increases in the mean total genotypic value for all three breeding schemes are summarised in Supplementary Table S1. Assuming an average cycle length of 10 years for all three breeding schemes, the average rate of genetic gain per year for the three breeding programs was 1.4%, 2.6% and 2.7% for PS, GS1 and GS2. The rate of genetic gain for the PS scheme was similar to that reported by Sugar Research Australia (SRA, Performance Report 2015–2016, page 25).

Genetic gain and additive genetic variance over 10 selection cycles for three different breeding schemes assuming a purely additive trait genetic architecture. B1–B5 = burn-in cycles of phenotypic selection as outlined in Fig. 2. C1–C5 = breeding cycles for the three breeding schemes phenotypic selection (PS), genomic selection 1 (GS1) and genomic selection 2 (GS2) as outlined in Figs. 2, 3 and 4. Light lines represent the single replicates per breeding scheme, bold lines represent averages across 100 replicates. (a) Average true breeding value among the 83 clones that are selected as parents. (b) Average additive genetic variance among the best 83 clones after each cycle. Base = randomly sampled initial 83 parents from the base population
The development of the total (additive) genetic variance after the last burn-in cycle showed strong differences between the three breeding programs (Fig. 5b, Supplementary Table S1). The conventional PS scheme gradually reduced the genetic variance to a total reduction after the five PS cycles of − 40%, compared to the starting point. Conversely, the GS breeding schemes both drastically increased diversity after the first breeding cycle to + 370% and + 111% compared to the starting point (after burn-in) for GS1 and GS2, respectively. After the five breeding cycles, the genetic variance in GS1 was 34% above the genetic variance measured at the starting point (after burn-in), whereas under GS2 the reduction in genetic variance at the end-point was similar to PS ( − 40%).
Genomic selection schemes increase genetic gain and genetic variance
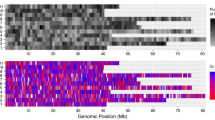
Under both GS schemes, there was a very strong increase in additive genetic variance after the first breeding cycle (Fig. 5b, Supplementary Table S1), along with substantial increases in genetic gain of + 32% and + 33% for GS1 and GS2, respectively. To investigate what could have caused this substantial increase in the additive genetic variance we recorded the changes in allele frequencies for each of the 1000 QTL after each of the 10 selection cycles (5 × burn-in plus 5 × breeding cycles) compared to the previous cycle. The changes of the QTL alleles are summarised in Fig. 6. For the PS scheme, there was an observable relationship between QTL frequency change and sampled QTL effect, i.e. QTL with large absolute effects were constantly increased in frequency at higher rates than QTL with low effects. Similarly, QTL with negative effects were decreased at stable rates across the breeding cycles. For both GS schemes, there was a notable, strong change in QTL allele frequencies after the first breeding cycle, which corresponds to the additive genetic variance peak shown in Fig. 5b. Notably, there was a substantial increase in allele frequencies of QTL with moderate effects, implying that under GS those QTL are stronger selected for than under phenotypic selection. This trend flattened out as the simulation progressed over the next breeding cycles (Fig. 6).

Allele frequency changes of the 1000 QTL under the purely additive genetic model. B1–B5 = burn-in cycles of phenotypic selection as outlined in Fig. 2. C1–C5 = breeding cycles for the three breeding schemes phenotypic selection (a), genomic selection 1 (GS1, b) and genomic selection 2 (GS2, C) as outlined in Figs. 2, 3 and 4. Each coloured line represents one QTL. The colours indicate the initially sampled ‘true’ QTL effect. Allele frequency shifts are averaged across 100 replicates
To investigate the relationship between allele frequency changes and QTL effects, and to put this into context with the strong increase in genetic gain in GS-featured breeding schemes, we calculated average effects for the favourable QTL alleles (i.e. for QTL with negative effects for allele \(Q\), we calculated the average effect for allele q). Neglecting the dominance term, we calculated the average effect as \(\alpha = \left( {1 - {\text{freq}}\_Q} \right)*{\text{eff}}\_Q\) where \(\alpha\) is the average effect, \({\text{freq}}\_Q\) is the frequency of allele \(Q\) and \(eff\_Q\) is the ‘true’ QTL effect, which was sampled as described above. The relationship between the average effect \(\alpha\) and the true QTL effect \({\text{eff}}\_Q\) in the three compared breeding schemes is shown in Supplementary Figure S3. Because frequencies of QTL with high absolute effects were increased at higher rates than QTL with lower absolute effects throughout the selection cycles, their average effect decreases gradually. Both GS schemes drove a large portion of QTL with the largest ‘true’ effects to fixation, hence their average effect was zero after the five breeding cycles. Under PS, fixation of those QTL was not reached after five breeding cycles (Supplementary Fig. 3b). We then investigated which breeding scheme achieved the highest realisation of improvement for the 1000 QTL after the first breeding cycle by calculating the product of the average allele effect after burn-in and the allele frequency change after one cycle of selection, and plotting this against the average allele effect after burn-in (Fig. 7). This clearly shows that both GS schemes increase frequencies of QTL alleles with high average effects at much higher rates than the PS scheme. Figure 7 also shows a higher downward trend for QTL alleles with lower average effects, which could potentially explain the peak in additive genetic variance for both GS-based breeding schemes after one cycle of breeding (Fig. 5b).

