
Abstract
Key message
A dominantly inherited major-effect QTL for powdery mildew resistance in cucumber was fine mapped. Two tandemly arrayed cysteine-rich receptor-like protein kinase genes were identified as the most possible candidates.
Abstract
Powdery mildew (PM) is one of the most severe fungal diseases of cucumber (Cucumis sativus L.) and other cucurbit crops, but the molecular genetic mechanisms of powdery mildew resistance in cucurbits are still poorly understood. In this study, through marker-assisted backcrossing with an elite cucumber inbred line, D8 (PM susceptible), we developed a single-segment substitution line, SSSL0.7, carrying 95 kb fragment from PM resistance donor, Jin5-508, that was defined by two microsatellite markers, SSR16472 and SSR16881. A segregating population with 3600 F2 plants was developed from the SSSL0.7 × D8 mating; segregation analysis confirmed a dominantly inherited major-effect QTL, Pm1.1 in cucumber chromosome 1 underlying PM resistance in SSSL0.7. New molecular markers were developed through exploring the next generation resequenced genomes of Jin5-508 and D8. Linkage analysis and QTL mapping in a subset of the F2 plants delimited the Pm1.1 locus into a 41.1 kb region, in which eight genes were predicted. Comparative gene expression analysis revealed that two concatenated genes, Csa1M064780 and Csa1M064790 encoding the same function of a cysteine-rich receptor-like protein kinase, were the most likely candidate genes. GFP fusion protein-aided subcellular localization indicated that both candidate genes were located in the plasma membrane, but Csa1M064780 was also found in the nucleus. This is the first report of dominantly inherited PM resistance in cucumber. Results of this study will provide new insights into understanding the phenotypic and genetic mechanisms of PM resistance in cucumber. This work should also facilitate marker-assisted selection in cucumber breeding for PM resistance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cucumber, Cucumis sativus L., is an economically important vegetable crop, globally ranking 4th in quantity of world vegetable production (FAO STAT 2013, http://faostat3.fao.org). However, frequent occurrence of powdery mildew (PM) causes serious losses in yield and quality. PM is mainly caused by Podosphaera fusca (synonym Podosphaera xanthii) (Perez-Garcia et al. 2009), which occurs more frequently in subtropical and tropical areas, because of the optimum temperature for conidial germination (25–30 °C) and the availability of a wide range of hosts including a variety of cucurbit crops (Sitterly 1978; Zitter et al. 1996; Lebeda et al. 2011; He et al. 2013). At present, in most parts of cucumber production areas aiming for fresh market consumption, fungicide application is the major method of disease control, which, however, is less effective under a disease outbreak since fruits are harvested almost daily (Fukino et al. 2013). In addition, long-term use of fungicides may cause the pathogen to develop resistance or insensitivity to them, rendering the chemicals ineffective. Thus, breeding cultivars resistant or tolerant to PM is a desirable strategy for disease control.
Marker-assisted selection (MAS) is more economic and more efficient than traditional breeding based on phenotypic selection (Lande and Thompson 1990). In recent years, due to the use of next generation sequencing technologies and the availability of genetic and genomics resources, a lot of PMR genes and quantitative trait loci (QTL) have been reported in cucumber. Sakata et al. (2006) identified six QTL on cucumber chromosomes 1, 5, 6 and 7 for powdery mildew resistance derived from PI 197088. Fukino et al. (2013) conducted further QTL mapping of PMR from this PI line with 111 recombinant inbred lines (RIL) and identified nine QTL for PM resistance that were distributed in six cucumber chromosomes. Hofstede et al. (2008) and de Ruiter et al. (2008) identified two major-effect QTL for PM resistance in the leaves (pm-l) and hypocotyls (pm-h), both of which were located in cucumber chromosome 5. Zhang et al. (2011) identified three QTL (pm5.1, pm5.2 and pm5.3) in chromosome 5, and one (pm6.1) in chromosome 6 for PM resistance from a North China cucumber line K8. He et al. (2013) identified six QTL in chromosomes 1, 3, 4 and 5 for PM resistance in WI 2757 with major-effect QTL in chromosome 5. More recently, Nie et al. (2015) conducted fine genetic mapping of the major-effect QTL, pm5.1 that was identified previously by de Ruiter et al. (2008) and He et al. (2013), and found a MLO-like gene as the most likely candidate for the pm5.1 locus. The resistance conferred by pm5.1 was due to insertion of a retrotransposon sequence into the exonic region of the Mlo-like gene (Nie et al. 2015). While these findings have provided insights into the genetic control of PM resistance in cucumber, it is far from satisfactory to understand the molecular mechanisms of host resistance against cucumber PM. In addition, most PMR genes or QTL in cucumber reported to date exhibit recessive inheritance, which is not convenient to use in cucumber breeding. Munger et al. (1979) and El-Jack and Munger (1983) found dominant inheritance of PMR in ‘Spartan Salad 77-717’ and PI 197088. Later, Morishita et al. (2003) suggested that PMR in197088-5, a selection from PI 197088, was controlled by two genes, one recessive and the other incompletely dominant. Expression of PMR in 197088-5 depends on interactions of the two genes and the temperature (Morishita et al. 2003).
