
Abstract
Key message
A novel R gene was mapped to a locus on chromosome 11 from 30.42 to 30.85 Mb, which was proven to be efficient in the improvement of rice blast resistance.
Abstract
Rice blast is a devastating fungal disease worldwide. The use of blast resistance (R) genes is the most important approach to control the disease in rice breeding. In the present study, we finely mapped a novel resistance gene Pi65(t), conferring a broad-spectrum resistance to the fungus Magnaporthe oryzae, using bulked segregant analysis in combination with next-generation sequencing technology. Segregation in a doubled haploid (DH) population and a BC1F2 population suggested that resistance to blast in Gangyu129 was likely conferred by a single dominant gene, designated Pi65(t); it was located on chromosome 11 from 30.20 to 31.20 Mb using next-generation sequencing. After screening recombinants with newly developed molecular markers, the region was narrowed down to 0.43 Mb, flanked by SNP-2 and SNP-8 at the physical location from 30.42 to 30.85 Mb based on the Nipponbare reference database in build 5. Using the software QTL IciMapping, Pi65(t) was further mapped to a locus between InDel-1 and SNP-4 with genetic distances of 0.11 and 0.98 cM, respectively. Within this region, 4 predicted R genes were found with nucleotide binding site and leucine-rich repeat (NBS-LRR) domains. We developed molecular markers to genotype 305 DH lines and found that InDel-1 was closely linked with Pi65(t). Using InDel-1, a new rice variety Chuangxin1 containing Pi65(t) was developed, and it is highly resistant to rice blast and produces a high yield in Liaoning province of China. This indicated that Pi65(t) could play a key role in the improvement of rice blast resistance.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice blast, caused by Magnaporthe oryzae (M. oryzae), is the most important fungal disease in rice (Oryza sativa) worldwide (Ou 1985). An outbreak of blast can devastate rice yields, completely destroying crops in the most extreme cases. The use of host resistance has been proven to be the most effective and economical method to control rice blast (Fukuoka et al. 2009; Jeung et al. 2007).
To date, 84 rice blast R loci have been identified (Huang et al. 2011; Liu et al. 2010; Ma et al. 2014; Xu et al. 2014). Among them, 24 have been cloned (Pb1, Pia, Pib, Pid2, Pid3, Pik, Pikh, Pik-m, Pik-p, Pish, Pit, Pita, Piz-t, Pi1, Pi2, Pi5, Pi9, Pi21, Pi25, Pi36, Pi37, Pi54, Pi56 and PiCO39) (Chauhan et al. 2002; Liu et al. 2013a, b). The majority of rice blast R genes are associated with a hypersensitive response (HR), according to the gene-for-gene concept, and race specificity is the key feature of this R gene-mediated disease resistance (Fukuoka et al. 2009; Jia et al. 2000). Due to highly frequent variation in the M. oryzae population (Dean et al. 2005), the resistance of new rice varieties simply with a single R gene can be lost quickly, especially when such a variety is grown in large areas (Fukuoka et al. 2009; Hulbert et al. 2001; Qu et al. 2006). The way to solve this problem and control rice blast is pyramiding multiple R genes into a single rice variety, and deployment of rice varieties with broad-spectrum resistance is another practical means to control blast disease (Chen et al. 1995; Hittalmani et al. 2000). Therefore, it is more and more important to screen rice germplasm with broad-spectrum resistance and identify broad-spectrum R genes.
Molecular mapping has been extensively used for the identification of R genes; it can provide the starting point for gene cloning and marker-assisted selection in rice breeding (Hittalmani et al. 2000; Jeung et al. 2007). This is usually conducted by genotyping segregating populations derived from bi-parental crosses; however, it is time-consuming and laborious (Michelmore et al. 1991; Salvi and Tuberosa 2005). The bulked segregant analysis (BSA) provides a simple approach to identify genes by genotyping a pair of bulked DNA samples from two sets of individuals with contrasting extreme phenotypes (Michelmore et al. 1991). Over the past decades, BSA technologies have been used frequently to map important traits in many crops (Cheng et al. 2015; Jeon et al. 2003; Liu et al. 2005). The rapid development of next-generation sequencing (NGS) technologies gives a further push to BSA strategies for gene mapping. The technique for gene mapping using NGS in combination with BSA has been developed (Das et al. 2015; Fekih et al. 2013; Sun et al. 2013; Takagi et al. 2015; Takagi et al. 2013; Takagi 2013) and demonstrated in the identification of genes in maize (Zea mays L.) (Xia et al. 2014), cucumber (Cucumis sativus L.) (Lu et al. 2014), cotton (Gossypium spp.) (Chen et al. 2015), wheat (Triticum aestivum L.) (Trick et al. 2012), rice (Oryza sativa L.) (Abe et al. 2012; Yang et al. 2013), and sunflower (Helianthus annuus L.) (Livaja et al. 2013).
