
Abstract
Key message
A major QTL conditioning high degree of femaleness in cucumber was identified by marker analysis and next generation sequencing.
Abstract
Cucumber (Cucumis sativus L.) is a model species for sex determination studies, and its yield is associated with the degree of femaleness. Subgynoecy represents a sex form with a high degree of femaleness for which the genetic basis remains elusive. In this study, genetic analysis in the F2 and BC1 populations developed from a cross between subgynoecious S-2-98 and monoecious M95 suggested a quantitative nature of subgynoecy. Application of simple sequence repeat markers between subgynoecious and monoecious bulks constructed from BC1 plants identified three QTLs: sg3.1, sg6.1, and sg6.2. The major QTL sg3.1 contributed to 54.6 % of the phenotypic variation, and its presence was confirmed by genome-wide comparison of SNP profiles between parental lines and a subgynoecious bulk constructed from BC6 plants. Using PCR-based markers developed from the SNP profile, sg3.1 was further delimited to a genomic region of 799 kb. The genetic basis of subgynoecy revealed here shall shed light on the development of elite cultivars with high yield potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cucumber (Cucumis sativus L.) has been a model system to study the biological processes of sex determination (Malepszy et al. 1991), which is of tremendous significance in plant breeding and evolutionary studies. Cucumber can be divided into seven sex types based on the distribution of male, female and bisexual flowers along the main stem. Of these types, the monoecious and gynoecious types are most frequently used in cucumber production. Monoecious cucumber produces unisexual male flowers at the basal part of the stem, followed by both male and female flowers at later growth stages. In contrast, gynoecious cucumber only bears female flowers. The process of sex determination is regulated by the combination of genes, hormones as well as environmental conditions, together resulting in multiple sexual types (Kubicki 1969, 1974; Shifriss 1961; Trebitsh et al. 1987). It has been well established that ethylene is the major regulator of sex determination of cucurbit species (Rudich et al. 1972; Byers et al. 1972; Papadopoulou et al. 2005; Salman-Minkov et al. 2008; Yin and Quinn 1995). Application of ethylene or its precursor ACC promotes the formation of female flowers in monoecious cucumber (Yamasaki et al. 2003), while interfering with ethylene synthesis or signalling using chemical inhibitors induces male flowers in gynoecious cucumber (Takahashi and Jaffe 1983). High endogenous ethylene concentrations as well as elevated expression of the ethylene biosynthesis gene 1-aminocyclopropane-1-carboxylic acid synthase 2 (CsACS2) are in correlation with the formation of female flower (Saito et al. 2007). In addition, auxin also enhances the formation of female flowers, possibly by induction of ethylene production, whereas gibberellic acid suppresses female flower formation (Trebitsh et al. 1987; Yamasaki et al. 2003). It has been reported that sex determination of cucumber is controlled by three major genes: F/f, M/m, and A/a. The monoecious allele (m) confers the formation of bisexual flowers, while the dominant M allele suppresses stamina development and thus results in female flowers (Kubicki 1969; Li et al. 2009). The Female (F) locus controls the degree of femaleness, and plants that are homozygous for the F allele exhibit gynoecy (Shiber et al. 2008; Trebitsh et al. 1997). The androecious locus (a) promotes maleness, and plants which are homozygous for both the a and f allele display androecy (Kubicki 1969; Perl-Treves 1999). Genetic studies revealed that the M gene encodes CsACS2 (Li et al. 2008, 2009) and the F locus is genetically associated with another ACS gene (CsACS1G) (Trebitsh et al. 1997; Mibus and Tatlioglu 2004; Knopf and Trebitsh 2006; Shiber et al. 2008). A genome-wide structural variation study of cucumber reveals that a 30.2-kb duplication involving four genes gave rise to the F locus (Zhang et al. 2015). Furthermore, the sex expression of cucumber is modulated by several modifier genes (Galun 1962; Kubicki 1974; Pitrat 1994).