Among the types of segregated populations, the single-segment substitution line (SSSL) is a powerful tool for functional genomics, and molecular breeding and genetic dissection of QTLs, which has been used in many crops (Shen et al. 2014; Liu et al. 2008). With high level of uniformity of the genetic background except for a single substituted segment, all the phenotypic variations among SSSLs could be associated with the substituted segments in those lines. In our previous study (Lin et al. 2012), we developed a serious of SSSLs with PM resistance introgressed from Jin5-508 in the genetic background of PM susceptible D8 (PM susceptible, dwarf type plant architecture) which was facilitated by marker-assisted selection with simple sequence repeat (SSR) and sequence characterized amplified regions (SCAR) markers. At the BC11 generation, one such SSSL, namely SSSL0.7 exhibited high resistance to the powdery mildew pathogen. We developed a large segregating population with SSSL0.7. The objectives of the present study were to identify and fine map the major-effect QTL conferring resistance to PM in SSSL0.7. We developed a secondary F2 segregating population from a cross between SSSL0.7 and the recurrent parent, D8. The analysis of recombination events in the PM region with SNP markers delimitated the PM locus to a 41.1-kb region. Candidate genes were predicted and analyzed by qRT-PCR. The markers linked to the substituted segment detected in our study might be useful for selection of PM resistance in different cucumber accessions.
Materials and methods
Plant materials
The PM-resistant single-segment substitution line SSSL0.7 was derived from marker-assisted backcrossing (Lin et al. 2012) with a North China type, indeterminate cucumber inbred line Jin5-508 as the donor of PM resistance and a American type, dwarf cucumber inbred line D8 as the recipient (recurrent parent, PM susceptible). Briefly, from 2004 to 2009, the F1 of D8 × Jin5-508 was backcrossed with D8 for 11 generations (two generations per year). Starting from BC2, at each generation, a SCAR marker linked with PMR in Jin5-508 was used to select a plant for backcrossing with D8 (Alfandi et al. 2009). At BC11, 17 plants were self-pollinated to generate 17 families with a total of 449 plants. These chromosome segment introgression lines (CSILs) were near isogenic at the PMR loci as compared with the recurrent parent, D8. Microsatellite marker analysis of these CSIL identified a SSSL, SSSL0.7, that carried a chromosome fragment from Jin5-508 and was highly resistant to powdery mildew (Lin et al. 2012). SSSL0.7 was delimited by two SSR markers SSR16472 and SSR16881 in cucumber chromosome 1, which were 0.7 cM apart on the genetic map developed by Ren et al. (2009). In this study, to clone the PM resistance locus carried by SSSL0.7, an F2 population consisting of 3600 plants was developed by crossing SSSL0.7 with D8.
Powdery mildew screening
Powdery mildew resistance of parental lines Jin5-508, D8 and SSSL0.7 was repeatedly evaluated in three seasons (fall 2011 and 2012, spring 2013) in the greenhouses of Yangzhou University (Yangzhou, China). In 2014 spring greenhouse season, Jin5-508, D8, SSSL0.7 and (SSSL0.7 × D8) F1 (50 plants each), and 3600 F2 plants were screened for PMR. PM conidia were collected from naturally infected cucumber plants in the greenhouse. A spore suspension at 106 spores per ml was made by soaking heavily infected leaves in tap water (containing 0.01 % Tween-20 as surfactant). Inoculation was performed at the 3rd true-leaf stage by spraying the spore suspension evenly on the surface of the seedlings according to Morishita et al. (2003). After inoculation, the plants were maintained in a controlled growth chamber at 25 °C day/20 °C night with a 16-/8-h (light/dark) photoperiod. Fifteen days after inoculation, the percentage of infected area of each leaf of each sample plant was determined with the following visual rating scale: 0 = no symptom; 1 = infection area <30 %; 2 = 30–60 %; 3 = >60–80 %; 4 = >80 %; and 5 = leaf senesced. Disease index (DI) was calculated using the following equation: DI = ∑(Disease scale × number of leaves of that specific scale)/(number of leaves inoculated × the highest disease grade) × 100.
DNA extraction and SSR identification
Genomic DNA was extracted from young leaf tissues using the CTAB method. DNA concentration was estimated on a 0.8 % agarose gel. Each 25 µl PCR reaction contained 19.0 µl water, 2.5 µl 10 × buffer, 2 µl dNTPs (10 mM), 0.5 µl Taq DNA polymerase (10 U/µl), 0.25 µl each of left and right primers (50 ng/µl), and 0.5 µl DNA (10 ng/µl). The PCR amplifications were performed using a GeneAmp PCR system 9700 (Applied Biosystems Inc., Foster City, CA) with the following program: 94 °C for 4 min; 35 cycles of 94 °C for 15 s, 55 °C for 15 s, and 72 °C for 30 s; and a final extension at 72 °C for 5 min. Subsequently, 2 µl of the PCR product mixed with 1 µl of 6 × loading buffer was subjected to electrophoresis in a 6 % polyacrylamide gel following Sambrook and Russell (2001).
