
Abstract
Reperfusion after acute myocardial infarction further exaggerates cardiac injury and adverse remodeling. Irrespective of cardiac cell types, loss of specifically the α isoform of the protein kinase GSK-3 is protective in chronic cardiac diseases. However, the role of GSK-3α in clinically relevant ischemia/reperfusion (I/R)-induced cardiac injury is unknown. Here, we challenged cardiomyocyte-specific conditional GSK-3α knockout (cKO) and littermate control mice with I/R injury and investigated the underlying molecular mechanism using an in vitro GSK-3α gain-of-function model in AC16 cardiomyocytes post-hypoxia/reoxygenation (H/R). Analysis revealed a significantly lower percentage of infarct area in the cKO vs. control hearts post-I/R. Consistent with in vivo findings, GSK-3α overexpression promoted AC16 cardiomyocyte death post-H/R which was accompanied by an induction of reactive oxygen species (ROS) generation. Consistently, GSK-3α gain-of-function caused mitochondrial dysfunction by significantly suppressing mitochondrial membrane potential. Transcriptomic analysis of GSK-3α overexpressing cardiomyocytes challenged with hypoxia or H/R revealed that NOD-like receptor (NLR), TNF, NF-κB, IL-17, and mitogen-activated protein kinase (MAPK) signaling pathways were among the most upregulated pathways. Glutathione and fatty acid metabolism were among the top downregulated pathways post-H/R. Together, these observations suggest that loss of cardiomyocyte-GSK-3α attenuates cardiac injury post-I/R potentially through limiting the myocardial inflammation, mitochondrial dysfunction, and metabolic derangement. Therefore, selective inhibition of GSK-3α may provide beneficial effects in I/R-induced cardiac injury and remodeling.
Key messages
-
GSK-3α promotes cardiac injury post-ischemia/reperfusion (I/R).
-
GSK-3α regulates inflammatory and metabolic pathways post-hypoxia/reoxygenation (H/R).
-
GSK-3α overexpression upregulates NOD-like receptor (NLR), TNF, NF-kB, IL-17, and MAPK signaling pathways in cardiomyocytes post-H/R.
-
GSK-3α downregulates glutathione and fatty acid metabolic pathways in cardiomyocytes post-H/R.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute myocardial infarction (MI) is a common cause of heart failure which is characterized by loss of cardiomyocytes, infarct size expansion, left ventricle wall thinning, and cardiac remodeling [1]. Percutaneous coronary intervention (PCI) can effectively re-perfuse the infarcted cardiac tissue; however, reperfusion further exacerbates the cardiac tissue injury due to oxidative stress and cell death. No effective treatment is currently available to limit PCI-induced cardiac injury, progressive cardiac remodeling, and heart failure.
Glycogen synthase kinase-3 (GSK-3), a Ser/Thr kinase downstream of Akt, has been reported to play key roles in a variety of pathological conditions including cancers, infertility, Alzheimer’s disease, diabetes, obesity, cardiac development as well as cardiovascular diseases like MI and pressure-overload (PO)-induced cardiac remodeling and heart failure [2,3,4,5,6,7]. The two isoforms of GSK-3, α and β, are largely similar (~97%) in their catalytic domain but display overlapping as well as unique functions [8,9,10]. GSK-3 was initially identified as one of the key regulators of glycogen metabolism by modulating the rate-limiting enzyme glycogen synthase [11]. Later, once cloned, GSK-3 was recognized as a regulator of a variety of cell signaling pathways and cellular processes including proliferation, differentiation, metabolism, and cell death [12,13,14]. Apoptosis and necrosis are the most common types of cell death reported in ischemic cardiac diseases. Ischemia/reperfusion (I/R) promotes cell death largely through apoptosis and necrosis. Apoptosis is a well-explored, programmed cell death mechanism that is regulated by signaling pathways including caspase signaling and a variety of mitochondrial components. In contrast, necrosis is characterized by cell swelling and a subsequent rupture followed by releasing the intracellular contents [15, 16].