Gynoecy and monoecy are frequently exploited in cucumber breeding. Gynoecious cucumber markedly boosts yield compared to monoecious cucumber; however, optimal growth conditions are required to realize the full fruit production potential of gynoecious cucumber. This presents a challenge for cucumber breeders in China, as it is difficult to maintain such optimal growth conditions. As a result, under the regular growth conditions in China not all female flowers develop into a marketable cucumber, especially when long fruits are produced. Therefore, cultivars with a percentage of female flowers in between that of monoecious and gynoecious cucumber would be more suitable for cultivation. Subgynoecy can be regarded as a special type of monoecy, though with a higher percentage of female flowers. Furthermore, subgynoecious cucumber exclusively develops female flowers at later growth stages. Chen et al. (2011) identified two subgynoecious loci, one acting recessively and another one dominantly, in inbred lines 97-17 and S-2-98, respectively. The inheritance analyses indicated that the subgynoecious trait of S-2-98 is independent of the F and M genes. However, the genetic basis of the subgynoecious trait in S-2-98 remains unclear.
In this study, we carried out a thorough investigation of the genetic architecture of the subgynoecious trait in S-2-98. Quantitative trait loci (QTLs) were identified by simple sequence repeat (SSR) and sequencing-based analyses. A major QTL was further narrowed down using PCR-based markers and breeding materials harbouring this QTL were created.
Materials and methods
Plant materials and phenotypic evaluation
A line with high degree of femaleness emerged from the monoecious cultivar “DongHuzao”. After nine generations of selfing, it was developed into a stable subgynoecious line, referred to as S-2-98. S-2-98 initially displays a phase in which only male flowers are formed, followed by a phase in which both male and female flowers are formed and terminates into a phase in which sequential female flowers are formed. S-2-98 was used as the maternal parent in the cross with M95. M95 is a monoecious inbred line developed from monoecious variety “JingChun 4”. F1 plants were self-pollinated to produce the F2 population consisting of 192 plants. To create subgynoecious breeding materials with M95 background, a single plant with the highest degree of femaleness was backcrossed to M95 in each backcross generation. The BC1 population consisted of 188 plants. Crossing was carried out in the experimental fields in Hunan, China.
Phenotypic evaluation was carried out when the majority of plants in the population developed at least 25 nodes on the main stem. Flower sex type of each node of each plant was recorded. In case flowers were abscised in basal nodes, the flower sex type was determined based on the difference in size between that of the male and female stalks. The percentage of female flowers (R female) of each individual was calculated to determine the degree of femaleness using the formula R female = N female/(N female + N male) × 100 %, where N female stands for the number of nodes bearing female flowers and N male stands for the number of nodes bearing male flowers. Subgynoecious cucumber displays continuous female flower nodes at the later growth stage, a feature differing from monoecious cucumbers. Therefore, the number of continuous female flower nodes at later growth stage, designated N cf, was also taken into account when selecting subgynoecious individuals to carry out backcrossing or to construct bulked DNA pools.
QTL analysis with SSR markers
Two DNA pools, a monoecious pool (M pool) and a subgynoecious pool (S pool), were constructed using BC1 plants. Genomic DNA was isolated from ten plants with R female ranging from 85 to 95 % for the S pool and ten plants with R female ranging from 1 to 25 % for the M pool. A total of 2112 SSR primers, developed from the whole genome sequence of the cucumber inbred line 9930 (Huang et al. 2009), were screened for polymorphisms in the parental lines and subsequently in the S pool and M pool. Polymorphic markers between the two pools (Supplementary material S1) were subjected to linkage analysis using JoinMap 3.0 software with a Log of odds (LOD) of 3.0 and the Kosambi mapping function (Lander and Botstein 1989). The QTL analysis was performed with Interval Mapping and Multiple-QTL model Mapping (MQM) using MapQTL 4.0 software (Jansen 1993; Jansen and Stam 1994; Van Ooijen 2006).
QTL analysis with next generation sequencing data
In total, 108 BC6 and 87 BC7 plants were grown in the autumn of 2012 in the greenhouses in Beijing, China. After phenotypic evaluation, seven plants (BC6-1, BC6-2, BC6-3, BC6-4, BC6-5, BC6-6 and BC6-7) with high R female and N cf were selected from the BC6 population (Supplementary material S2). Genomic DNA was isolated from selected individuals using the CTAB method. Equal amounts of genomic DNA of each individual were mixed to construct the subgynoecious BC6 pool (SBC6 pool). Parental lines S-2-98 and M95, as well as SBC6 pool were subjected to whole genome sequencing using an Illumina GAIIx Sequencer1. Reads with a length of 100 bp were generated from paired-end sequencing.