Genome resequencing and SNP marker development
The genomes of the two parental lines, Jin5-508 and D8 were resequenced with Illumina HiSeq 2500. In brief, 5 µg genomic DNAs were used for preparation of paired-end sequencing libraries with insert sizes of ~200 to 500 bp following manufacturer’s instructions. A total of 54.9 and 61.7 million clean reads were obtained for Jin5-508, and D8 with an average sequencing depth 30× and 35×, respectively. Single nucleotide polymorphisms (SNPs) between the two lines were detected with the GATK software package (McKenna et al. 2010). Local realignment and base recalibration were performed to improve the accuracy of SNP genotyping. The 9930 cucumber draft genome [V2.0, http://www.icugi.org/cgi-bin/ICuGI/index.cgi)] was used as the reference. SNPs were filtered using the criteria SNP quality value >30 and base quality >30. All SNPs in the region delimited by SSR16472 and SSR16881 in chromosome 1 were considered for design of new markers. SNP-based dCAPS markers were designed with the dCAPS Finder 2.0 program (http://helix.wustl.edu/dcaps/dcaps.html), and Primer Premier 5.0 (http://www.premierbiosoft.com/). For genotyping with the dCAPS markers, following PCR reactions as described above, the appropriate restriction enzyme was added to the PCR reaction and incubated for 2 h at the temperature according to the manufacturer’s instructions (New England Biolabs, Ipswich, MA, USA). Two microliters of digested PCR products were then mixed with 1 µl of 10 × loading buffer, and subjected to electrophoresis in a 6 % polyacrylamide gel following Sambrook and Russell (2001).
Linkage analysis
The phenotypic data of PM inoculation responses among the 3600 F2 plants were subjected to quantitative and qualitative linkage analyses with molecular markers. From 3600 F2 plants, 121 individuals were randomly selected for linkage analysis (PMR as a single gene) or QTL mapping (using DI data). All marker alleles derived from D8 were assigned as ‘a’ allele; those from SSSL0.7 as ‘b’ allele; heterozygous as ‘h’. In linkage mapping, χ 2 test for goodness-of-fit was performed against the expected 1:2:1 segregation ratio for each marker. Linkage analysis was performed with the Kosambi mapping function using JoinMap 3.0 (Van Ooijen and Voorrips 2001) with the threshold LOD score of 2.5. QTL analysis was performed with WinQTL Cartographer (V2.5) using the CIM (composite interval mapping) model (Wang et al. 2012).
Gene prediction and sequence annotation
The genomic DNA region harboring the PMR locus was annotated with FGENESH (http://sunl.softberry.com/) and InterProScan (http://www.ebi.ac.uk/InterProScan). Genes in this region were predicted with BLASTx at the NCBI (National Center for Biotechnology Information) website (http://blast.ncbi.nlm.nih.gov).
Quantitative reverse-transcription PCR (qRT-PCR) analysis of candidate gene
Total RNA was isolated from leave samples of Jin5-508, D8 and SSSL0.7 at 0, 12, 24, and 48 h after inoculation of powdery mildew pathogen using RNAiso Plus (Takara, China). The concentration of RNA samples was adjusted to 1000 µg/mL using Biophotometer Plus (Expander, Germany). RNA was reverse-transcribed using a Takara PrimeScript® RT reagent kit with gDNA eraser according to the manufacturer specifications. RT-PCR was performed using a RealMasterMix (SYBR Green) kit (TIANGEN, China) on an iQ™ 5 Multicolor real-time PCR detection system (Bio-RAD, USA) in 20 µL reactions. The PCR primers were designed with Primer Premier 5.0 (Premier Biosoft International, Palo Alto, CA, USA). The sequence information for all primers used in the present study is presented in Table 1. Three replications of each treatment and control were used for real-time RT-PCR. The relative mRNA expression was based on the 2−△△Ct method. Each expression profile was independently verified in three technical replications under identical conditions.
Subcellular localization of Csa1M064780.1 and Csa1M064790.1 proteins
The coding region of two possible candidate genes, Csa1M064780.1 and Csa1M064790.1, were amplified by PCR and subcloned into the PGEM-T Easy vector (Promega, USA). The DNA fragments were digested with Xba I and Kpn I, and then inserted into the pUC-GFP vector, which was verified by restriction and sequencing analysis. The verified recombinant plasmid was transformed into living onion epidermal cells by particle bombardment following Kinkema et al. (2000). The transformants were incubated on 1/2 MS medium for 24 h at 26 °C in dark. The fluorescence of GFP was visualized using a Leica TCS SP2/MP confocal laser scanning microscope (Leica Microsystems, Mannheim, Germany).