Accumulating evidence suggests that GSK-3α plays a crucial role in heart diseases [3, 4]. In vivo studies employing loss-of-function and gain-of-function models have shown that GSK-3α plays a unique role in cardiac pathophysiology [9, 10, 17,18,19]. These in vivo studies, employing chronic ischemia and pressure overload (PO) models, have demonstrated that GSK-3α limits cardiomyocyte proliferation and promotes cardiac cell death and fibrosis, with consequent adverse effects on remodeling. Most importantly, we recently reported a key role of GSK-3α in mitochondrial dysfunction and cardiomyocyte apoptosis. Loss of GSK-3α in cardiomyocytes limits mitochondrial permeability transition pore (mPTP) opening, cardiomyocyte death, fibrosis, adverse cardiac remodeling, and dysfunction under chronic PO [20].
Cardiomyocyte-specific conditional targeting of GSK-3β is protective against ischemic cardiac disease [21]; however, this has a detrimental cardiac effect in a diet-induced obesity model [5]. The loss of GSK-3β in cardiac fibroblasts promotes ischemia-induced fibrotic remodeling and heart failure [22]. In stark contrast to GSK-3β, a recent study has reported that loss of GSK-3α in cardiac fibroblasts limited cardiac fibrosis post-MI [19]. Surprisingly, cardiomyocyte-specific conditional double deletion of GSK-3α and GSK-3β in the adult heart leads to cardiac fibrosis and fatal dilated cardiomyopathy (DCM) even without cardiac stressors [23, 24]. These studies strongly suggest that targeting both GSK-3 isoforms chronically is not biologically useful, but irrespective of cardiac cell type, loss of GSK-3α is protective.
Available GSK-3 inhibitors target both isoforms α and β non-selectively and have been validated in phase II clinical trials for neurological disorders (ClinicalTrials.gov:NCT01096082, NCT01543724) and used in the clinic. Studies from double knockout raise concerns that chronic use of non-selective inhibitors could lead to fibrotic cardiac remodeling, DCM, and heart failure in patients. These studies provide a strong rationale for the selective targeting of GSK-3α to limit I/R-induced cardiac injury.
Herein, we investigated the unknown role of GSK-3α in I/R-induced cardiac injury by employing a cardiomyocyte-specific GSK-3α knockout mouse model. To study the underlying mechanisms, transcriptomic studies were performed employing the GSK-3α gain-of-function model in human AC16 cardiomyocyte cells challenged with hypoxia/reoxygenation (H/R). We identified, for the first time, that loss of GSK-3α in cardiomyocytes limits I/R-induced cardiac injury. Consistently, GSK-3α overexpression promotes cardiac cell death post-H/R through excess ROS generation and mitochondrial dysfunction. Mechanistically, GSK-3α was identified to enhance inflammatory pathways including NOD-like receptor (NLR), TNF, NF-κB, IL-17, and mitogen-activated protein kinase (MAPK) signaling pathways and metabolic derangement in cardiomyocyte post-ischemic stressor.
Methods
Cardiomyocyte-specific conditional GSK-3α knockout animal
Tamoxifen-inducible cardiomyocyte-specific GSK-3α knockout mice were generated as described previously [18]. Briefly, mice with a floxed exon 2 of GSK3A gene (GSK3Afl/fl) were crossed with alpha-myosin heavy chain (a-MHC) promoter-driven MerCreMer mouse for two generations to obtain GSK3Afl/flCre(+/−) genotype. At 12 weeks, both GSK3Afl/flCre(+/−)and littermate control GSK3Afl/fl male mice were treated with tamoxifen through feeding with a tamoxifen containing chow diet for 2 weeks, followed by two additional weeks on a regular diet to clear the tamoxifen from the circulation to minimize confounding factors. All animal procedures were carried out as per current NIH guidelines. The Institutional Animal Care and Use Committee (IACUC) of Vanderbilt University Medical Center approved all animal procedures and treatments, and animal-related experiments were performed at the Vanderbilt University Medical Center.