The japonica rice variety Guangyu 129 is resistant to many Chinese M. oryzae races/isolates and was predicted to carry novel blast R gene (Wang et al. 2015). The objectives of the current study were to finely map a rice blast R gene using a BSA-NGS approach in the DH and BC1F2 populations derived from the crosses of Gangyu129/Liaoxing1 and DH181/Liaoxing1, respectively. Results will certainly benefit the improvement of rice blast resistance in breeding program.
Materials and methods
Plant materials
A mapping population of 305 DH lines was developed from the Gangyu129/Liaoxing1 cross. Gangyu129 and its parent Danjing4 have been highly resistant to rice blast for many years in China. Gangyu129 was resistant to 8 isolates (ZA1, ZA9, ZB1, ZB13, ZC1, ZE1, ZF1 and ZG1) (Fig. S1a), whereas Liaoxing1 was susceptible to these isolates (Fig. S1b). The line DH181 of the DH population is highly resistant to rice blast, whereas its agronomical traits are similar to Liaoxing1. A BC1F2 population of 850 individuals was developed from the DH181/Liaoxing1 cross.
Inoculation and disease evaluation
Field and greenhouse inoculations were used to evaluate blast reactions of the DH and BC1F2 populations. Natural inoculation was conducted in the field, in Dagushan town of Dandong, Liaoning province, China, where rice blast is very serious every year. Field trials were conducted in randomized complete blocks with three replicates. Plots of each line consisted of two 2 m rows of 20 plants with 30 cm spacing between rows. Two rows of control variety (Lijiangxintuanheigu) were planted every 10 rows. No fungicide was used during the entire growth period. The seeds were sown in mid-April, and seedlings were transplanted by the end of May. Incidents of rice blast infection were scored at seedling and tillering stages using a disease assessment scale of 0–5 (Pan et al. 1996). Artificial inoculation was conducted in greenhouse. At 56 days after sowing (5−6 leaves stage), seedlings were spray-inoculated with a mixture of 8 isolates (ZA1, ZA9, ZB1, ZB13, ZC1, ZE1, ZF1 and ZG1) that were isolated from the field in 2012 (Wang et al. 2013), and these are prevalent isolates of M. oryzae in China. After being sprayed with a suspension of the mixture, the inoculated plants were placed in darkness in dew chambers at 100 % relative humidity and 26 °C for 24 h, and then transferred to a greenhouse and maintained at 26 °C. The inoculated seedlings were scored 7 day after inoculation following Pan et al. (1996).
Genetic analysis
Genetic analysis of resistance to rice blast was performed on the DH and BC1F2 populations. A Chi-square test (χ 2) was used to evaluate the goodness of fit of observed and expected segregation ratios in the DH and BC1F2 populations.
Specific-locus amplified fragment (SLAF) library construction and high-throughput sequencing
First, we carried out a SLAF pre-design experiment (Sun et al. 2013 ). The enzymes and sizes of restriction fragments were evaluated using training data. To maintain the sequence depth uniformity of different fragments, a tight length range was selected (about 30–50 bp); a pilot PCR amplification was performed to check the reduced representation library features within this target range, which would ordinarily include fragments with similar amplification features on the gel. When non-specifically amplified bands appeared on the gel, we then repeated the pre-design step to produce a new scheme. We constructed the SLAF library in accordance using the pre-designed scheme. Then, two DNA pools were constructed, one R-pool and one S-pool, by mixing an equal amount of DNA from 50 resistant and 50 susceptible DH lines, respectively. Finally, pair-end sequencing was performed according to the selected SLAFs using an Illumina high-throughput sequencing platform, followed by SNP genotyping and evaluation.
Sequence clustered and association analysis
All SLAF pair-end reads with clear index information were clustered based on sequence similarity. To reduce computing requirements, identical reads were merged, and sequence similarity was detected using one-to-one alignment by BLAST (Kent 2002). SLAFs with two, three, or four tags were considered to be polymorphic markers.