All reads were aligned to the 9930 reference genome with the BWA software (Li and Durbin 2009). SNP calling was conducted using SAM tools software (Li and Durbin 2009). Because both parents are homozygous lines, only homozygous SNPs in both parents were retained. The Illumina phred-like quality score and mapping score were set to larger than 30. The read depth was set between 3 and 40. The SNP-index was calculated for SBC6 pool by QTL-seq and MutMap (Abe et al. 2012; Takagi et al. 2013). The SNP-index was determined by the number of reads showing the same genotype as the parental line S-2-98 divided by the total number of reads mapped to this SNP site. An average SNP-index of SNPs located at a given genomic interval was computed using a sliding window analysis of 1 Mb window size and 10 kb increment. The SNP-index graph for SBC6 pool was plotted.
The dCAPS and InDel markers were developed based on the SNP profile. Primers for dCAPS markers were designed using web-based designer software (http://helix.wustl.edu/dcaps/dcaps.html). Primers for InDel markers were designed using Primer 5 program.
Development of near-isogenic subgynoecious lines and fine mapping of the major QTL
Genotypic analysis with dCAPS and InDel markers was undertaken for BC7 plants. Three plants (BC7-7, BC7-16 and BC7-17) harbouring only the major QTL on chromosome 3 and showing high R female and N cf (Supplementary material S2) were selected from BC7 populations and backcrossed with male parent M95 to create the BC8 population. The phenotypic analysis of the three BC8 populations was carried out in the winter of 2012 in Hainan, China. Five BC8 plants (NS7-1, NS7-20, NS7-24, NS16-13 and NS17-23) showing high R female and N cf (Supplementary material S2) were self-crossed, producing a total of 496 BC8S1 plants, which were subsequently grown in the spring of 2013 in the experimental fields in Beijing, China. Based on genotypic analysis, the 496 BC8S1 plants were divided into four groups: homozygous dominant plants, homozygous recessive plants, heterozygous plants and recombinants. The difference in R female among these four groups was used to verify the inheritance pattern of the subgynoecious trait. Genotypic and phenotypic investigation was further carried out for recombinants using PCR-based markers to narrow down the genomic region underlying the major QTL. All the primers for dCAPs and InDel markers used for genotyping are presented in Supplementary material S4.
Results
Inheritance of the subgynoecious trait in S-2-98
To determine the inheritance pattern of the subgynoecious trait of S-2-98, S-2-98 was crossed to the monoecious parent M95. In the resulting F1, F2 and BC1 populations, the phenotype was scored on the basis of the R female, which corresponds to the percentage of female flowers on the main stem. The R female reached 87 % for maternal parent S-2-98 and 20 % for M95. All F1 individuals produced sequential female flowers at the later growth stage, similar to maternal parent S-2-98. The average R female of the F1 population was 71 %, which suggests that the subgynoecious trait of S-2-98 is (semi-) dominant to the monoecious trait of M95. In the BC1 and F2 populations the R female displayed a continuous variation, ranging from 7 to 100 % in the BC1 population and from 3 to 100 % in the F2 population (Fig. 1), indicative of the polygenic nature of the subgynoecious trait.

Distribution of R female of BC1 and F2 populations. Arrows point the R female of S-2-98 and M95
Identification of three QTLs through SSR analysis
In order to identify loci underlying the subgynoecious trait of S-2-98, we conducted SSR mapping on the BC1 population. Among 2112 pairs of SSR markers that were screened, 440 pairs were polymorphic between both parents. Of these 440, 39 were polymorphic between the M pool and the S pool (Supplementary material S1). These 39 markers were subsequently applied to the BC1 population to map QTLs underlying the subgynoecious trait. Three QTLs were identified; sg3.1 located on chromosome 3, which is tightly linked to SSR13466, and sg6.1 and sg6.2 both located on chromosome 6, which are tightly linked to SSR01308 and SSR02123, respectively (Fig. 2; Table 1). These three QTLs together explained 62.9 % of the phenotypic variation, of which 54.6 % was explained by sg3.1 alone (Table 1). These results indicated that the subgynoecious trait of S-2-98 was controlled by a major QTL (sg3.1) and two minor QTLs.