Cardiac ischemia/reperfusion surgery
Myocardial ischemia/reperfusion (I/R) surgery was performed as described previously [25]. Briefly, post-tamoxifen treatment and washout period, GSK3Afl/flCre(knockout) and littermate GSK3Afl/fl(control) mice were anesthetized with 2% isoflurane inhalation using an isoflurane delivery system (Viking Medical, Medford, NJ). A skin cut of ~1.2 cm was made on the left chest, and the heart smoothly popped out. The left anterior descending coronary artery (LAD) was ligated with a slipknot for 30 min. After ischemia, the slipknot was released gently to re-perfuse the myocardium. Post-24 h reperfusion, the chest was re-opened in anesthetized mice, and the LAD was re-ligated using the same suture. Evan’s blue dye was injected through the aorta, and the heart was quickly excised and frozen.
Triphenyl tetrazolium chloride staining
Triphenyl tetrazolium chloride (TTC) staining was performed using transverse heart sections as described previously [18]. The frozen whole heart was sliced into five 1-mm-thick sections and then incubated with 1% TTC (Sigma #T8877) solution for 15 min at room temperature. The imaging was done under an inverted Nikon microscope using a 4X objective. The area at risk was calculated by measuring Evan’s blue negative and TTC positive area, and the infarcted area was calculated by measuring the TTC negative area with NIS-Elements software. Myocardial infarct size is expressed as a percentage of the total LV area.
Cell culture and GSK-3α plasmid transfection and treatment
AC16 cardiomyocytes were purchased from Sigma-Aldrich (SCC109) and cultured as per vendor instructions. Briefly, AC16 cells were maintained in Dulbecco’s Modified Eagle’s Medium/F12 nutrient mixture (Sigma-Aldrich #D6434) containing 2 mM L-glutamine supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin antibiotic cocktail (Sigma-Aldrich #P4333). The culture flasks were incubated in a 37 °C CO2 humidified incubator containing 5% CO2 and 95% air. Cells were split at 80–90% confluency using 0.05% trypsin-EDTA (Sigma-Aldrich #T4299).
AC16 cells were transfected as described previously [26]. Briefly, 0.32 × 106 AC16 cells were seeded onto 6-well tissue culture plates. At ~60% confluency, cells were serum-starved with 1X Opti-MEM (Gibco #00448) for 2 h prior to transfection with Flag control or Flag-GSK-3α WT human plasmid using Fugene 6 (Promega #E2693) in a 3:1 ratio and incubated for 4 h in a CO2 incubator. After 4 h of transfection, an equal volume of complete DMEM containing 2X FBS and 2X penicillin-streptomycin cocktail was added and the cells incubated for a further 24 h in a CO2 incubator.
Hypoxia and reoxygenation
The AC16 cardiomyocytes were challenged with H/R as described previously [27]. Briefly, H/R was performed using a regulated Whitley H45 hypoxic chamber and incubator. Prior to challenging cells with hypoxia, media was replaced with serum-free media and cells were incubated for 3 h in a CO2 incubator for acclimatization. Cells were then transferred to the hypoxic chamber containing 95% N2, 5% CO2, and 1% O2 concentrations, which was maintained throughout the hypoxia period. Hypoxia incubation was done either chronically (overnight) for cell death/viability assays or acutely (6 h) for cell signaling studies, if otherwise specified. To simulate reperfusion conditions, cells were taken out from the hypoxia chamber and media were replaced with pre-warmed complete DMEM supplemented with 10% FBS and 1% antibiotic cocktail and were incubated in a CO2 incubator for reoxygenation accordingly.
Annexin V and 7-AAD labeling
To assess the level of cell death, Annexin V (Ann V) and 7-amino-actinomycin D (7-AAD) staining was performed as described previously [28]. Briefly, an equal number of AC16 cardiomyocytes were seeded on 6 well culture plates, and following transfection with GSK-3α or control plasmid, H/R was performed. AC16 cells, including floating ones, were harvested and collected through centrifugation at 600 g for 5 min. The supernatant was discarded, and cells were resuspended in a complete DMEM medium. A total of 0.4 × 106 cells were re-suspended in 100 µl of Annexin binding buffer (Biolegend #B266205) containing Ann V antibody (Biolegend 640906) and 7-AAD reagent (Biolegend 420403) and incubated for 30 min at room temperature in the dark. After 30 min, 100 µl of PBS was added to each tube and vortexed quickly. Results were acquired using BD FACS Aria III Flow Cytometer and data analyzed using FlowJo software.