The parameters SNP-index and Δ(SNP-index) (Abe et al. 2012; Takagi 2013) were calculated to identify candidate regions for rice blast disease resistance. An SNP-index is the proportion of reads harboring the SNPs that are different from the reference sequence. We calculated Δ(SNP-index) by subtracting the SNP-index of R-pool from that of S-pool. Thus, SNP-index is equal to 0 if all short reads contain genomic fragments from Liaoxing1, while SNP-index is equal to 1 if all short reads were from Gangyu129. The SNP-index graphs for R-pool and S-pool, and the corresponding Δ(SNP-index), were plotted.
The Δ(SNP-index) value should not be significantly different from 0 in a genomic region that has no target genes (Takagi 2013). We calculated statistical confidence intervals of Δ(SNP-index) for all the SNP positions with given read depths under the null hypothesis of no R genes, and plotted them along with Δ(SNP-index). If the Δ(SNP-index) value of a marker was bigger than the threshold at the 99 % level of significance, the marker was called diff_marker, and regions with three or more consecutive diff_markers were identified as trait-related candidate regions.
Marker development and recombinants screening
To screen recombinants from the BC1F2 population, polymorphic markers between the two parental lines were developed in the predicted region of rice chromosome 11. All SSR markers within the mapping region were used for polymorphism identification. To develop more polymorphic markers between Gangyu129 and Liaoxing1, we downloaded the sequence of predicted genes (http://rapdblegacy.dna.affrc.go.jp/) in the mapped region and designed SNP markers based on the sequence of those genes (Table S1). After PCR and sequencing, we observed much difference in sequences between the two varieties—based on that difference, we developed one InDel and nine SNP markers to screen the recombinants to finely map the locus for rice blast resistance in Gangyu129. Primers for the InDel and SNP markers were designed using Primer 5. (http://www.PromerBiosoft.com).
The fine mapping strategy
We investigated 850 BC1F2 plants for their genotypes using polymorphic markers. Based on the difference between the genotypes as assessed using polymorphic markers, six recombinants were screened, and comparison of the donor region to the phenotypes among multiple recombinants enabled us to narrow down the location of the Pi65(t) region. This mapping strategy, based on recombinant-derived progeny, effectively minimizes experimental errors that may result from genetic background noise and environmental variation (Zuo et al. 2014).
Linkage analysis and marker-assisted selection
Besides the polymorphic markers in the predicted region, we screened some SSR markers in other region of rice chromosome 11. The linkage analysis was performed using the software QTL IciMapping (Li et al. 2008) and recombination values were converted to centiMorgens using the Kosambi mapping function (Kosambi 1943). Using the tightly linked marker InDel-1, we selected individuals that carried R gene from the DH or BC1F2 population, and then backcrossed with the susceptible parent Liaoxing1, an adapted variety in Liaoning province. Based on phenotypic and genotypic identification, the homozygous resistant plants were selected, with agronomical traits similar to the recurrent parent from the backcross population.
Results
Phenotypic and genetic analysis
To investigate the inheritance of the resistance to M. oryzae in Gangyu129, reciprocal crosses between Gangyu129 and Liaoxing1 were performed. Both F1 plants were resistant to rice blast; the DH population showed a 1:1 segregation in both the natural and artificial inoculation tests, with \(\chi_{1:1}^{2} = 0. 7 3 8 {\text{ and }}0. 3 9 7\), respectively (Table 1); the BC1F2 population showed a 3:1 segregation in both the natural and artificial inoculations, with \(\chi_{3:1}^{2} = 1. 8 2 2 {\text{ and }}0. 5 6 6\), respectively (Table 1). These indicated that the resistance to M. oryzae in Gangyu129 was likely conferred by a single dominant gene, temporarily designated Pi65(t).
Sequencing and SNP identification
After SLAF library construction and high-throughput sequencing, a total of 7.53 Gb of data, including 88,652,415 valid single-end reads, was procured with an averaged read length of 80 bp (Table 2). The total number of SLAF tags was 172,419 with an average coverage of 339.78 folds. The average sequence depths of SNP markers were tenfold in parents and 50-fold in pools. The Q20 ratio ranged from 80.41 to 85.13 %. Based on the results of SLAF positioning on the genome, the SLAF tag numbers on each chromosome were calculated (Table 3), and a distribution diagram of SLAF tags on each chromosome is shown in Fig. 1a. The SLAF tags were distributed equally on each chromosome and the rice genome has been successfully simplified. A total of 172,419 high-quality SLAF tags were developed, 9092 of which were polymorphic according to analysis of allele numbers and the differences between gene sequences. The polymorphic SLAF tags were referred to as markers with a polymorphism rate of 5.27 %. Summary statistics for marker numbers are shown in Table S2, and a distribution diagram of markers on each chromosome is shown in Fig. 1b.