Results
Performance of powdery mildew resistance in SSSL0.7
Fifteen days after inoculation, no symptoms could be observed on the resistant parent Jin5-508 (DI = 0) in any of the four environments tested. The DI for D8 was 35.9, 36.7, 30.6 and 37.7 in 2011, 2012, 2013 and 2014, respectively. SSSL0.7 also showed consistent and high resistance to PM inoculation with DI of 6.3, 5.2, 3.7 and 3.3, respectively, in the 4 years. The level of resistance in SSSL0.7 was slightly lower than its original donor Jin5-508, which was reasonable because we found some other introgression lines among the backcross derivatives that also exhibited low level PM resistance. These results suggested that a major-effect QTL from Jin5-508 was successfully introgressed to the D8 genetic background (Fig. 1).

Morphology and powdery mildew (PM) natural infestation responses of D8 (PM susceptible recurrent parent, left), Jin5-508 (PM resistance donor, right), and the derived single-segment substitution line SSSL0.7 (PM resistant, middle)
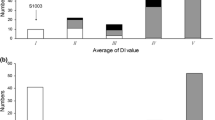
In 2014 greenhouse screening tests, 3600 F2 plants derived from D8 × SSSL0.7 were tested for responses to PM inoculation. Frequency distribution of the 3600 plants based on DI is shown in Fig. 2, which showed a largely normal distribution. The average DI for SSSL0.7, D8 and their F1 was 3.3, 37.7 and 12.3, respectively. These results indicated that SSSL0.7 carried a dominant major-effect QTL for powdery mildew resistance, which, to our knowledge, was the first reported dominant gene to powdery mildew resistance in cucumber. We designated this locus as Pm1.1. The categorical data (R, S) of 121 selected individuals among the 3600 plants were used in linkage analysis to map Pm1.1 (below).

Frequency distribution of disease index of powdery mildew inoculation responses among 3600 F2 plants derived from the cross between D8 (PM susceptible recurrent parent) and SSSL0.7 (PM-resistant)
Molecular delimitation of substituted segment length in SSSL0.7
The introgressed fragment carrying Pm1.1 in SSSL0.7 was delimited by two SSR markers SSR16472 and SSR16881, but the exact boundaries of breakpoints for this introgression were not known due to the limited number of available SSR markers. To gain a more precise estimation of the length of the introgressed fragment, 10 SNP-derived dCAPS markers were designed to further characterize the length of introgression in SSSL0.7. Among the 10 markers, five (SNP01 W to SNP05 W) were located in the region outside SSR16472, and five (SNP06 W to SNP10 W) were outside SSR16881. All 10 markers were polymorphic between D8 and Jin5-508, but monomorphic between SSSL0.7 and D8 indicating that the substituted segment in SSSL0.7 was correctly defined by SSR16472 and SSR16881. The physical distance of this Jin5-508-derived region was about 94.7 kb on scaffold000059 of 9930 (Huang et al. 2009).
Fine mapping of Pm1.1 locus
Twenty SNP-derived dCAPS markers were designed within the 94.7 kb region to narrow down the substituted segment carrying the Pm1.1 locus. Three of the markers, SNP02, SNP09, and SNP20, were polymorphic between Jin5-508 and D8. Primer sequence information of the three dCAPS markers together with the two flanking SSR markers SSR16472 and SSR16881 is presented in Table 1. QTL analysis was performed with the five markers and DI data from 121 randomly selected F2 plants with the CIM model in WinQTL Cartographer 2.5. The result is shown in Fig. 3a. A QTL peak, Pm1.1, was present between two dCAPS markers SNP09 and SNP20 with a LOD score of 2.6. This QTL accounted for 18.4 % of the total phenotypic variations (Fig. 3a, b). Consistent with this, when categorical data were used in linkage analysis with the 121 plants, the Pm1.1 locus was placed in between SNP09 and SNP20 suggesting Pm1.1 was indeed located in the interval between SNP09 and SNP20.

Genetic and physical maps of powdery mildew resistance major-effect QTL Pm1.1. In a LOD curve in chromosome 1 from the QTL analysis based on 121 F2 plants. b A linkage map for Pm1.1 on chromosome 1 based on categorical data of disease inoculation responses among the 121 F2 plants. Numbers below the chromosome are map locations in cM. c Analysis of the overlap mapping of the Pm1.1 locus with 7 recombinants indicated the Pm1.1 locus was residing in the 41.1-kb region flanked by SNP09 and SNP20. The right is the disease index (DI) of each recombinant and two parents. Filled, gray and open bars represent homozygous fragments from SSSL0.7, possible heterozygous interval and homozygous fragments from D8. Genotypes of five markers of each recombinant plant are also shown under each chromosome map
To further verify the position of Pm1.1, all 3600 secondary F2 plants were genotyped with SSR16472 and SSR1688, from which seven recombinants (SD1 to SD7) were identified. Then, the seven plants were genotyped with three dCAPS markers. Genotypic data of five markers in the seven recombinant plants, and the four intervals defined by these markers are illustrated in Fig. 3c. Two of the seven recombinants, SD01 and SD06 were highly susceptible to PM, and the rest were highly resistant. Comparison of the marker-defined intervals and the PMR of the seven plants led to the conclusion that Pm1.1 must reside in the 41.1 kb region flanked by SNP09 and SNP20.