Reactive oxygen species measurement
To assess the total reactive oxygen species (ROS) generation post-H/R, AC16 cells were seeded on 24 well tissue culture plates and transfected with GSK-3α plasmid followed by H/R for 6 h/2 h, respectively. 2′,7′-Dichlorofluorescin diacetate (DCFDA) (Sigma-Aldrich #D6883) was added to the culture plates at a concentration of 10 μM for 30 min. Results were obtained using the spectrophotometer (Glomax) at an excitation wavelength of 485 nm and an emission wavelength of 535 nm. For cellular ROS generation, assay cells were harvested, washed, resuspended in 100 μl PBS and stained with 10 μM 2′,7′-dichlorofluorescin diacetate (DCFDA) for 30 min. Results were acquired under BD FACS Aria III Flow Cytometer, and data was analyzed using FlowJo.
Mitochondrial membrane potential assay
Mitochondrial membrane potential was assessed using TMRE-mitochondrial membrane potential assay kit (Abcam #113852) following the manual instructions. Briefly, 2 × 105 AC16 cells/well were seeded in 6 well plates and transfected with either Flag or Flag-GSK-3α plasmid as described above. Post-H/R, a cardiomyocyte group was treated with 20 μM FCCP (a compound that eliminates the mitochondrial membrane potential) as a positive control for 10 min followed by treatment with 200 nM TMRE for 20 min. Other groups were treated with only either DMSO or TMRE. Cells were harvested by trypsinization, and TMRE was detected in the resuspended cells through flow cytometry.
Cell lysate preparation and immunoblotting
LV tissue lysate was prepared as described previously [10]. For AC16 cardiomyocytes, post-H/R treatment cells were placed on ice and washed twice with ice-cold PBS (Sigma-Aldrich #8537). After PBS aspiration from culture plates, 1X lysis buffer (Thermo Scientific TB263248), containing phosphatase and protease inhibitor cocktail (Abcam #ab201114), was added to the culture plate. Cells were collected through scrapers into microcentrifuge tubes. The lysate was centrifuged at 14,000 RPM at 4 °C for 20 min. The supernatant was transferred to a fresh tube, and protein estimation was done using a colorimetric protein assay (Bio-Rad #5000006).
Immunoblotting was performed as described previously [27]. Briefly, an equal amount of protein was loaded on 4–12% gradient Tris-glycine SDS-polyacrylamide gel (Invitrogen #NP0321BOX) and electrophoresed. The gel was then transblotted onto nitrocellulose membranes using a semi-dry method (Bio-Rad Trans-Blot Turbo). The membrane was then blocked with 5% skimmed milk in 1X TBST for 1 h. The membrane was incubated with primary antibodies for GSK-3α/β (Cell Signaling #5676S), p-GSK-3α (Cell Signaling #9327), COX IV (3E11) (Cell Signaling #4850), β-actin (Sigma #A5441-2ML), and GAPDH (Fitzgerald #10R-G109a) overnight at 4 °C. The next day membrane was washed 3X with TBST solution for 5 min each and incubated with a secondary antibody of Donkey anti-mouse IgG (HRP) (Abcam #ab205724) or Goat anti-rabbit IgG (HRP) antibody (Abcam #ab6721) for 1 h on a shaker at room temperature. After washing with TBST, the membrane was quickly incubated with a Clarity Western ECL Substrate Kit (Bio-Rad #170-5060) and the blot developed under a gel documentation system (Bio-Rad Chemidoc Touch Imaging System). Blot quantification was done using ImageJ software, and all samples were normalized with respective loading controls.