Realised additive genetic gain for the 1000 QTL under the purely additive genetic model after one breeding cycle. The three breeding schemes phenotypic selection (PS, A), genomic selection 1 (GS1, B) and genomic selection 2 (GS2, C) as outlined in Figs. 2, 3 and 4 are shown. Each coloured dot represents one QTL. X-axes show the average allele effect for each QTL after 5 burn-in phenotypic selection cycles. Y-axes show the product of the average allele effect and the corresponding shift in allele frequency after the first breeding cycle
We investigated if LD between QTL that were targeted in the GS schemes and other QTL on the same chromosome was the reason for the peak in additive genetic variance after one GS cycle, i.e. LD was causal for the contrasting frequency changes of the different QTL. Therefore, we selected the top 20 QTL that were increased or decreased at the highest rates after the first GS1 cycle (see GS1 in Fig. 6) and calculated pairwise LD as r2 between them and all other QTL on the same chromosome. Then, we measured allele frequency changes of those co-located QTL and plotted this against the corresponding pairwise r2 values, to investigate if there were observable patterns (Supplementary Figure S4). As shown in Supplementary Figure S4, there were no detectable trends of LD between the top 20 increased/decreased QTL and the frequency changes of QTL on the same chromosome. It is not clear if co-location of QTL on the same chromosome could be the reason for the contrasting allele frequency changes (instead of LD), but since the effect was so strong it seems unlikely that this is simply a consequence of sampling error.
To investigate if there is an association between the strong increase in additive genetic variance after one GS cycle and the genome size, we repeated the above described simulations for a simulated organism in which the 10,000 SNP markers and 1000 QTL were distributed on only 10 chromosomes of 110 cM (instead of 100). As shown in Supplementary Figure S5, the increase in genetic variance in both GS schemes was much smaller than for the simulations in which a genome with 100 chromosomes was used. This implies that the genome size could be important for the development of genetic variance over breeding cycles when GS is implemented. Interestingly, the difference in the rate of genetic gain between the GS schemes and the PS scheme was larger when a genome consisting of 10 chromosomes was considered (Supplementary Figure S5), compared to the initial simulations in which 100 chromosomes were assumed (Fig. 5).
Genetic gain and genetic variance for a quantitative trait with a non-additive genetic architecture
The development of genetic improvement and the total genetic variance for the ADE_model is shown in Fig. 8. Here, the rates of gain were substantially lower compared to the A_model, particularly for the two GS schemes. The average gains per year were 1.1%, 1.5% and 1.6% for the PS, GS1 and GS2 breeding schemes, respectively (Supplementary Table S2). Further, the variability between the 100 replicates for each breeding simulation has increased substantially compared to the A_model (Fig. 5). After the first cycle of both GS-based breeding schemes, there was a substantial drop in genetic gain which was not observed for the PS scheme (Fig. 8a). The rank of the two GS-based breeding schemes changed over the course of the breeding cycles, so GS2 yielded about 3% more gain after five cycles than GS1. The average trend of the total genetic variance over time was very similar to the trend observed under the A_model (Fig. 8b), i.e. there was a substantial increase in total genetic variance under the two GS-based breeding schemes. However, under the ADE_model, no difference in the average total genetic variance between GS1 and GS2 was observed.

Genetic gain and total genetic variance over 10 selection cycles for three different breeding schemes assuming a non-additive trait genetic architecture. B1–B5 = burn-in cycles of phenotypic selection as outlined in Fig. 1. C1–C5 = breeding cycles for the three breeding schemes phenotypic selection (PS), genomic selection 1 (GS1) and genomic selection 2 (GS2) as outlined in Figs. 2, 3 and 4. Light lines represent the single replicates per breeding scheme, bold lines represent averages across 100 replicates. (a) Average total genotypic value among the 83 clones that are selected as parents. (b) Average total genetic variance among the best 83 clones after each cycle. Base = randomly sampled initial 83 parents from the base population. See materials and methods and Supplementary Figure S1 for detailed description of the simulation of non-additive QTL effects
Cost–benefit of genomic selection under an additive genetic model
The results of comparing the cost–benefit ratio of two GS schemes with different numbers of genotyped clones under the A_model are summarised in Fig. 9. Here, 1000 QTL additively contribute to the simulated quantitative trait with no non-additive gene action. After the first breeding cycle, gains achieved from GS2 when only 500 clones were genotyped were below the gains achieved through phenotypic selection (Fig. 9). While GS1 delivers more gain throughout the five breeding cycles for relatively low numbers of genotype clones, both GS1 and GS2 converge when 10,000 clones are genotyped per rapid cycling step.

Cost–benefit of the two genomic selection schemes using different numbers of genotyped clones in the rapid recurrent genomic selection step under the additive ‘A_model’. Here, it is assumed that the quantitative trait is affected by additive QTL effects only. Five different scenarios with 500, 1000, 2500, 5000 and 10,000 genotyped clones per RRGS step are compared. Because both GS schemes deploy three RRGS steps, the total number of genotyped clones per cycle is 1500, 3000, 7500, 15,000 and 30,000. Genetic gain is compared to the genetic merit of the base population after the 5 × burn-in cycles. The solid purple line represents genetic gain achieved through phenotypic selection. A value of 0.1% (solid lines, left Y-axis) means that for every $1000 dollar that were invested in genotyping, a relative increase in genetic gain of 0.1% could be achieved compared to standard phenotypic selection. Assuming a 1% increase equates to $12.5 M annual net profit in Australian sugarcane, a value of 0.1% (solid lines, left Y-axis) means that for every $1000 invested in genotyping the return is $12,500. This value is already corrected for the genetic gain achievable through phenotypic selection
Overall, there is an increase in genetic gain when increasing the number of genotyped clones in the GS breeding scheme (Fig. 9, dashed lines). This trend increased with the number of breeding cycles, i.e. the difference between the dashed lines and the solid purple line (Fig. 9, phenotypic selection) is larger in cycle 5 than in cycle 1. Relative gains per $1000 invested for genotyping increased from less than 0.05% in cycle 1 to almost 0.2% in cycle 5 (Fig. 9, solid lines). A value of 0.1% means that for every $1000 that were invested in genotyping, a relative increase in genetic gain of 0.1% could be achieved compared to standard phenotypic selection. Based on the average total cane harvested in Australia (31.37MT) and the average CCS content of 13.72% between 2006 and 2015 (Sugar Research Australia 2016), and assuming a sugar price of $450 per tonne, a 1% improvement in cane yield and CCS would deliver $19.37 M more revenue to the industry per annum. Net profits from 1% improvement would be $12.5 M annually, considering harvesting and milling costs of $9.5 and $12.5 per tonnes. Assuming that a 1% increase equates to $12.5 M annual net profit for the Australian sugarcane industry, a relative increase of 0.1% through GS means that for every $1000 that were invested in genotyping the return is $12,500. This value is already corrected for the genetic gain achievable through phenotypic selection. In cycle 5 under GS1, every $1000 invested in genotyping resulted in an increase in genetic gain with an approximate return of investment of more than $35,000. The return of investment associated with increased genotyping is, however, diminishing. To investigate the effect of substantial investments into increasing the size of a conventional selection scheme we simulated a double-sized PS scheme in which we simulated 500 crosses to generate 50,000 offspring, a CAT stage with 5000 tested clones and a FAT stage in which the best 300 were assessed. Under the A_model, doubling the size of the PS scheme only resulted in an extra 5% in genetic gain at the end of breeding cycle 5 (Supplementary Figure S6).