a SLAF tags (black lines) distributed on 12 rice chromosomes. b Polymorphic markers (black lines) distributed on 12 rice chromosomes
Association analysis
The SNP-index was calculated for each SNP identified. SNP-index graphs were generated for the R-pool (Fig. 2a) and S-pool (Fig. 2b) by plotting the average SNP-index against the position of each sliding window in the Nipponbare genome assembly. By combining the information of SNP-index in the R-pool and S-pool, Δ(SNP-index) was calculated and plotted against the genome positions (Fig. 2c). It was expected that the SNP-index graphs of the R-pool and S-pool would be identical for the genomic regions that are irrelevant to the phenotypic difference, whereas the genomic region(s) harboring R genes would exhibit unequal contributions from R and S parental genomes. In addition, the SNP-index of these regions for R- and S-pools would appear as mirror images (Takagi 2013).

SNP-index graphs of R-pool (a), S-pool (b) and Δ(SNP-index) graph (c) from SLAF-seq analysis. X-axis represents the position of 12 rice chromosomes and Y-axis represents the SNP-index. A candidate gene (Pi65(t)) location was identified in rice chromosome 11 (30.2–31.2 Mb interval) with the criteria that a the SNP-index in R-pool was near 1, b SNP-index in L-pool was near 0, and c the Δ(SNP-index) was over the confidence value (P < 0.01)
The Δ(SNP-index) value should be significantly different from 0 if a genomic region harbors a major QTL of the target genes. In this study, the threshold of Δ(SNP-index) value was 0.8122 at the 99 % significance level. Further, the Δ(SNP-index) values of 37 markers (diff-marker) were bigger than 0.8122, and they were found on Chr10 and Chr11 (Fig. 2c; Table 2). Among them, only 1 diff-marker was mapped on Chr10, whereas the other 36 were all located on Chr11, from 30.20 to 31.20 Mb based on the Nipponbare reference database in build 5 (http://rapdblegacy.dna.affrc.go.jp/). These demonstrated that a major QTL/gene conferring rice blast disease resistance was present at the 30.20–31.20 Mb region on chromosome 11 in the rice variety Gangyu129.
Developing SSR, Indel and SNP markers to narrow down the region of Pi65(t)
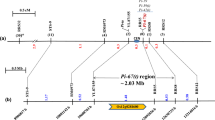
Based on the annotation of Nipponbare reference database in build 5 (http://rapdblegacy.dna.affrc.go.jp/), about 101 predicted genes were located within the region on chromosome 11 from 30.20 to 31.20 Mb. To screen the recombinants and refine the Pi65(t) gene, 42 SSR markers were selected on chromosome 11 to identify the polymorphism between Gangyu129 and Liaoxing1. The results showed that RM286, RM332, RM27181 and RM27364 were polymorphic between the two varieties. Based on the next-generation sequencing data and the sequence of mapped genes in Nipponbare from 30.20 to 31.20 Mb, we designed 9 SNP markers to amplify DNA from Gangyu129 and Liaoxing1 (the sequence and location of the SNP markers are shown in Table S1). Using SNP-3, we obtained two PCR products for 928 and 947 bp from Liaoxing1 and one PCR product for 947 bp from Gangyu129. The sequence alignment indicated an InDel for 19 bp between Gangyu129 and Liaoxing1 (Fig. S2a). We developed an InDel marker (InDel-1) based on the sequence difference, with the result of polyacrylamide gel electrophoresis using InDel-1 (Fig. S2b). Three primers (SNP-1, InDel-1 and RM27364) were used for a recombination test by polyacrylamide gel electrophoresis in the BC1F2 populations derived from DH181/Liaoxing1. Six recombinants were recovered within the region on chromosome 11 from 30.20 to 31.20 Mb. To find the position where the recombinant events occurred, SNP-1 to SNP-9 were used to amplify DNA from the recombinants and two parents. Comparing the sequence, graphic genotypes and disease reaction, we found six recombinants, and they were products of single-crossovers. No recombinants were detected using SNP-3, SNP-4, SNP-5, SNP-6 and SNP-7. Together, the mapping data indicated that the Pi65(t) locus falls within an interval of 0.43 Mb on chromosome 11 flanked by SNP-2 (30.42 Mb) and SNP-8 (30.85 Mb) (Fig. 3). Based on the Nipponbare reference database in build 5, 20 predicted genes were located within the interval of 0.43 Mb; among them, 19 were related to disease resistance (Table 4).