Mitochondrial extraction
A total of 1 × 106 AC16 cells were seeded and reoxygenated for 6 h post-hypoxia. Cells were harvested and rinsed with ice-cold PBS and pelleted. Cell pellets were then frozen overnight and thawed the following day to weaken the cell membranes. Mitochondrial extraction was carried out as per manufacturer’s protocol (Abcam ab110170). Briefly, cells were resuspended in a hypotonic buffer, reagent A, to a concentration of 5 mg/ml and incubated on ice for 10 min, followed by homogenization. After centrifugation, the supernatant was collected and labeled as “1.” The remaining pellet was resuspended in reagent B and centrifuged, and the supernatant was collected as “2.” Supernatants 1 and 2 were combined, mixed vigorously, and centrifuged. The mitochondrial pellet was resuspended in reagent C, supplemented with protease and phosphatase inhibitors.
For cytosolic extraction, acetone precipitation was carried out. Ice-cold acetone was added to the remaining supernatant from the samples, vortexed, incubated for 90 min at −20 °C, and then centrifuged. The pellet was air dried for 30 min and then subjected to lysis using RIPA buffer supplemented with protease and phosphatase inhibitors. After centrifugation, the supernatant was collected and used as the cytosolic fraction for Western blotting.
RNA extraction and cDNA preparation
Total RNA was isolated from AC16 cells using RNeasy kit 74106 (Qiagen), and complementary DNA (cDNA) was prepared as described previously [27]. Briefly, cells were lysed directly with lysis buffer containing β-mercaptoethanol followed by ethanol washing. RNA was eluted and dissolved in RNase-free water. The quality of total RNA was assessed by OD260/OD280 and OD260/OD230 ratios using a NanoDrop. cDNA was synthesized using iScript™ cDNA Synthesis Kit (Bio-Rad #170-8891).
Library construction, quality control, and sequencing
For sequencing, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. The first strand cDNA was synthesized, and the library was checked with Qubit and real-time PCR for quantification and bioanalyzer for size distribution detection. Quantified libraries were pooled and sequenced on Illumina platforms. Clustering of the index-coded samples was performed according to the manufacturer’s instructions, and paired-end reads were generated.
For analysis, raw reads of FASTQ format were processed through in-house Perl scripts and clean reads were obtained by removing reads containing adapter, reads containing poly-N, and low-quality reads from raw data. Q20, Q30, and GC contents for the clean data were calculated. The quality of produced data was determined by Q20 at each cycle.
RNA-seq data analysis
Transcriptomic analysis was performed as described previously [29, 30]. Briefly, the expected number of fragments per kilobase of transcript sequence per million (FPKM) base pairs sequenced was determined with consideration of the effect of sequencing depth and gene length for the reads counted at the same time. FPKM of each gene was calculated based on the length of the gene and read count mapped to this gene. Differential expression analysis of two conditions/groups (triplicates per condition) was performed using the DESeq2 R package (1.20.0), which determines differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting p-values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate (FDR). Genes with an adjusted p-value <0.05 found by DESeq2 were assigned as differentially expressed. In gene set enrichment analysis (GSEA), the genes were ranked according to the degree of differential expression in the two samples, and then, the predefined gene sets were tested to see if they were enriched at the top or bottom of the list. Enrichment was performed with Gene Ontology (GO) analysis of differentially expressed genes which was implemented by the clusterProfiler R package and GO terms with adjusted p-values <0.05 considered significantly enriched, differentially expressed genes. KEGG, DisGeNET, and Reactome were the databases used for enrichment analysis. clusterProfiler software was used to test the statistical enrichment of differentially expressed genes in the KEGG, Reactome pathway, and DisGeNET pathway.
Quantitative real-time PCR (qRT-PCR)
A set of top dysregulated genes were randomly selected for qRT-PCR to validate the transcript data. qRT-PCR was performed as described previously [31]. Briefly, total RNA was extracted using the RNeasy kit (Qiagen), and cDNA was synthesized using SuperScript II reverse-transcriptase (Life Technologies). qRT-PCR was performed using SYBR Green PCR master mix and appropriate primers (Sup Table 1). The calculations were performed using the delta-delta-Ct method, and results were normalized with GAPDH.