Annotation and genes prediction in the 41.1 kb region
Annotation of the 41.1 kb genomic region delimited by SNP09 and SNP20 predicted 8 genes in this region. The predicted functions and associated information of the eight genes are presented in Table 2. Two of the genes (Csa1M064720.1 and Csa1M064750.1) encoded proteins with unknown functions. Csa1M064730.1 was predicted to encode a protein with gibberellin 2-oxidase activity that acts specifically on C-20 gibberellins. Csa1M064740.1 was predicted to encode a pyruvate orthophosphate dikinase (PPDK) regulatory protein that has both kinase and phosphatase activities towards PPDK. Csa1M064760.1 and Csa1M064770.1 were predicted to encode proteins belong to late embryogenesis abundant (LEA) family. Csa1M064780.1 and Csa1M064790.1 were both predicted to encode a cysteine-rich receptor-like protein kinase (Table 1).
Expression analysis of Pm1.1 candidate genes in response to PM inoculation
Expression dynamics of all eight genes in D8, SSSL0.7 and Jin5-508 at 0, 12, 24 and 48 h after inoculation of PM pathogen were investigated to identify possible candidate gene(s) for Pm1.1 in this 41.1 kb interval. The results are presented in Fig. 4 and supplemental Fig. S1. Two of the genes, Csa1M064780.1 and Csa1M064790.1, exhibited consistent patterns in the two PMR lines (Jin5-508 and SSL0.7) and the susceptible D8. That is, the relative expressions of both genes were low and stable in D8 before and after inoculation; both were up-regulated in SSSL0.7 and Jin5-508 12 h after inoculation. The expression level Csa1M064790.1 kept increasing after inoculation at all three time points, whereas that of Csa1M064780.1 did not show any significant increase 12 h after inoculation (Fig. 4). On the other hand, no consistent trends in expression levels were found among the three cucumber lines for the remaining six genes (Fig. S1) suggesting that both Csa1M064780.1 and Csa1M064790.1 were good candidates for the Pm1.1 locus in cucumber.

Expression level of Pm1.1 candidate genes Csa1M064780.1 and Csa1M064790.1 in D8 (susceptible, black), Jin5-508 (resistant, blue) and SSS0.7 (resistant, red) parental lines. Data are the means of three replicates (±SD). The cucumber β-actin gene was used as an internal control. Each value denotes the mean relative level of expression of three replicates. Means with the same lowercase letter do not significantly differ by the least significant difference (LSD) test at p ≤ 0.05 with a completely randomized design (color figure online)
Subcellular localization of Csa1M064780.1 and Csa1M064790.1 proteins
The cellular location of Csa1M064780.1 and Csa1M064790.1 proteins was investigated in onion epidermal cells transiently expressing gene fusions to the green fluorescent protein (GFP) by particle bombardment to provide further evidence for the potential roles of Csa1M064780.1 and Csa1M064790.1 in transcriptional regulation. Csa1M064780.1-GFP fluorescence was observed in plasma membrane and nucleus, but Csa1M064790.1-GFP fluorescence was observed exclusively in the plasma membrane (Fig. 5).

Membrane localization of theCsa1M064780.1 and Csa1M064790.1-GFP fusion protein. Full coding sequences of Csa1M064780.1 and Csa1M064790.1 were fused with green fluorescent protein (GFP) gene. The recombinant plasmids of Csa1M064780.1-GFP and Csa1M064790.1-GFP were then transformed into onion epidermal cells by particle bombardment, and viewed by confocal microscopy
Discussion
We used a single-segment substitution line (SSSL0.7) for fine genetic mapping of a major-effect QTL, Pm1.1, for powdery mildew resistance in cucumber. Several types of mapping populations have been used in previous QTL mapping studies of PM resistance in cucumber (Sakata et al. 2006; Liu et al. 2008; Zhang et al. 2011; He et al. 2013; Fukino et al. 2013; Nie et al. 2015) including F2, F2:3 or RIL populations. A limitation of these populations is the genetic background noises that sometimes may mask the true QTL effects, especially for minor-effect QTLs (Wei et al. 2015). SSSL0.7 possessed a small fragment (~94.7 kb) from the donor of PM resistance (Jin5-508) that had a simple genetic background, which allowed more precise mapping of the target QTL (Qi et al. 2013). In screening tests over 4 years, the powdery mildew resistance of SSSL0.7 was highly consistent and stable, indicating the 94.7-kb region truly carried a PMR locus. On the other hand, the DI of SSSL0.7 was slightly higher than the donor line Jin5-508 (less resistant) indicating SSSL0.7 carried a major-effect QTL (Pm1.1) and additional QTL may exist in Jin5-508 for complete resistance to powdery mildew. This was consistent with our observations that other SSSL carrying different Jin5-508 chromosome fragments were also resistance to PM although the performance of SSSL0.7 was the best among all SSSLs we developed from the Jin5-508 × D8 cross (Lin et al. 2012). Characterization of other powdery mildew-resistant SSSL will also allow us to have a complete picture of the genetic structure of powdery mildew resistance in Jin5-508.