Identification of three QTLs (sg3.1, g6.1, and sg6.2) by SSR analysis in BC1 population
QTL-seq identified four QTLs, including a new QTL
To refine the regions harbouring the QTLs identified through SSR mapping, QTL-seq was conducted to compare the SNP profiles of both parental lines and the SBC6 pool on a genome-wide scale. The SBC6 pool consisted of seven plants (from BC6-1 to BC6-7) selected from the BC6 population on the basis of an R female ranging from 50 to 81 % and an N cf ranging from 10 to 17 (Supplementary material S2). The SNP-index plot of the SBC6 pool revealed four regions harbouring QTLs on chromosomes 3, 4 and 6 (Fig. 3). One region corresponding to the major QTL sg3.1 was located at the interval between 8.4 and 24.7 Mb on chromosome 3. Two other regions corresponding to the two minor QTLs sg6.1 and sg6.2 were located at the intervals of 9.2–11.3 Mb and 24.2–24.8 Mb on chromosome 6, respectively. The fourth region, referred to sg4.1, was situated at the interval between 2.9 and 6.1 Mb on chromosome 4. QTL sg4.1 was not identified through SSR mapping probably owing to the shortage of polymorphic SSR markers on chromosome 4 between the parental lines (Supplementary material S3). SSR markers were populated at a density of 13 per Mb on chromosome 4, whereas across other chromosomes the density ranged from 16 to 34 per Mb. Compared with chromosomes 3, 4 and 6, the SNP-index of chromosomes 1, 2, 5 and 7 nearly approached 0, indicating that these chromosomes were rich in alleles from the paternal parent M95.

Identification of four QTLs by QTL-seq in BC6 population. X-axis represents the position of seven chromosomes and Y-axis represents the SNP-index. The grey line represents the lowest SNP-index value within statistical confidence intervals under the null hypothesis of no QTL (P < 0.05)
Refinement of the major QTL sg3.1 to a 799-kb interval
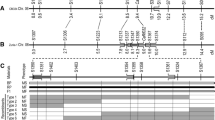
To speed up the application of the subgynoecious trait in breeding as well as fine mapping of the major QTL, we isolated plants harbouring only the major QTL sg3.1 using marker-assisted selection. In the BC7 population, three individuals (BC7-7, BC7-16 and BC7-17) harbouring the major QTL sg3.1 (Fig. 4a) were used to create advanced lines by one round of backcrossing to M95, followed by one round of selfing. The resulting BC8S1 families (NS7-1, NS7-20, NS7-24, NS16-13 and NS17-23) consisted of 496 individuals (Fig. 4b). These individuals were grouped into four types according to genotype: homozygous dominant, homozygous recessive, hetereozygous and recombinants. The average R female was 68.9 % for homozygous dominant plants and 46.9 % for heterozygous plants, both of which were significantly higher than the average R female (27.6 %) for homozygous recessive plants (Fig. 4b). This evidence further supported that the subgynoecious trait of S-2-98 was (semi-) dominant and mainly controlled by sg3.1. Selfing was performed for homozygous dominant plants to create near isogenic lines aiming at high yield.

Refinement of the major QTL sg3.1. a Three BC7 individuals harbouring the major QTL sg3.1. b The average R female of dominant homozygous, heterozygous and recessive homozygous plants of five BC8S1 families for recombinant screening. Asterisks represent significant difference between average R female of plants with different genotypes (P < 0.05). c The major QTL sg3.1 was narrowed down to a 799-kb interval between dCAPs markers C3D2 and C3D4. R female higher than 30 % was a criterion to assess whether the region harboured sg3.1. The grey region carries heterozygous alleles. The white region carries recessive homozygous alleles as M95. The joints of grey and white regions mean that a crossover occurs somewhere between the neighbouring markers
To narrow down the major QTL, recombinants from BC7 and BC8S1 populations were analysed using dCAPs markers. BC7-17 and BC7-7 had high R female (75.0 and 48.2 %, respectively) indicating the presence of the S-2-98 allele, which pinpointed sg3.1 to the heterozygous genomic region on the right side of marker C3D2. Recombinant NS7-20-64 showed low R female (20.0 %) and was homozygous recessive at the left side of marker C3D1. This, together with the data from BC7-17 and BC7-7, delimits sg3.1 to the interval between C3D2 and C3D1. Recombinants NS7-24-82 and NS7-1-71 showed a moderately high R female (57.7 % and 37.5 %, respectively) indicating the presence of the S-2-98 allele, which pinpointed sg3.1 to the left side of marker C3D5. This delimited interval is supported by the data from NS16-13-58, which showed low R female (20.7 %) and was homozygous recessive at the left side of marker C3D5. NS7-1-46 depicted a moderately high R female (47.6 %) indicating the presence of the S-2-98 allele, which pinpointed sg3.1 to the left side of marker C3D4. This delimited region was supported by the data from NS16-13-29 and NS17-23-57, which showed low R female (28.6 and 30.4 %, respectively) and were homozygous recessive at the left side of C3D4. Altogether, combination of the genotypic and phenotypic data from these recombinants delimited sg3.1 to a 799-kb genomic region between markers C3D2 and C3D4. (Fig. 4c; Supplementary material S4).