Cost–benefit of genomic selection under a complex non-additive genetic model
The results of comparing the cost–benefit ratio in two GS schemes with different numbers of genotyped clones under the ADE_model are summarised in Fig. 10. Gains achieved in the GS schemes after cycle 1 are negative for all genotyping scenarios, except for GS1 when 10,000 clones are genotyped per RRGS step (i.e. 30,000 per cycle). Even there, gains are well below the PS scheme (Fig. 10). In cycle 2, GS1 slightly overtakes the PS in terms of genetic gain when 10,000 clones per RRGS step are genotyped. However, relative %-gain per $1000 invested is very close to zero (cycle 2 in Fig. 10, solid lines). From cycle 4 on, GS1 overtakes phenotypic selection even when only 500 clones are genotyped per RRGS step, i.e. 1500 per cycle. From then on, relative %-gains per $1000 invested in genotyping are positive for GS1, also when only 1500 clones are genotyped per cycle (Fig. 10). In cycle 5, the relative cost–benefit of genotyping only 1500 clones per cycle is higher than genotyping 30,000 clones. The rank of the two GS schemes changed depending on how many clones are assumed to be genotyped in the breeding scheme, for both the relative cost–benefit (Fig. 10, solid lines) and the absolute %-gains that were realised (Fig. 10, dashed lines). GS1 tends to perform better when lower numbers of clones are genotyped, whereas GS2 realises higher gains when higher numbers of clones are genotyped. Even in the complex ADE_model scenario, an average $1000 investment in genotyping resulted in a $5,000 return through an increase in genetic gain, again assuming that a 1% increase in genetic gain equates to $12.5 M annual net profit for the Australian sugarcane industry. As opposed to the situation under an additive genetic model, the increases in gain in the doubled PS scheme were very substantial under the complex ADE_model. It took until the end of breeding cycle 5 until the two GS schemes with 30,000 clones genotyped per cycle reached similar levels of genetic gain as the doubled PS scheme (Supplementary Figure S6).

Cost–benefit of the two genomic selection schemes using different numbers of genotyped clones in the rapid recurrent genomic selection step under the non-additive ‘ADE model’. Here, it is assumed that the quantitative trait is affected by additivity, dominance and epistasis (average h2 = 0.3). Five different scenarios with 500, 1000, 2500, 5000 and 10,000 genotyped clones are compared. Because both GS schemes deploy three RRGS steps, the total numbers of genotyped clones per cycle is 1500, 3000, 7500, 15,000 and 30,000. Genetic gain is compared to the genetic merit of the base population after the 5 × burn-in cycles. The solid purple line represents genetic gain achieved through phenotypic selection. A value of 0.1% (solid lines, left Y-axis) means that for every $1000 dollar that were invested in genotyping, a relative increase in genetic gain of 0.1% could be achieved compared to standard phenotypic selection. Assuming a 1% increase equates to $12.5 M annual net profit in Australian sugarcane, a value of 0.1% (solid lines, left Y-axis) means that for every $1000 invested in genotyping the return is $12,500. This value is already corrected for the genetic gain achievable through phenotypic selection
Discussion
Recent studies that investigated genomic prediction for complex traits in sugarcane show promising prediction accuracies (Deomano et al. 2020; Hayes et al. 2021). This shows the potential of the technology for commercial breeding programs. Here, we assessed two alternate strategies for implementing GS in commercial sugarcane breeding. To cover a range of traits that could be the breeding target, we simulated an additive quantitative trait (A_model) and a complex quantitative trait that is substantially affected by non-additive gene action (ADE_model). In our study, we made simplified assumptions regarding the simulation of the genome which is therefore unlikely to reflect the full complexity observed in sugarcane. We consider our study as a first attempt to assess the potential of GS for improving genetic gain for a range of complex traits (additive to non-additive) while noting that doubtlessly further work is required that addresses the limitations of our study. Key areas for further investigation are a detailed assessment of the effect of allele dosage on complex trait improvement through breeding and the investigation of different parameterisations of additive and non-additive trait genetic architectures. This includes the exploration of different effect distributions for assigning genetic component effects (i.e. other distributions than the U-shape distribution used in our study), and the consideration of directional dominance which is likely to be important in sugarcane. Our results provide a first assessment of potential increases in genetic gain that could be realised under the assumptions made in our simulations.