To fine map the Pm1.1 locus in SSSL0.7, we developed a secondary, large F2 mapping population from the cross between the near isogenic SSSL0.7 and susceptible parent, D8, that segregated only at the Pm1.1 locus. The high level of resistance exhibited by the F1 indicated the dominant nature of PMR in SSSL0.7 (Fig. 1). Despite of the fact that only one gene (Pm1.1) was segregating in the F2 population, the frequency distribution of plants based on DI was still largely normal (Fig. 2) suggesting effects of environmental factors in expression of the resistance. Nevertheless, both QTL mapping and linkage analysis with 121 F2 plants placed Pm1.1 into the 41.1 kb interval defined by the two markers SNP09 and SNP20 (Fig. 3). The LOD support score (2.6) and the phenotypic variations explained by this QTL (R 2 = 18.4 %) (Fig. 3) were unexpectedly low. A similar phenomenon was found during map-based cloning of the PMR QTL pm5.1 (Nie et al. 2015). The reasons for this were probably due to the relatively small population and few markers employed in linkage analysis.
Through fine genetic mapping, we delimited the Pm1.1 locus into a 41.1 kb region in which eight genes were predicted (Table 2). Through comparative expression analysis, two tandem arrayed CRK genes, Csa1M064780.1 and Csa1M064790.1 were shown to be the candidate genes for Pm1.1 (Fig. 4, supplemental Fig. S1). While their expression remained low in the susceptible D8, both genes were up-regulated upon PM pathogen inoculation in the resistance SSSL0.7 and Jin5-508 (Fig. 4). Both of Csa1M064780.1 and Csa1M064790.1 proteins were expressed in the plasma membrane, but Csa1M064780.1-GFP fluorescence was also observed in the nucleus (Fig. 5) suggesting possible different roles of Csa1M064780.1 and Csa1M064790.1 in PM inoculation responses. At this time, it is not known if the PM resistance conferred by Pm1.1 was the result of functions of Csa1M064780.1 or Csa1M064790.1, or both. Functional analysis of Csa1M064780.1 and Csa1M064790.1 by complementation tests and other strategies is underway.
In plants, the receptor-like kinases (RLK) play fundamental roles in perceiving external stimuli, activating downstream signaling pathways, and regulating cellular behavior in response to pathogen infection. RLK contain an extracellular signal-sensing domain connected by a single transmembrane domain to an intracellular protein kinase domain (Shiu and Bleecker 2003; Osakabe et al. 2013). In the cucumber Gy14 draft genome, Wang et al. (2014) identified 192 homologs of the leucine-rich repeat (LRR) type RLK. One of the largest RLK subfamilies is the CRK which are transmembrane proteins characterized by the presence of two domains of unknown function 26 (DUF26) in their ectodomain. In the Arabidopsis thaliana genome, among the ~600 RLK, there are 44 CRK (Wrzaczek et al. 2010). The biological functions of CRK are largely uncharacterized. Recently, several studies have showed that CRK control important aspects of plant development and stress adaptation in response to biotic and abiotic stimuli in a non-redundant fashion, especially in reactive oxygen species/redox signaling and sensing (e.g., Czernic et al. 1999; Niina et al. 2014; Bourdais et al. 2015). In barley, a cysteine-rich receptor-like protein kinase gene HvCRK1 was observed to accumulate transiently following powdery mildew pathogen inoculation in barley (Rayapiram et al. 2012). In the present study, we provided evidence that one of the two cysteine-rich receptor-like protein kinase genes or both are the best possible candidate gene(s) for the dominantly inherited major-effect QTL for PMR in cucumber. This work added a new horizon for CRK in host resistance against fungal pathogens. Our work also provided the starting point in elucidating the regulatory mechanisms and enzyme kinetics underlying the CRK gene in the PMR pathway in cucumber.
In this study, for the first time, we identified a dominantly inherited major-effect QTL, Pm1.1, for PM resistance in cucumber. Previous QTL mapping studies identified recessively inherited loci for PM resistance (e.g., Kooistra 1968; Fujieda and Akiya 1962; Shanmugasundaram et al. 1971; He et al. 2013) with major-effect QTLs located, in most studies, in chromosome 5. Nie et al. (2015) cloned a candidate gene, pm5.1, for the major QTL in cucumber chromosome 5. The resistance conferred by pm5.1 was due to an insertion of retrotransposon into MLO-like gene (Nie et al. 2015). The relationship of Pm1.1 and pm5.1 in contributing to PMR in cucumber merits further investigation. Several PMR QTL were identified in the cucumber line WI2757 (He et al. 2013). While the major-effect QTL in WI2757 was consistent with the location of pm5.1 cloned by Nie et al. (2015), He et al. (2013) also identified QTL in chromosome 1 which seems to be co-localized with Pm1.1 identified in the present study (based on a common marker SSR16472). It will be interesting to see whether or not the powdery mildew-resistant line Jin5-508 possesses pm5.1 (retrotransposon insertion). If so, it may facilitate the understanding of interactions of different QTL in contributing to the host resistance to the powdery mildew pathogen in cucumber.