Discussion
The resource-limited cultivation practice in China impedes the expansion of gynoecious cucumbers. Subgynoecious cucumbers bearing a higher percentage of female flowers can enhance fruit production at low cost. Therefore, it is important to investigate the genetic basis of subgynoecious traits and create desirable breeding materials. In this study, we conducted genetic analysis of the subgynoecious trait of S-2-98 and identified four QTLs underlying this trait. Furthermore, we applied marker-assisted selection and created subgynoecious near isogenic lines of the major QTL sg3.1.
Four subgynoecious QTLs were identified in this study, and the major QTL sg3.1 explaining 54.6 % of the phenotypic variation was further delimited to a 799-kb genomic region on chromosome 3. Fazio et al. (2003) reported three QTLs (sex1.1, sex1.2 and sex6.1) associated with the number of female nodes on the main stem. The peak marker for sex6.1 was positioned at 63.8 cM on chromosome 6, adjacent to the marker SSR02123 tightly linked to sg6.2 identified in this study. Yuan et al. (2008) identified three QTLs for female flower ratio, two located on chromosome 2 and one located on chromosome 6 distant from either sex6.1 or sg6.2. The major QTL on chromosome 2 contributed to over 40 % of the phenotypic variation, and has not been fine-mapped. Of note, the major QTL on chromosome 2 was not found in Fazio et al. (2003) or our mapping results, likely because cucumber materials were derived from different backgrounds.
A total of 110 putative genes were predicted in the 799-kb region underlying QTL sg3.1. Genes Csa3g180260 and Csa3g180265 both contain one copy of AP2/ERF domain found in members of the AP2/ERF family. In Arabidopsis, the AP2/ERF gene family is divided into three groups (AP2, ERF and RAV) based on sequence similarity and the number of AP2/ERF domains (Sakuma 2002; Nakano et al. 2006). The AP2 class, containing two copies of the AP2 domain, is required for regulating temporal and spatial expression of flower homeotic genes (Drews et al. 1991; Jofuku et al. 1994). Genes belonging to the other two groups contain only one copy of the AP2 domain. They function in biotic or abiotic stress signalling by regulating the expression of downstream ethylene responsive genes by binding to the GCC box found in ethylene response elements (Lee et al. 2015; Okamuro et al. 1997). Ethylene is a major regulator of sex determination in cucumber. Csa3g180260 and Csa3g180265 might be involved in regulating sex determination of S-2-98 via interacting with ethylene-responsive genes. Another gene, Csa3g179160 is a potential homolog of an Arabidopsis SOC1-like gene, AGL42. SOC1 is a classic MIKC-type MADS box gene, which plays an important role in the regulation of flower induction and development by integrating multiple flowering pathways (Lee and Lee 2010). AGL42 is involved in floral transition and acts through a gibberellin-dependent pathway (Dorca-Fornell et al. 2011). To examine whether these genes were associated with the subgynoecious trait, we performed an association study using 115 cucumber accessions (containing 9 gynoecious and 106 monoecious accessions) focusing on the genomic interval of sg3.1. However, no significant associations were found in this region (data not shown). This might be explained by the absence of subgynoecious accessions with the same feature as S-2-98 among the 115 accessions used in this analysis. To elucidate the genetic basis of the subgynoecious trait, further effort is needed to map the major QTL sg3.1 at high resolution and functionally characterize candidate genes.
Author contribution statement
The experiment was conceived by SH. FB and QS performed molecular experiments. HC performed the phenotypic assay. QZ and ZZ conducted the bioinformatics analysis. DG, FB and SH wrote the paper.