Statistical analysis
Differences between data groups were evaluated for significance using an unpaired t-test or one-way/two-way ANOVA followed by Tukey’s post hoc test for multiple comparisons (GraphPad Prism Software Inc., San Diego, CA). Data are presented as mean ± SEM. For all tests, a p-value <0.05 was considered statistically significant.
Results
GSK-3α promotes ischemia/reperfusion-induced cardiac injury
Post-tamoxifen treatment, cardiomyocyte-specific conditional KO mice were characterized for levels of GSK-3α expressed in the heart. Immunoblotting of LV lysates revealed ~90% reduction in GSK-3α protein expression in the cKO vs. littermate controls whereas the level of GSK-3β was unchanged, as expected. Irrespective of genotype, the protein levels of GSK-3β were significantly higher in comparison to GSK-3α in the LV tissues (Fig. 1A, B). To identify the specific role of GSK-3α in ischemia/reperfusion (I/R)-induced cardiac injury, cKO and littermate controls were subjected to I/R. Following TTC staining, the area at risk and infarcted area were measured and the LV percentage was calculated. The area at risk was comparable between cKOs vs. control hearts, but there was significantly less infarcted area in GSK-3α cKOs compared to the control hearts subjected to I/R (Fig. 1C, D). These data suggest that GSK-3α promotes I/R-mediated cardiac tissue loss and deletion of GSK-3α in cardiomyocytes limits I/R-induced cardiac injury.

Cardiomyocyte-GSK-3α inactivation promotes I/R-induced cardiac injury. A Immunoblotting using LV lysates from cardiomyocyte-specific conditional KO and littermate control mice and B quantification of levels of GSK-3α and β. C Representative images of TTC-stained heart section from cardiomyocyte-specific GSK-3α KO (GSK3Afl/fl Cre) and littermate control (GSK3Afl/fl) mice subjected to ischemia-reperfusion (I/R). D Quantification of the area at risk (AAR) and infarct size show comparable AAR and significantly attenuated infarct size in the KO animals. n = 9–11; *p < 0.05
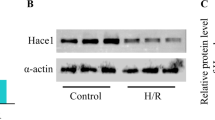
GSK-3α overexpression promotes AC16 cardiomyocyte death post-hypoxia/reoxygenation
To better understand the potential role of GSK-3α in a model of I/R, AC16 cardiomyocytes were employed. First, we assessed the GSK-3α activity in AC16 cardiomyocytes challenged with H/R. A significantly increased level of GSK-3α phosphorylation at Serine 21 (associated with inhibition) was observed in cardiomyocytes post-H/R (6 h/4 h) (Fig. 2A, B). We then transiently transfected AC16 cells with either Flag control or wild-type human Flag-GKS-3α plasmid to generate a gain-of-function model. AC16 cells were transfected for two different time points (24 and 48 h). Transfection efficiency of the plasmid was verified by assessing the total GSK-3α protein expression level compared to their respective Flag control group. The total level of GSK-3α protein was found to increase by ~4 fold after 24 h of transfection (Fig. 2C, D). This time point was selected for further experiments. The H/R model was used to simulate in vivo acute I/R conditions. Flag or Flag-GSK-3α transfected cell groups were exposed to either normoxia or H/R, and the level of cardiomyocyte death was assessed using Annexin-V and 7-AAD labeling (Fig. 2E). The cell viability was significantly lower in GSK-3α overexpressing cardiomyocytes post-H/R (Fig. 2F). We observed significantly higher cell death both in controls and GSK-3α overexpressing cardiomyocytes subjected to H/R when compared to normoxia groups. The level of necrotic cell death was comparable between GSK-3α overexpressing vs. control cardiomyocyte groups challenged with H/R (Fig. 2G). However, a significantly higher degree of cell death (measured by both early and late apoptosis) was observed in GSK-3α overexpressing vs. control cells subjected to H/R (Fig. 2H, I). Most importantly, among the apoptotic cell population, late apoptotic cells (dual positive for Annexin-V and 7-AAD) were more prevalent than early apoptotic cells (Fig. 2H, I). These observations suggest that GSK-3α promotes cardiomyocyte death post-H/R.