References
Alfandi M, Shen L, Qi X, Xu Q, Chen X (2009) A SCAR marker linked to powdery mildew resistance for selection of the near-isogenic lines in cucumber. In: Abstracts of 4th international cucurbitaceae symposium, pp 20–21
Bourdais G, Burdiak P, Gauthier A, Nitsch L, Salojärvi J, Rayapuram C et al (2015) Large- scale phenomics identifies primary and fine-tuning roles for CRKs in responses related to oxidative stress. PLoS Genet 11:e1005373
Czernic P, Visser B, Sun W et al (1999) Characterization of an Arabidopsis thaliana receptor-like protein kinase gene activated by oxidative stress and pathogen attack. Plant J 18:321–327
de Ruiter W, Hofstede R, de Vries J, van den Heuvel H (2008) Combining QTL for resistance to CYSDV and powdery mildew in a single cucumber line. In: Pitrat M (ed) Proceedings of the 9th EUCARPIA meeting on genetics and breeding of Cucurbitaceae, INRA, Avignon, France, 21–24 May 2008, pp 181–188
El-Jack A, Munger HM (1983) Two sources conferring partial dominant resistance to powdery mildew (Sphaerotheca fuliginea Poll.) in cucumber. Cucurbit Genet Coop 6:7–8
Fujieda K, Akiya R (1962) Genetic study of powdery mildew resistance and spine color on fruit in cucumber. J Jpn Soc Hort Sci 31:30–32
Fukino N, Yoshioka Y, Sugiyama M, Sakata Y, Matsumoto S (2013) Identification and validation of powdery mildew (Podosphaera xanthii)-resistant loci in recombinant inbred lines of cucumber (Cucumis sativus L.). Mol Breed 32:267–277
He XM, Li YH, Pandey S, Yandell BS, Pathak M, Weng Y (2013) QTL mapping of powdery mildew resistance in WI 2757 cucumber (Cucumis sativus L.). Theor Appl Genet 126:2149–2161
Hofstede R, de Ruiter W, de Vries RJ, van den Heuvel H (2008) Disease resistant cucumber plants. US Patent # US 2008/0307540 A1
Huang SW, Li RQ, Zhang ZH, Li L, Gu XF, Fan W et al (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281
Kinkema M, Fan W, Dong X (2000) Nuclear localization of NPR1 is required for activation of PR genes expression. Plant Cell 12:2339–2350
Kooistra E (1968) Powdery mildew resistance in cucumber. Euphytica 17:236–242
Lande R, Thompson R (1990) Efficiency of marker-assisted selection in the improvement of quantitative traits. Genetics 124:743–756
Lebeda A, Kristkova E, Sedlakova B, Coffey MD, McCreight JD (2011) Gaps and perspectives of pathotype and race determination in Golovinomyces cichoracearum and Podosphaera xanthii. Mycoscience 52:159–164
Lin XJ, Xu XW, Qian HM, Qi XH, Xu Q, Chen XH (2012) Analysis of cucumber chromosome segment introgression lines with powdery mildew resistance based on SSR markers. Acta Hort Sin 39:485–492 (in Chinese with English abstract)
Liu LZ, Yuan XJ, Cai R, Pan JS, He HL, Yuan LH, Guan Y, Zhu LH (2008) Quantitative trait loci for resistance to powdery mildew in cucumber under seedling spray inoculation and leaf disc infection. J Phytopathol 156:691–697
McKenna A, Hanna M, Banks E, Sivachenko A et al (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Morishita M, Sugiyama K, Saito T, Sakata Y (2003) Review: powdery mildew resistance in cucumber. JARQ 37:7–14
Munger HM, Morales A, Omara S (1979) Dominant genes for resistance to powdery mildew in cucumber. Cucurbit Genet Coop 2:10
Nie JT, He HL, Peng JL, Yang XQ, Bie BB, Zhao JL, Wang YL, Si LT, Pan JS, Cai R (2015) Identification and fine mapping of pm5.1: a recessive gene for powdery mildew resistance in cucumber (Cucumis sativus L.). Mol Breed 35:7
Niina I, Adrien G, Jarkko S, Riccardo S, Mikael B, Hannes K, Ari PM, Jaakko K, Michael W (2014) The Arabidopsis thaliana cysteine-rich receptor-like kinases CRK6 and CRK7 protect against apoplastic oxidative stress. Biochem Biophy Res Commun 445:457–462
Osakabe Y, Yamaguchi-Shinozaki K, Shinozaki K et al (2013) Sensing the environment: key roles of membrane-localized kinases in plant perception and response to abiotic stress. J Exp Bot 64:445–458
Perez-Garcia A, Romero D, Fernandez-Ortuno D, Lopez-Ruiz F, De Vicente A, Tores JA (2009) The powdery mildew fungus Podosphaera fusca (synonym Podosphaera xanthii), a constant threat to cucurbits. Mol Plant Pathol 10:153–160
Qi HH, Huang J, Zheng Q, Huang YQ, Shao RX, Zhu LY, Zhang ZX, Qiu FZ, Zhou GC, Zheng YL, Yue B (2013) Identification of combining ability loci for five yield-related traits in maize using a set of testcrosses with introgression lines. Theor Appl Genet 126:369–377