References
Abe A, Kosugi S, Yoshida K, Natsume S, Takagi H, Kanzaki H, Matsumura H, Yoshida K, Mitsuoka C, Tamiru M (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat Biotech 30:174–178
Byers R, Baker L, Sell H, Herner R, Dilley D (1972) Ethylene: a natural regulator of sex expression of Cucumis melo L. Proc Natl Acad Sci USA 69:717–720
Chen H, Tian Y, Lu X, Liu X (2011) The inheritance of two novel subgynoecious genes in cucumber (Cucumis sativus L.). Sci Hortic 127:464–467
Dorca Fornell C, Gregis V, Grandi V, Coupland G, Colombo L, Kater MM (2011) The Arabidopsis SOC1-like genes AGL42, AGL71 and AGL72 promote flowering in the shoot apical and axillary meristems. Plant J 67:1006–1017
Drews GN, Bowman JL, Meyerowitz EM (1991) Negative regulation of the Arabidopsis homeotic gene AGAMOUS by the APETALA2 product. Cell 65:991–1002
Fazio G, Staub J, Stevens M (2003) Genetic mapping and QTL analysis of horticultural traits in cucumber (Cucumis sativus L.) using recombinant inbred lines. Theor Appl Genet 107:864–874
Galun E (1962) Study of the inheritance of sex expression in the cucumber. The interaction of major genes with modifying genetic and non-genetic factors. Genetica 32:134–163
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281
Jansen RC (1993) Interval mapping of multiple quantitative trait loci. Genetics 135:205–211
Jansen RC, Stam P (1994) High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136:1447–1455
Jofuku KD, Den Boer B, Van Montagu M, Okamuro JK (1994) Control of Arabidopsis flower and seed development by the homeotic gene APETALA2. Plant Cell 6:1211–1225
Knopf RR, Trebitsh T (2006) The female-specific Cs-ACS1G gene of cucumber. A case of gene duplication and recombination between the non-sex-specific 1-aminocyclopropane-1-carboxylate synthase gene and a branched-chain amino acid transaminase gene. Plant Cell Physiol 47:1217–1228
Kubicki B (1969) Investigations on sex determination in cucumber (Cucumis sativus L.). Genet Pol. 10:5–143
Kubicki B (1974) New sex types in cucumber and their uses in breeding work. In: Able ST (ed), XIX International Horticultural Congress 1974 September 11–18 Warszawa, pp 475–485
Lander ES, Botstein D (1989) Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121:185–199
Lee J, Lee I (2010) Regulation and function of SOC1, a flowering pathway integrator. J Exp Bot 61:2247–2254
Lee S-Y, Hwang EY, Seok H-Y, Tarte VN, Jeong MS, Jang SB, Moon Y-H (2015) Arabidopsis AtERF71/HRE2 functions as transcriptional activator via cis-acting GCC box or DRE/CRT element and is involved in root development through regulation of root cell expansion. Plant Cell Rep 34:223–231
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760
Li Z, Pan J, Guan Y, Tao Q, He H, Si L, Cai R (2008) Development and fine mapping of three co-dominant SCAR markers linked to the M/m gene in the cucumber plant (Cucumis sativus L.). Theor Appl Genet 117:1253–1260
Li Z, Huang S, Liu S, Pan J, Zhang Z, Tao Q, Shi Q, Jia Z, Zhang W, Chen H (2009) Molecular isolation of the M gene suggests that a conserved-residue conversion induces the formation of bisexual flowers in cucumber plants. Genetics 182:1381–1385
Malepszy S, Niemirowicz-Szczytt K (1991) Sex determination in cucumber (Cucumis sativus L.) as a model system for molecular biology. Plant Sci 80:39–47
Mibus H, Tatlioglu T (2004) Molecular characterization and isolation of the F/f gene for femaleness in cucumber (Cucumis sativus L.). Theor Appl Genet 109:1669–1676
Nakano T, Suzuki K, Fujimura T, Shinshi H (2006) Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol 140:411–432
Okamuro JK, Caster B, Villarroel R, Van Montagu M, Jofuku KD (1997) The AP2 domain of APETALA2 defines a large new family of DNA binding proteins in Arabidopsis. Proc Natl Acad Sci USA 94:7076–7081
Papadopoulou E, Little HA, Hammar SA, Grumet R (2005) Effect of modified endogenous ethylene production on sex expression, bisexual flower development and fruit production in melon (Cucumis melo L.). Sex Plant Reprod 18:131–142