Under the A_model, GS drives genetic gain at an almost doubled rate of that achieved by PS. This accelerated genetic gain from the GS schemes is contributed by the three additional rounds of crossing, recombination and selection during the three RRGS steps. This allows a much more rapid frequency increase in favourable alleles of the additive QTL. Notably, both GS schemes were much more efficient in driving the allele frequencies of QTL with moderate ‘true’ effect but relatively high average effect, implying that GS can capture and increase favourable alleles which are at comparatively low frequency in the breeding population. Other simulation studies have shown that selection that is purely based on GEBVs can reduce genetic variance and increase the rate of inbreeding (Cros et al. 2015; Jannink et al. 2010). Interestingly, strong increases in genetic variance under GS could be observed in our study. Since this trend was much weaker when an organism with only 10 chromosomes was considered, we hypothesise that trends in genetic gain and genetic variance under GS-based breeding schemes could be quite different for crops with very large and complex genomes, compared to well-established major crops with much smaller and less complex genomes, such as maize or rice. This could be associated with the fact that QTL are spread across more chromosomes. While we were not able to explain the increase in genetic variance with LD between favourable and unfavourable QTL for the 20 QTL with the strongest frequency changes after the first breeding cycle (Fig. 6a, b), this could potentially rather be explained by an accumulated effect of many small-scale covariances between genome-wide QTL. This is also implied by the work of Gorjanc et al. (2018) who also found a substantial increase in genetic variance when transitioning from a phenotypic selection to a genomic selection scheme. Notably, Gorjanc et al. (2018) show that while the genetic variance increases in the first few cycles of the transition to GS-based breeding schemes, the genic variance decreases gradually.
Interestingly, similar trends for genetic variance were observed in an empirical study that investigated the genomic impacts of adapting tropical maize to temperate climates, through ten cycles of recurrent selection for flowering time (Wisser et al. 2019). In this study, major flowering genes with the largest effects on flowering time showed the strongest allele frequency shifts in the first four generations of recurrent selection. After that, an enrichment of alleles with smaller-sized phenotypic effects was observed, which was associated with an increase in both, heterozygosity and additive genetic variance in the population under consideration (Wisser et al. 2019). In our study, GS1 reduced the genetic variance at much slower rates than GS2, which implies that GS1 could deliver more long-term gains. In addition, this GS-based breeding strategy could be implemented in current conventional sugarcane breeding programs because it does not require substantial structural changes to the breeding program design. The first few stages of the GS1 scheme follow the same routines as in the PS scheme, i.e. generation of seedlings through crossing, selecting among and within families based on unreplicated row trials, and testing the best 2,500 clones in clonal assessment trials. The main difference of GS1 compared to the PS scheme is that instead of evaluating the best 150 clones from the CAT stage in FATs directly, three rounds of RRGS are included to rapidly increase frequencies of favourable alleles in this relatively strongly pre-selected material. After these three RRGS rounds, the best clones would be assessed in FAT-like trials for variety development, and also be recycled to initiate the next breeding cycle.
A major challenge in sugarcane is the complexity of the genome. With regard to GS, the importance of allele dosage for phenotypic trait variation has been demonstrated, e.g. for sugar content (Ming et al. 2001). It has been shown that general polysomy is important in chromosome assortment, with some cases of preferential pairing (Aitken et al. 2005, 2014; Jannoo et al. 2004), while meiosis mainly involves bivalent pairing (Price 1963). Furthermore, homologous gene conservation has been found to be prevalent (Garsmeur et al. 2011). In Saccharum spontaneum, it was shown that the numbers of alleles of the 35,525 annotated genes ranged from one to four with an average allele number of 2.3 (Zhang et al. 2018). To approximate this high complexity in our simulations, we made the simplified assumption of single-dose QTL, inspired by the recent Affymetrix SNP array for sugarcane for which 40,000 single-dose SNP were reported (Aitken et al. 2016) and which has recently been used for genomic prediction by Hayes et al. (2021). We acknowledge the limitations of our study and that the simplifications of the features underlying our meiosis simulation are unlikely to capture the full genomic and meiotic complexity as present in sugarcane. With regard to dosage effects, it could be that combining the results from these SNPs in our simulation study gives similar results as simulating higher-dose SNP markers if single-dose SNPs are present throughout the genome. If selection has resulted in parts of the genome being duplicated with low levels of polymorphism, then incorporating dosage would be more valuable. Detailed follow-up simulation work could aim at addressing these questions which are not covered by our simulation work presented here. In addition, further empirical research that improves the understanding of molecular recombination processes on a genome-wide scale is required. This will provide new possibilities for parameterisation of population genomic simulations beyond what is feasible based on current knowledge. This could enable future studies that focus more explicitly on the role of allele dosage in the context of GS-based breeding schemes, to provide further guidance for modern genetic improvement programs, especially with regard to non-additive gene action. However, our simulated scenarios matched observations from empirical data in terms of distributions of pairwise LD between SNPs, average narrow-sense heritability and genomic prediction accuracies (e.g. as reported by Hayes et al. 2021). Calibrating genetic and genomic properties of simulated populations with empirical data is critical to enable that the conclusions drawn are transferable and relevant to the targeted genetic improvement program, and that the full potential of simulations can be explored in the breeding optimisation process (Bernardo 2020).
Our results show that under the complex non-additive genetic ADE_model, the success rate of GS depends on the GS breeding scheme, the number of clones that are genotyped and the stage of the breeding program, likely reflecting how changes in QTL allele frequencies change available additive genetic variance and therefore the efficiency of selection. Given the relatively high accuracies in forward predictions that were reported in commercial sugarcane breeding trials by Hayes et al. (2021) (e.g. up to 0.33 for TCH, 0.55 for CCS and 0.55 for Fibre), it can be assumed that the simulated non-additive ADE_model scenario represents an extreme case and is perhaps more complex than what can be expected in a commercial breeding program. The fact that even under extreme non-additivity the relative cost–benefit of genotyping only 500 clones per rapid cycling step can be larger than genotyping 10,000 clones (cycle 4–5 in Fig. 10) is encouraging that GS can successfully be implemented to improve complex traits in sugarcane, like TCH, at moderate investment costs.