Rayapiram C, Michael KJ, Maiser F, Shanir JV, Hornshoj H, Rung JH, Gregersen PL, Schweizer P, Collingeb D, Lyngkjaer MF (2012) Regulation of basal resistance by a powdery mildew-induced cysteine-rich receptor-like protein kinase in barley. Mol Plant Pathol 13:135–147
Ren Y, Zhang Z, Staub J, Cheng Z, Li X, Lu J, Miao H, Kang H, Xie B, Gu X (2009) An integrated genetic and cytogenetic map of the cucumber genome. PLoS One 4:1–8
Sakata Y, Kubo N, Morishita M, Kitadani E, Sugiyama M, Hirai M (2006) QTL analysis of powdery mildew resistance in cucumber. Theor Appl Genet 112:243–250
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory, Cold Spring Harbor
Shanmugasundaram S, Williams PH, Peterson CE (1971) Inheritance of resistance to powdery mildew in cucumber. Phytopathology 61:1218–1221
Shen GJ, Zhan W, Chen HX, Xing YZ (2014) Dominance and epistasis are the main contributors to heterosis for plant height in rice. Plant Sci 215–216:11–18
Shiu SH, Bleecker AB (2003) Expansion of the receptor-like kinase/Pelle gene family and receptor-like proteins in Arabidopsis. Plant Physiol 132:530–543
Sitterly WP (1978) Powdery mildew of cucurbits. In: Spencer DM (ed) The powdery mildews. Academic Press, London, pp 359–379
Van Ooijen J, Voorrips R (2001) JoinMap 3.0, Software for calculation of genetic linkage maps. Plant Research International, Wageningen, The Netherlands
Wang SC, Basten CJ, Zeng ZB (2012) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm. Accessed 01 Aug 2012
Wang YH, VandenLangenberg K, Wehner TC, Weng Y (2014) QTLs for downy mildew resistance and their association with LRR-RLK resistance gene analogs in cucumber. In: Proceedings of the cucurbitaceae, Bay Harbor, MI, pp 17–20
Wei XY, Wang B, Peng Q, Wei F, Mao KJ, Zhang XG, Sun P, Liu ZH, Tang JH (2015) Heterotic loci for various morphological traits of maize detected using a single segment substitution lines test-cross population. Mol Breed 35:94
Wrzaczek M, Brosché M, Salojärvi J, Kangasjärvi S, Idänheimo N et al (2010) Transcriptional regulation of the CRK/DUF26 group of receptor-like protein kinases by ozone and plant hormones in Arabidopsis. BMC Plant Biol 10:95
Zhang SP, Liu MM, Miao H, Zhang SQ, Yang YH, Xie BY, Gu XF (2011) QTL mapping of resistance genes to powdery mildew in cucumber. Sci Agric Sin 44:3584–3593
Zitter TA, Hopkins DL, Thomas CE (1996) Compendium of cucurbits diseases. APS Press, Saint Paul
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 31171978), the National Key Basic Research Program of China (973 Program) (No. 2012CB113900), National Science & Technology Project for Rural Area (No. 2012AA100103), International Foundation for Science (No. C/5150-1) and Jiangsu Science & Technology Project (BE2012326).
Author contributions statement
X.C. and Y.W. conceived the experiment. X.X. and T.Y. performed the research. R.X., Y.S. and X.L. collected data. X.X., Y.W., Q.X. and X.Q. analyzed the data and wrote the manuscript. All authors reviewed and approved this submission.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Communicated by M. J. Havey.
Xuewen Xu and Ting Yu have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.

Fig. S1
Comparison of expression level of six predicted genes in the Pm1.1 candidate gene region (Csa1M064720.1, Csa1M064730.1, Csa1M064740.1, Csa1M064750.1, Csa1M064760.1 and Csa1M064770.1). Data are the means of three replicates (±SD). The cucumber β-actin gene was used as an internal control. Each value denotes the mean relative level of expression of three replicates. Means with the same lowercase letter do not significantly differ by the least significant difference (LSD) test at p ≤ 0.05 with a completely randomized design. (JPEG 2668 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Xu, X., Yu, T., Xu, R. et al. Fine mapping of a dominantly inherited powdery mildew resistance major-effect QTL, Pm1.1, in cucumber identifies a 41.1 kb region containing two tandemly arrayed cysteine-rich receptor-like protein kinase genes. Theor Appl Genet 129, 507–516 (2016). https://doi.org/10.1007/s00122-015-2644-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-015-2644-4