Graphical genotypes of the recombinants spanning the Pi65(t) region using SNP-1 to SNP-9. The black bar is the introgression region from Gangyu129 (R). The white bar is the Liaoxing1 genome (S). Nine SNP markers are localized on the physical map. Numbers below the markers are the physical distance from the top on chromosome 11 based on the Nipponbare reference database in build 5. Six recombinants were detected in the region defined by the nine polymorphic markers. Recombinant-1 was from a crossover between SNP-7 and SNP-8 with a resistant phenotype. Recombinant-2 and recombinant-4 were from crossovers between SNP-2 and SNP-3, with resistant and susceptible phenotypes, respectively. Recombinant-3 and recombinant-5 were from crossovers between SNP-8 and SNP-9, with resistant and susceptible phenotypes, respectively. Recombinant-6 was from a crossover between SNP-1 and SNP-2 with a resistant phenotype. Together, these recombinants indicated that the Pi65(t) locus is within an interval of 0.43 Mb on chromosome 11 flanked by SNP-2 (30.42 Mb) and SNP-8 (30.85 Mb)
Linkage analysis and marker-assisted selection
One InDel, nine SNP and four SSR markers on chromosome 11 showed polymorphisms between the resistant and susceptible parents. The 14 polymorphic markers were then used to analyze the genetic distance between Pi65(t) and the markers. Using the software QTL IciMapping, the R gene Pi65(t) was mapped to a locus between InDel-1 and SNP-4 with genetic distances of 0.11 and 0.98 cM, respectively (Fig. S3; Table S3), and its physical position is at 30.57–30.63 Mb. Within this region, 4 predicted R genes were found with nucleotide binding site and leucine-rich repeat (NBS-LRR) domains (Table 4). Using InDel-1, DH170 carrying Pi65(t) was screened and then backcrossed with Liaoxing1. Via phenotypic and genotypic identification, we got 14 homozygous resistant plants with similar agronomical traits to Liaoxing1 from the backcross population and they carried the rice blast R gene Pi65(t). Combined with yield and quality tests, we have successfully bred a new rice variety “Chuangxin1” that is highly resistant to rice blast in Liaoning province of China.
Discussion
Pi65(t) is a novel broad-spectrum R gene to rice blast
In the present study, we mapped a gene, Pi65(t), with broad-spectrum resistance to rice blast, into an interval of 0.43 Mb on chromosome 11. Firstly, Pi65(t) was located within the region on chromosome 11 from 30.20 to 31.20 Mb. Then, the markers SNP-1, RM27364 and InDel-1 were chosen to select recombinants and delimited the introgression region of Pi65(t). Secondly, nine SNP markers were developed to complete the chromosome walk to the target region. The Pi65(t) locus is located in a region of chromosome 11 with known blast R genes, including Pi54, Pi66(t), Pi-k, Pik-m and Pik-p. Pi54 was mapped to a 2-cM interval between RM224 and Y6855RA in the same region. Pi-k, Pik-m and Pik-p are located between the SSR markers RM1233 and RM224 on the long arm of chromosome 11, with respective distances of 1.6 and 0.2 cM to the linked markers (Ashikawa et al. 2008). Pi66(t) was mapped to an interval on Chr11 with the physical position from 27.7 to 27.84 Mb based on the Nipponbare reference database in MSU 7.0 (Liu et al. 2015). To differentiate these resistance genes, we constructed an integrated physical map via analysis of the positions of Pi54, Pi66(t), Pi-k, Pik-m and Pik-p (Fig. S4). In this physical map, the Pi65(t) locus was defined by SNP-2 and SNP-8, which were located from 30.42 to 30.85 Mb on chromosome 11; Pi54 is located at 27.46 Mb (Sharma et al. 2010); Pi66(t) is located at 29.9–30.0 Mb based on the Nipponbare reference database in build 5 and the Pi-k, Pik-m and Pik-p loci are located at the genomic positions 30.17 Mb. Further, in our previous study, the allele at Pik locus in Gangyu129 was proved to have no resistance function (Wang et al. 2015). Thus, the resistance in Gangyu129 is conferred by a novel broad-spectrum R gene because no other blast R genes were mapped to the same region on chromosome 11. However, it is possible that the broad-spectrum resistance was attributed to the synergy of multiple R genes that were linked very closely in the mapping interval. For this, we are performing gene transformation to confirm the function of the candidate genes.