Our results are encouraging for implementing GS in sugarcane, especially for quantitative traits that are substantially affected by additive genetic effects (e.g. CCS, Fibre, disease resistance). Genotyping using a SNP array is still relatively costly in sugarcane ($95/sample), in comparison with other crops. However, as shown in Fig. 9, substantial gains can be achieved when only 1500 clones are genotyped per GS breeding cycle (i.e. $142,500 per 10-year cycle). As genotyping costs drop, as has happened in other crop species, GS would become even more accessible. If GS was implemented in a way that it replaces the relatively costly CAT stage, the relative cost–benefits would likely be much larger than in the current simulations. Interestingly, doubling the size of the PS scheme only had a minimal positive effect on the rate of gain under the additive A_model. Under the complex ADE_model, however, a PS scheme of twice the size compared to the reference PS scheme in our study resulted in substantial increases in genetic gain. These results imply that investing in enhancing PS is particularly worthwhile if the trait is strongly affected by non-additive gene action.
The strong variation in genetic gain that was observed between the five breeding cycles in the non-additive ADE_model scenario implied that QTL allele frequencies are the main driver for additive and non-additive genetic variance, i.e. the frequencies of QTL alleles that are involved in epistatic networks determine how much additive genetic variance there is, and hence how effectively the quantitative trait can be improved via selection. This is in accordance with earlier work that investigated the role of epistasis for directional selection and genetic improvement (e.g. Cheverud and Routman 1996, 1995; Cooper et al. 2002; Podlich and Cooper 1998). Cheverud and Routman (1996) showed in a two-locus example that when strong additive x additive epistasis was present, additive variance is large when one of the two interacting QTL is at an extreme allele frequency (i.e. 0 or 1). In such a situation, the variation at the other locus is exposed as additive and can be explored through selection. This might explain the rapid increase in genetic gain for both GS-based breeding schemes after completion of breeding cycle 1.
The RRGS steps in the two GS-based breeding schemes prioritise parents with high additive genetic effects and therefore capture and improve general combining ability (GCA) in each generation. However, this practice is less efficient for crossbred populations in which specific combining ability (SCA) is likely to play a major role (in theory, SCA is affected by dominance and epistasis), as expected in sugarcane. This could also explain the negative trends in genetic gain for both GS schemes under the ADE_model after the first breeding cycle, i.e. that QTL with positive additive effects were assigned to epistatic interactions that have a strong negative effect on the simulated trait. This motivates the investigation of alternate GS-based breeding strategies that could help to overcome problems associated with strong non-additive gene action, for instance, based on reciprocal recurrent selection which aims to maximise both GCA and SCA (Comstock et al. 1949). A detailed description of a potential GS-based reciprocal recurrent selection program for sugarcane is given by Yadav et al. (2020). In theory, the combination of improving both, additive and non-additive genetic effects holds the potential to improve long-term genetic gain in hybrid sugarcane breeding but further investigations are needed. A GS-based breeding scheme like GS1 presented here might be a first step for implementing GS technology in sugarcane because it requires relatively few structural changes to the existing design of a conventional PS program. The results reported here motivate both further empirical and simulation work to develop improved strategies for GS implementation in sugarcane breeding that are tailored to specific breeding program contexts.
References
Aitken KS, Jackson PA, McIntyre CL (2005) A combination of AFLP and SSR markers provides extensive map coverage and identification of homo(eo)logous linkage groups in a sugarcane cultivar. Theor Appl Genet 110(5):789–801. https://doi.org/10.1007/s00122-004-1813-7
Aitken KS, McNeil MD, Hermann S, Bundock PC, Kilian A, Heller-Uszynska K, Henry RJ, Li J (2014) A comprehensive genetic map of sugarcane that provides enhanced map coverage and integrates high-throughput Diversity Array Technology (DArT) markers. BMC Genomics 15:152. https://doi.org/10.1186/1471-2164-15-152
Aitken K, Farmer A, Berkman P, Muller C, Wei X, Demano E, Jackson P, Magwire M, Dietrich B, Kota R (2016) Generation of a 345K sugarcane SNP chip. Proc Int Soc Cane Technol 29:1923–1930
Baker P, Jackson P, Aitken K (2010) Bayesian estimation of marker dosage in sugarcane and other autopolyploids. TAG. Theoretical and applied genetics. Theoretische und angewandte Genetik 120(8):1653–1672. https://doi.org/10.1007/S00122-010-1283-Z