Perl-Treves R (1999) Male to female conversion along the cucumber shoot: approaches to studying sex genes and floral development in Cucumis sativus. Bios Scientific Publishers, Oxford, pp 189–215
Pitrat M (1994) Gene list for Cucumis melo L. Cucurbit Genetic Coop Report 17:135–147
Rudich J, Halevy A, Kedar N (1972) Ethylene evolution from cucumber plants as related to sex expression. Plant Physiol 49:998–999
Saito S, Fujii N, Miyazawa Y, Yamasaki S, Matsuura S, Mizusawa H, Fujita Y, Takahashi H (2007) Correlation between development of female flower buds and expression of the CS-ACS2 gene in cucumber plants. J Exp Bot 58:2897–2907
Sakuma Y, Liu Q, Dubouzet JG, Abe H, Shinozaki K, Yamaguchi-Shinozaki K (2002) DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration-and cold-inducible gene expression. Biochem Bioph Res Co 290:998–1009
Salman-Minkov A, Levi A, Wolf S, Trebitsh T (2008) ACC synthase genes are polymorphic in watermelon (Citrullus spp.) and differentially expressed in flowers and in response to auxin and gibberellin. Plant Cell Physiol 49:740–750
Shiber A, Gaur R, Rimon-Knopf R, Zelcer A, Trebitsh T, Pitrat M (2008) The origin and mode of function of the female locus in cucumber. Cucurbitaceae 2008:263–270
Shifriss O (1961) Sex control in cucumbers. J Hered 52:5–12
Takagi H, Abe A, Yoshida K, Kosugi S, Natsume S, Mitsuoka C, Uemura A, Utsushi H, Tamiru M, Takuno S (2013) QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J 74:174–183
Takahashi H, Jaffe M (1983) Further studies of auxin and ACC induced feminization in the cucumber plant using ethylene inhibitors. Phyton 44:81–86
Trebitsh T, Rudich J, Riov J (1987) Auxin, biosynthesis of ethylene and sex expression in cucumber (Cucumis sativus). Plant Growth Regul 5:105–113
Trebitsh T, Staub JE, O’Neill SD (1997) Identification of a 1-aminocyclopropane-1-carboxylic acid synthase gene linked to the female (F) locus that enhances female sex expression in cucumber. Plant Physiol 113:987–995
Van Ooijen J (2006) JoinMap 4. Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen
Yamasaki S, Fujii N, Takahashi H (2003) Characterization of ethylene effects on sex determination in cucumber plants. Sex Plant Reprod 16:103–111
Yin T, Quinn JA (1995) Tests of a mechanistic model of one hormone regulating both sexes in Cucumis sativus (Cucurbitaceae). Am J Bot 82:1537–1546
Yuan X, Pan J, Cai R, Guan Y, Liu L, Zhang W, Li Z, He H, Zhang C, Si L (2008) Genetic mapping and QTL analysis of fruit and flower related traits in cucumber (Cucumis sativus L.) using recombinant inbred lines. Euphytica 164:473–491
Zhang Z, Mao L, Chen H, Bu F, Li G, Sun J, Li S, Sun H, Jiao C, Blakely R (2015) Genome-wide mapping of structural variations reveals a copy number variant that determines reproductive morphology in cucumber. Plant Cell 114:135848
Acknowledgments
We thank Dr. Yiqun Weng and Arjan van Zeijl for their help with revising the manuscript and Tao Lin for his help with SNP profiling. This study was funded by the National Natural Science Foundation of China (NSFC: 31225025), the National Program on Key Basic Research Projects in China (The 973 Program: 2012CB113900), the National High Tech Research Development Program in China (The 863 Program: 2010AA10A108, 2012AA100101), and the Chinese Ministry of Finance (1251610601001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
The authors declare that all experiments described in this manuscript comply with the current laws in China.
Additional information
Communicated by H. J. van Eck.
F. Bu, H. Chen and Q. Shi contributed equally.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bu, F., Chen, H., Shi, Q. et al. A major quantitative trait locus conferring subgynoecy in cucumber. Theor Appl Genet 129, 97–104 (2016). https://doi.org/10.1007/s00122-015-2612-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-015-2612-z