Development and utilization of molecular markers for Pi65(t)
The traditional phenotypic evaluation of resistance to M. oryzae in rice breeding is labor-intensive, time-consuming, and easily influenced by environmental factors. With high-resolution genetic and physical mapping strategies, molecular markers associated with disease resistance can be effectively deployed in crop breeding (Boyd et al. 2013). A number of PCR-based markers have also been developed for several blast R genes, such as Pita (Jia et al. 2002), Piz, Piz-t, Pit, Pik, Pik-m, Pik-p, Pita-2 (Hayashi et al. 2006). The DNA markers offer an efficient and rapid means to select blast R genes. However, because of sequence similarity and clustering of Pi genes, an effective marker must be highly specific. In this study, we developed an InDel marker (InDel-1) located at 30.57 Mb on Chr11 and it is tightly linked with Pi65(t) resistance genotype. Because the sequences of genes at 30.42 and 30.57 Mb on Chr11 are very similar, two fragments of 120 bp and 139 bp were amplified with Indel-1 from Liaoxing1, and one fragment of 139 bp from Gangyu129 (Fig. S2b). We were able to screen the homozygous resistant individuals by selecting those plants with only a 139 bp using Indel-1, and the individuals that had two bands were all heterozygotes or homozygous susceptible plants at the Pi65(t) locus.
Though Pi65(t) has not been cloned, we could improve the resistance of Liaoxing1 using Indel-1. To improve the resistance of Liaoxing1 to rice blast, we screened a resistant line (DH170) from the DH population derived from Gangyu129 (R)/Liaoxing1 (S). DH170 was identified to carry Pi65(t) using InDel-1, and we then backcrossed it with Liaoxing1. In the F6 population derived from DH170/Liaoxing1, via phenotypic and genotypic identification we screened 14 homozygous resistant plants with similar agronomical traits to Liaoxing1 and they carried the rice blast R gene Pi65(t). Combined with tests of yield and quality, we have thus bred a new rice variety “Chuangxin1” that is highly resistant to rice blast in Liaoning province of China, indicating that Pi65(t) would play a key role in the improvement of rice blast resistance in northern China.
Reliability and advantages of the gene mapping strategy using SLAF-seq and BSA
In the current study, a rice blast R gene Pi65(t) was finely mapped using SLAF-seq in combination with BSA; this has many advantages. Firstly, the SNP markers of the genetic map are much denser compared with SSR or other markers. Using SLAF-seq, we detected 9092 SNP markers in total and more than 700 markers were mapped to every chromosome, which is conducive to map a gene to a small region. Secondly, it saves the labors and shortens the process of the research. In view of the recent rapid development in sequencing technology, we foresee that the method will dramatically accelerate gene mapping and crop improvement in a cost-effective and time-saving manner.
Author contribution statement
W. J. Zheng and Y. Wang performed the experiments and wrote the paper. L. L. Wang, Z. B Ma, J. M. Zhao, P. W, L. X. Zhang and Z. H. Liu performed the experiments. X. C. Lu designed the experiments.
References
Abe A, Kosugi S, Yoshida K et al (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotechnol 30:174–178
Ashikawa I, Hayashi N, Yamane H, Kanamori H, Wu JZ, Matsumoto T, One K, Yano M (2008) Two adjacent nucleotide-binding site-leucine-rich repeat class genes are required to confer Pikm-specific rice blast resistance. Genetics 180:2267–2276
Boyd LA, Ridout C, O’Sullivan DM, Leach JE, Leung H (2013) Plant–pathogen interactions: disease resistance in modern agriculture. Trends Genet 29:233–240
Chauhan RS, Farman ML, Zhang HB, Leong SA (2002) Genetic and physical mapping of a rice blast resistance locus, Pi-CO39(t), that corresponds to the avirulence gene AVR1-CO39 of Magnaporthe grisea. Mol Genet Genom 267:603–612
Chen DH, Zeigler RS, Leung H, Nelson RJ (1995) Population structure of Pyricularia grisea at two screening sites in the Philippines. Phytopathology 85:1011–1020