Bernardo R (2020) Reinventing quantitative genetics for plant breeding: something old, something new, something borrowed, something BLUE. Heredity. https://doi.org/10.1038/s41437-020-0312-1
Bernardo R, Yu J (2007) Prospects for Genomewide Selection for Quantitative Traits in Maize. Crop Sci 47(3):1082. https://doi.org/10.2135/cropsci2006.11.0690
Bhuiyan SA, Croft BJ, Cox MC (2013) Breeding for sugarcane smut resistance in Australia and industry response: 2006–2011. Proceedings of the Australian Society of Sugar Cane Technologists 1–9
Cheverud JM, Routman EJ (1995) Epistasis and its contribution to genetic variance components. Genetics 139(3):1455–1461
Cheverud JM, Routman EJ (1996) Epistasis as a source of increased additive genetic variance at population bottlenecks. Evol Int J Org Evol 50(3):1042–1051. https://doi.org/10.1111/j.1558-5646.1996.tb02345.x
Comstock RE, Robinson HF, Harvey PH (1949) A breeding procedure designed to make maximum use of both general and specific combining ability 1. Agron J 41(8):360–367. https://doi.org/10.2134/agronj1949.00021962004100080006x
Cooper M, Podlich DW, Micallef KP, Smith OS, Jensen NM, Chapman SC, Kruger NL (2002) Complexity, quantitative traits and plant breeding: a role for simulation modelling in the genetic improvement of crops. In: Kang MS (ed) Quantitative genetics, genomics, and plant breeding. CABI Pub, Oxon, UK, New York, pp 143–166
Cooper M, Gho C, Leafgren R, Tang T, Messina C (2014) Breeding drought-tolerant maize hybrids for the US corn-belt. Discovery to product. J Exp Bot 65(21):6191–6204. https://doi.org/10.1093/jxb/eru064
Cros D, Denis M, Bouvet J-M, Sánchez L (2015) Long-term genomic selection for heterosis without dominance in multiplicative traits: case study of bunch production in oil palm. BMC Genomics 16:651. https://doi.org/10.1186/s12864-015-1866-9
de Bem Oliveira I, Resende MFR, Ferrão LFV, Amadeu RR, Endelman JB, Kirst M, Coelho ASG, Munoz PR (2019) Genomic prediction of autotetraploids; Influence of relationship matrices, allele dosage, and continuous genotyping calls in phenotype prediction. G3 Bethesda, Md 9(4):1189–1198. https://doi.org/10.1534/g3.119.400059
de Lara CLA, Santos MF, Jank L, Chiari L, Vilela MDM, Amadeu RR, Dos Santos JPR, Pereira GdS, Zeng Z-B, Garcia AAF (2019) Genomic selection with allele dosage in panicum maximum Jacq. G3 Bethesda, Md 9(8):2463–2475. https://doi.org/10.1534/g3.118.200986
de Morais LK, de Aguiar MS, Albuquerque e Silva P de, Câmara TMM, Cursi DE, Júnior ARF, Chapola RG, Carneiro MS, Bespalhok Filho JC (2015) Breeding of sugarcane. In: Cruz VMV, Dierig DA (eds) Industrial crops, vol 9. Springer, New York, NY, pp 29–42
de Oliveira EJ, de Resende MDV, da Silva SV, Ferreira CF, Oliveira GAF, da Silva MS, de Oliveira LA, Aguilar-Vildoso CI (2012) Genome-wide selection in cassava. Euphytica 187(2):263–276. https://doi.org/10.1007/s10681-012-0722-0
Deomano E, Jackson P, Wei X, Aitken K, Kota R, Pérez-Rodríguez P (2020) Genomic prediction of sugar content and cane yield in sugar cane clones in different stages of selection in a breeding program, with and without pedigree information. Mol Breed 40(4):445. https://doi.org/10.1007/s11032-020-01120-0
Endelman JB, Carley CAS, Bethke PC, Coombs JJ, Clough ME, da Silva WL, de Jong WS, Douches DS, Frederick CM, Haynes KG, Holm DG, Miller JC, Muñoz PR, Navarro FM, Novy RG, Palta JP, Porter GA, Rak KT, Sathuvalli VR, Thompson AL, Yencho GC (2018) Genetic variance partitioning and genome-wide prediction with allele dosage information in autotetraploid potato. Genetics 209(1):77–87. https://doi.org/10.1534/genetics.118.300685
Fernandes SB, Dias KOG, Ferreira DF, Brown PJ (2018) Efficiency of multi-trait, indirect, and trait-assisted genomic selection for improvement of biomass sorghum. Theor Appl Genet 131(3):747–755. https://doi.org/10.1007/s00122-017-3033-y
Gaffney J, Schussler J, Löffler C, Cai W, Paszkiewicz S, Messina C, Groeteke J, Keaschall J, Cooper M (2015) Industry-scale evaluation of maize hybrids selected for increased yield in drought-stress conditions of the US corn belt. Crop Sci 55(4):1608. https://doi.org/10.2135/cropsci2014.09.0654
Garcia AAF, Mollinari M, Marconi TG, Serang OR, Silva RR, Vieira MLC, Vicentini R, Costa EA, Mancini MC, Garcia MOS, Pastina MM, Gazaffi R, Martins ERF, Dahmer N, Sforça DA, Silva CBC, Bundock P, Henry RJ, Souza GM, van Sluys M-A, Landell MGA, Carneiro MS, Vincentz MAG, Pinto LR, Vencovsky R, Souza AP (2013) SNP genotyping allows an in-depth characterisation of the genome of sugarcane and other complex autopolyploids. Sci Rep 3:3399. https://doi.org/10.1038/srep03399
García-Ruiz A, Cole JB, VanRaden PM, Wiggans GR, Ruiz-López FJ, van Tassell CP (2016) Changes in genetic selection differentials and generation intervals in US Holstein dairy cattle as a result of genomic selection. Proc Natl Acad Sci USA 113(28):E3995-4004. https://doi.org/10.1073/pnas.1519061113
Garsmeur O, Charron C, Bocs S, Jouffe V, Samain S, Couloux A, Droc G, Zini C, Glaszmann J-C, van Sluys M-A, D’hont A, (2011) High homologous gene conservation despite extreme autopolyploid redundancy in sugarcane. New Phytol 189(2):629–642. https://doi.org/10.1111/j.1469-8137.2010.03497.x
Garsmeur O, Droc G, Antonise R, Grimwood J, Potier B, Aitken K, Jenkins J, Martin G, Charron C, Hervouet C, Costet L, Yahiaoui N, Healey A, Sims D, Cherukuri Y, Sreedasyam A, Kilian A, Chan A, van Sluys M-A, Swaminathan K, Town C, Bergès H, Simmons B, Glaszmann JC, van der Vossen E, Henry R, Schmutz J, D’hont A, (2018) A mosaic monoploid reference sequence for the highly complex genome of sugarcane. Nat Commun 9(1):2638. https://doi.org/10.1038/s41467-018-05051-5
George AW, Aitken K (2010) A new approach for copy number estimation in polyploids. J Heredit 101(4):521–524. https://doi.org/10.1093/jhered/esq034
Gorjanc G, Gaynor RC, Hickey JM (2018) Optimal cross selection for long-term genetic gain in two-part programs with rapid recurrent genomic selection. Theor Appl Genet. https://doi.org/10.1007/s00122-018-3125-3
Gouy M, Rousselle Y, Bastianelli D, Lecomte P, Bonnal L, Roques D, Efile J-C, Rocher S, Daugrois J, Toubi L, Nabeneza S, Hervouet C, Telismart H, Denis M, Thong-Chane A, Glaszmann JC, Hoarau J-Y, Nibouche S, Costet L (2013) Experimental assessment of the accuracy of genomic selection in sugarcane. Theor Appl Genet 126(10):2575–2586. https://doi.org/10.1007/s00122-013-2156-z
Haldane JBS (1919) The combination of linkage values and the calculation of distances between the loci of linked factors. Genetics 8:299–309
Hayes BJ, Wei X, Joyce P, Domano E, Yue J, Nguyen L, Ross E, Cavellero T, Aitken KS, Voss-Fels KP (2021) Accuracy of genomic prediction of complex traits in sugarcane. (companion paper, submitted)
Heffner EL, Jannink J-L, Sorrells ME (2011) Genomic selection accuracy using multifamily prediction models in a wheat breeding program. Plant Genome 4(1):65. https://doi.org/10.3835/plantgenome2010.12.0029
Hickey JM, Chiurugwi T, Mackay I, Powell W (2017) Genomic prediction unifies animal and plant breeding programs to form platforms for biological discovery. Nat Genet 49(9):1297–1303. https://doi.org/10.1038/ng.3920
Hunt CH, van Eeuwijk FA, Mace ES, Hayes BJ, Jordan DR (2018) Development of genomic prediction in sorghum. Crop Sci 58(2):690. https://doi.org/10.2135/cropsci2017.08.0469
Jannink J-L, Lorenz AJ, Iwata H (2010) Genomic selection in plant breeding. From theory to practice. Brief Funct Genomics 9(2):166–177. https://doi.org/10.1093/bfgp/elq001
Jannoo N, Grivet L, David J, D’Hont A, Glaszmann J-C (2004) Differential chromosome pairing affinities at meiosis in polyploid sugarcane revealed by molecular markers. Heredity 93(5):460–467. https://doi.org/10.1038/sj.hdy.6800524
Karlin S, Liberman U (1978) Classifications and comparisons of multilocus recombination distributions. Proc Natl Acad Sci USA 75(12):6332–6336. https://doi.org/10.1073/pnas.75.12.6332
Lorenz AJ, Smith KP, Jannink J-L (2012) Potential and optimization of genomic selection for fusarium head blight resistance in six-row barley. Crop Sci 52(4):1609. https://doi.org/10.2135/cropsci2011.09.0503
Ly D, Hamblin M, Rabbi I, Melaku G, Bakare M, Gauch HG, Okechukwu R, Dixon AGO, Kulakow P, Jannink J-L (2013) Relatedness and genotype × environment interaction affect prediction accuracies in genomic selection. Stud Cassava Crop Sci 53(4):1312. https://doi.org/10.2135/cropsci2012.11.0653
Meuwissen TH, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819–1829
Ming R, Liu SC, Moore PH, Irvine JE, Paterson AH (2001) QTL analysis in a complex autopolyploid: genetic control of sugar content in sugarcane. Genome Res 11(12):2075–2084. https://doi.org/10.1101/gr.198801
Ming R, Moore PH, Wu K-K, D'hont A, Glaszmann JC, Tew TL, Mirkov TE, da Silva J, Jifon J, Rai M, Schnell RJ, Brumbley SM, Lakshmanan P, Comstock JC, Paterson AH (2005) Sugarcane improvement through breeding and biotechnology. In: Janick J (ed) Plant breeding reviews, vol 30. Wiley, Oxford, pp 15–118
Osborn TC, Chris Pires J, Birchler JA, Auger DL, Jeffery Chen Z, Lee H-S, Comai L, Madlung A, Doerge RW, Colot V, Martienssen RA (2003) Understanding mechanisms of novel gene expression in polyploids. Trends Genet 19(3):141–147. https://doi.org/10.1016/s0168-9525(03)00015-5
Park S, Jackson P, Berding N, Inman-Bamber G (2007) Conventional breeding practices within the Australian sugarcane breeding program. In: Proceedings of the International Society of Cane Technologists
Piperidis N, D’hont A (2020) Sugarcane genome architecture decrypted with chromosome-specific oligo probes. Plant J Cell Mol Biol. https://doi.org/10.1111/tpj.14881
Podlich DW, Cooper M (1998) QU-GENE: a simulation platform for quantitative analysis of genetic models. Bioinform Oxford Engl 14(7):632–653
Poland J, Endelman J, Dawson J, Rutkoski J, Wu S, Manes Y, Dreisigacker S, Crossa J, Sánchez-Villeda H, Sorrells M, Jannink J-L (2012) Genomic selection in wheat breeding using genotyping-by-sequencing. Plant Genome 5(3):103. https://doi.org/10.3835/plantgenome2012.06.0006
Price S (1963) Cytogenetics of modern sugar canes. Econ Bot 17(2):97–106. https://doi.org/10.1007/BF02985359