Chen W, Yao JB, Chu L, Yuan ZW, Li Y, Zhang YS (2015) Genetic mapping of the nulliplex-branch gene (gb_nb1) in cotton using next-generation sequencing. Theor Appl Genet 128:539–547
Cheng LR, Yang AG, Jiang CH, Ren M, Zhang Y, Feng QF, Wang SM, Guan YS, Luo CG (2015) Quantitative trait loci mapping for plant height in tobacco using linkage and association mapping methods. Crop Sci 55:641–647
Das S, Upadhyaya H, Bajaj D et al (2015) Deploying QTL-seq for rapid delineation of a potential candidate gene underlying major trait-associated QTL in chickpea. DNA Res. doi:10.1093/dnares/dsv004
Dean RA, Talbot NJ, Ebbole DJ et al (2005) The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 434:980–986
Fekih R, Takagi H, Tamiru M et al (2013) MutMap+: genetic mapping and mutant identification without crossing in rice. PLoS ONE 8:e68529
Fukuoka S, Saka N, Koga H et al (2009) Loss of function of a proline-containing protein confers durable disease resistance in rice. Science 325:998–1001
Hayashi K, Yoshida H, Ashikawa I (2006) Development of PCR-based allele-specific and InDel marker sets for nine rice blast resistance genes. Theor Appl Genet 113:251–260
Hittalmani S, Parco A, Mew TV, Zeigler RS, Huang N (2000) Fine mapping and DNA marker-assisted pyramiding of the three major genes for blast resistance in rice. Theor Appl Genet 100:1121–1128
Huang HM, Huang L, Feng GP et al (2011) Molecular mapping of the new blast resistance genes Pi47 and Pi48 in the durably resistant local rice cultivar Xiangzi 3150. Phytopathology 101:620–626
Hulbert SH, Webb CA, Smith SM, Sun Q (2001) Resistance gene complexes: evolution and utilization. Annu Rev Phytopathol 39:285–312
Jeon JS, Chen D, Yi GH, Wang G, Ronald P (2003) Genetic and physical mapping of Pi5 (t), a locus associated with broad-spectrum resistance to rice blast. Mol Genet Genom 269:280–289
Jeung JU, Kim BR, Cho YC, Han SS, Moon HP, Lee YT, Jena KK (2007) A novel gene, Pi40(t), linked to the DNA markers derived from NBS-LRR motifs confers broad spectrum of blast resistance in rice. Theor Appl Genet 115:1163–1177
Jia Y, McAdams SA, Bryan GT, Hershey HP, Valent B (2000) Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. EMBO J 19:4004–4014
Jia YL, Wang ZH, Singh P (2002) Development of dominant rice blast Pi-ta resistance gene markers. Crop Sci 42:2145–2149
Kent WJ (2002) BLAT—the BLAST-like alignment tool. Genome Res 12:656–664
Kosambi DD (1943) The estimation of map distance from recombination values. Ann Eugen 12:172–175
Li HH, Ribaut JM, Li ZL, Wang JK (2008) Inclusive composite interval mapping (ICIM) for digenic epistasis of quantitative traits in biparental populations. Theor Appl Genet 116:243–260
Liu XQ, Wang L, Chen S, Lin F, Pan QH (2005) Genetic and physical mapping of Pi36 (t), a novel rice blast resistance gene located on rice chromosome 8. Mol Genet Genomics 274:394–401
Liu JL, Wang XJ, Mitchell T, Hu YJ, Liu XL, Dai LY, Wang GL (2010) Recent progress and understanding of the molecular mechanisms of the rice–Magnaporthe oryzae interaction. Mol Plant Pathol 11:419–427
Liu WD, Liu JL, Ning YS, Ding B, Wang XL, Wang ZL, Wang GL (2013a) Recent progress in understanding PAMP- and effector-triggered immunity against the rice blast Fungus Magnaporthe oryzae. Mol Plant 6:605–620
Liu Y, Liu B, Zhu XY, Yang JY, Bordeos A, Wang GL, Leach JE, Leung H (2013b) Fine-mapping and molecular marker development for Pi56(t), a NBS-LRR gene conferring broad-spectrum resistance to Magnaporthe oryzae in rice. Theor Appl Genet 126:985–998
Liu Y, Qi XS, Young ND, Olsen KM, Caicedo AL, Jia YL (2015) Characterization of resistance genes to rice blast fungus Magnaporthe oryzae in a “Green Revolution” rice variety. Mol Breeding 35:1–8
Livaja M, Wang Y, Wieckhorst S, Haseneyer Grit et al (2013) BSTA: a targeted approach combines bulked segregant analysis with next-generation sequencing and de novo transcriptome assembly for SNP discovery in sunflower. BMC Genom 14:628
Lu HF, Lin T, Klein J, Wang SH, Qi JJ, Zhou Q, Sun JJ, Zhang ZH, Weng YQ, Huang SW (2014) QTL-seq identifies an early flowering QTL located near flowering locus T in cucumber. Theor Appl Genet 127:1491–1499
Ma J, Jia M, Jia Y (2014) Characterization of rice blast resistance gene Pi61 (t) in rice germplasm. Plant Dis 98:1200–1204
Michelmore RW, Paran I, Kesseli R (1991) Identification of markers linked to disease-resistance genes by bulked segregant analysis: a rapid method to detect markers in specific genomic regions by using segregating populations. Proc Natl Am Sci USA 88:9828–9832
Ou SH (1985) Rice diseases, 2nd edn. Commonwealth Mycological Institute, Kew
Pan QH, Wang L, Ikehashi H, Tanisaka T (1996) Identification of a new blast resistance gene in the indica rice cultivar Kasalath using Japanese differential cultivars and isozyme markers. Phytopathology 86:1071–1075
Qu SH, Liu GF, Zhou B, Bellizzi M, Zeng L, Dai LY, Han B, Wang GL (2006) The broad-spectrum blast resistance gene Pi9 encodes a nucleotide-binding site–leucine-rich repeat protein and is a member of a multigene family in rice. Genetics 172:1901–1914
Salvi S, Tuberosa R (2005) To clone or not to clone plant QTLs: present and future challenges trends in plant. Science 10:297–304
Sharma T, Rai A, Gupta S, Singh N (2010) Broad-spectrum blast resistance gene Pi-kh cloned from rice line tetep designated as Pi54. J Plant Biochem Biot 19:87–89
Sun XW, Liu DY, Zhang XF et al (2013) SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 8:e58700
Takagi H (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Takagi H, Uemura A, Yaegashi H et al (2013) MutMap-Gap: whole-genome resequencing of mutant F2 progeny bulk combined with de novo assembly of gap regions identifies the rice blast resistance gene Pii. New Phytol 200:276–283
Takagi H, Tamiru M, Abe A et al (2015) MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat Biotechnol 33:445–449
Trick M, Adamski NM, Mugford SG, Jiang CC, Febrer M, Uauy C (2012) Combining SNP discovery from next-generation sequencing data with bulked segregant analysis (BSA) to fine-map genes in polyploid wheat. BMC Plant Biol 12:14
Wang SW, Zheng WJ, Zhao JM, Wei SH, Wang Y, Zhao BH, Liu ZH (2013) Identification and analysis of Magnaporthe oryzae avirulence genes in Liaoning Province. Scientia Agricultura Sinica 47:462–472 (In Chinese)
Wang Y, Zhao JM, Zhang LX, Wang P, Wang SW, Wang H, Wang XX, Liu ZH, Zheng WJ (2015) Analysis of the diversity and function of the alleles of the rice blast resistance genes Piz-t, Pita and Pik in 24 rice cultivars. J Integr Agr. doi:10.1016/S2095-3119(15)61207-2
Xia C, Chen LL, Rong TZ, Li R, Xiang Y, Wang P, Liu CH, Dong XQ, Liu B, Zhao D, Wei RJ, Lan H (2014) Identification of a new maize inflorescence meristem mutant and association analysis using SLAF-seq method. Euphytica 202:35–44
Xu X, Hayashi N, Wang CT, Fukuoka S, Kawasaki S, Takatsuji H, Jiang CJ (2014) Rice blast resistance gene Pikahei-1 (t), a member of a resistance gene cluster on chromosome 4, encodes a nucleotide-binding site and leucine-rich repeat protein. Mol Breed 34:691–700
Yang ZM, Huang DQ, Tang WQ, Zheng Y, Liang KJ, Cutler AJ, Wu WR (2013) Mapping of quantitative trait loci underlying cold tolerance in rice seedlings via high-throughput sequencing of pooled extremes. PLoS ONE 8:e68433
Zuo WL, Chao Q, Zhang N et al (2014) A maize wall-associated kinase confers quantitative resistance to head smut. Nat Genet 2015 47:151–157
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No.31571993 and 31301636), and Natural science foundation of Liaoning Province in China (2014027027).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest in regard to this manuscript.
Ethical standards
We declare that these experiments comply with the ethical standards in China, where they were performed.
Additional information
Communicated by M. Thomson.
W. Zheng and Y. Wang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zheng, W., Wang, Y., Wang, L. et al. Genetic mapping and molecular marker development for Pi65(t), a novel broad-spectrum resistance gene to rice blast using next-generation sequencing. Theor Appl Genet 129, 1035–1044 (2016). https://doi.org/10.1007/s00122-016-2681-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-016-2681-7