R Core Team (2018) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Riedelsheimer C, Czedik-Eysenberg A, Grieder C, Lisec J, Technow F, Sulpice R, Altmann T, Stitt M, Willmitzer L, Melchinger AE (2012) Genomic and metabolic prediction of complex heterotic traits in hybrid maize. Nat Genet 44(2):217–220. https://doi.org/10.1038/ng.1033
Rutkoski J, Benson J, Jia Y, Brown-Guedira G, Jannink J-L, Sorrells M (2012) Evaluation of genomic prediction methods for fusarium head blight resistance in wheat. Plant Genome 5(2):51. https://doi.org/10.3835/plantgenome2012.02.0001
Slater AT, Cogan NOI, Forster JW, Hayes BJ, Daetwyler HD (2016) Improving genetic gain with genomic selection in autotetraploid potato. Plant Genome. https://doi.org/10.3835/plantgenome2016.02.0021
Spindel J, Begum H, Akdemir D, Virk P, Collard B, Redoña E, Atlin G, Jannink J-L, McCouch SR (2015) Genomic selection and association mapping in rice (Oryza sativa). Effect of trait genetic architecture, training population composition, marker number and statistical model on accuracy of rice genomic selection in elite, tropical rice breeding lines. PLoS Genet 11(2):e1004982. https://doi.org/10.1371/journal.pgen.1004982
Sugar Research Australia (2016) Annual Report 2015/16, https://sugarresearch.com.au/wp-content/uploads/2017/03/250694_SRA_Annual_Report_2015_2016.pdf, Indooroopilly QLD, Australia
Wei X, Jackson P (2016) Addressing slow rates of long-term genetic gain in sugarcane. Proc Int Soc Cane Technol 29:1923–1930
Werner CR, Qian L, Voss-Fels KP, Abbadi A, Leckband G, Frisch M, Snowdon RJ (2018) Genome-wide regression models considering general and specific combining ability predict hybrid performance in oilseed rape with similar accuracy regardless of trait architecture. Theor Appl Genet 131(2):299–317. https://doi.org/10.1007/s00122-017-3002-5
Wisser RJ, Fang Z, Holland JB, Teixeira JEC, Dougherty J, Weldekidan T, de Leon N, Flint-Garcia S, Lauter N, Murray SC, Xu W, Hallauer A (2019) The genomic basis for short-term evolution of environmental adaptation in maize. Genetics 213(4):1479–1494. https://doi.org/10.1534/genetics.119.302780
Wu KK, Burnquist W, Sorrells ME, Tew TL, Moore PH, Tanksley SD (1992) The detection and estimation of linkage in polyploids using single-dose restriction fragments. Theor Appl Genet 83(3):294–300. https://doi.org/10.1007/BF00224274
Yadav S, Jackson P, Wei X, Ross EM, Aitken K, Deomano E, Atkin F, Hayes BJ, Voss-Fels KP (2020) Accelerating genetic gain in sugarcane breeding using genomic selection. Agronomy 10(4):585. https://doi.org/10.3390/agronomy10040585
Zhang J, Zhang X, Tang H, Zhang Q, Hua X, Ma X, Zhu F, Jones T, Zhu X, Bowers J, Wai CM, Zheng C, Shi Y, Chen S, Xu X, Yue J, Nelson DR, Huang L, Li Z, Xu H, Zhou D, Wang Y, Hu W, Lin J, Deng Y, Pandey N, Mancini M, Zerpa D, Nguyen JK, Wang L, Yu L, Xin Y, Ge L, Arro J, Han JO, Chakrabarty S, Pushko M, Zhang W, Ma Y, Ma P, Lv M, Chen F, Zheng G, Xu J, Yang Z, Deng F, Chen X, Liao Z, Zhang X, Lin Z, Lin H, Yan H, Kuang Z, Zhong W, Liang P, Wang G, Yuan Y, Shi J, Hou J, Lin J, Jin J, Cao P, Shen Q, Jiang Q, Zhou P, Ma Y, Zhang X, Xu R, Liu J, Zhou Y, Jia H, Ma Q, Qi R, Zhang Z, Fang J, Fang H, Song J, Wang M, Dong G, Wang G, Chen Z, Ma T, Liu H, Dhungana SR, Huss SE, Yang X, Sharma A, Trujillo JH, Martinez MC, Hudson M, Riascos JJ, Schuler M, Chen L-Q, Braun DM, Li L, Yu Q, Wang J, Wang K, Schatz MC, Heckerman D, van Sluys M-A, Souza GM, Moore PH, Sankoff D, VanBuren R, Paterson AH, Nagai C, Ming R (2018) Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat Genet 50(11):1565–1573. https://doi.org/10.1038/s41588-018-0237-2
Zhao D, Li Y-R (2015) Climate change and sugarcane production: potential impact and mitigation strategies. Int J Agronomy 2:1–10. https://doi.org/10.1155/2015/547386
Zhao Y, Gowda M, Liu W, Würschum T, Maurer HP, Longin FH, Ranc N, Reif JC (2012) Accuracy of genomic selection in European maize elite breeding populations. Theor Appl Genet 124(4):769–776. https://doi.org/10.1007/s00122-011-1745-y
Zhong S, Dekkers JCM, Fernando RL, Jannink J-L (2009) Factors affecting accuracy from genomic selection in populations derived from multiple inbred lines. A barley case study. Genetics 182(1):355–364. https://doi.org/10.1534/genetics.108.098277
Funding
This research was funded by Sugar Research Australia (Project No.: 2017/02).
Author information
Authors and Affiliations
Contributions
KPVF, MC and BJH conceived the simulation study. XW and KSA provided extensive support with breeding scheme designs and simulation parameter settings. KPVF, MC, EMR, MF and BJH performed the simulations. KPVF, BJH and MC analysed the data and interpreted the results. KPVF wrote the manuscript. All authors edited and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Martin Boer.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Voss-Fels, K.P., Wei, X., Ross, E.M. et al. Strategies and considerations for implementing genomic selection to improve traits with additive and non-additive genetic architectures in sugarcane breeding. Theor Appl Genet 134, 1493–1511 (2021). https://doi.org/10.1007/s00122-021-03785-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-021-03785-3