GSK-3α promotes cardiomyocyte death post-H/R. A Immunoblotting and B quantification show time-dependent increases of GSK-3α phosphorylation (inhibition) under H/R. C Representative immunoblot shows endogenous expression levels of GSK-3α/β and expression of transfected Flag-GSK-3α in AC16 cardiomyocytes. D Quantification shows significantly increased GSK-3α expression in transfected cardiomyocytes. E Representative flow cytometry scatter dot plots show Annexin-V-FITC and 7-AAD staining in control and GSK-3α overexpressing cardiomyocytes under normoxia or hypoxia-reoxygenation (H/R) conditions. The bar diagrams show the percentage of F viable cells, G total necrotic cells, H total apoptotic cells, and I cells in late apoptosis. Upon H/R, cells overexpressing GSK-3α exhibited significant decreases in cell viability and increases in apoptosis compared to normoxia control groups. n = 6 each group; *p < 0.05, **p < 0.005, ***p < 0.0005
GSK-3α promotes ROS production in AC16 cardiomyocytes post-H/R
Levels of ROS are known to increase upon stresses such as I/R. An increased level of ROS is damaging to the cell and can induce cell death leading to cardiac injury upon I/R. Since overexpression of GSK-3α exacerbates cardiomyocyte apoptosis (Fig. 2E–I), we hypothesized that GSK-3α promotes cell death through induction of ROS generation post-H/R. In this series of experiments, GSK-3α overexpressing AC16 cardiomyocytes and control groups were subjected to either normoxia or H/R. Total and cellular ROS levels were measured using DCFDA staining. Total ROS generation was assessed by measuring the fluorescence intensity of DCFDA in the culture plate containing cells and medium. Irrespective of GSK-3α overexpression, ROS generation was increased in H/R compared to normoxia groups. However, consistent with the level of cell death, ROS generation was significantly elevated in the GSK-3α overexpressing vs. control cells post-H/R (Fig. 3A).

GSK-3α induces reactive oxygen species and mitochondrial dysfunction in cardiomyocytes post-H/R. A Bar diagrams show the mean fluorescence intensity (MFI) of dichlorodihydrofluorescein diacetate (DCFDA) in control and GSK-3α overexpressing cardiomyocytes post-hypoxia-reoxygenation (H/R) measuring total reactive oxygen species (ROS) generation. n = 6 for each group. B ROS analysis by FACS indicates that GSK-3α overexpression significantly increases the percentage of DCFDA-positive cells compared to normoxia and H/R control groups. C Mitochondrial membrane potential (MMP) was measured in AC16 cells using TMRE red, and representative histograms show the MMP levels in DMSO, FCCP, Flag controls, and GSK-3α overexpressing cell groups. D TMRE (MMP) quantification reveals a low signal in the DMSO only control compared to FCCP (positive control) in the AC16 cells. GSK-3α overexpression in AC16 cells significantly reduced MMP in comparison to the Flag-transfected control group. n = 3; **p < 0.005, ***p < 0.0005, ****p < 0.0001
Since intracellular ROS has a direct effect on mitochondrial function, we sought to assess the level of specifically cellular ROS in AC16 cardiomyocytes post-H/R. Transfected AC16 cells were subjected to H/R along with appropriate vehicle and normoxia controls. Post-H/R, cells were stained with DCFDA, and flow cytometry analysis was done to measure DCFDA-positive cardiomyocytes. Consistent with results that displayed increased levels of cell death in the GSK-3α overexpression group, a significantly higher number of DCFDA-positive cells were identified in GSK-3α overexpressing cells vs. control groups post-H/R (Fig. 3B). These findings strongly suggest that increased level of GSK-3α induces ROS generation post-H/R.
Mitochondrial GSK-3α overexpression suppresses mitochondrial membrane potential post-H/R
I/R-induced cardiac cell deaths are largely regulated by an aberrant mitochondrial function. The mitochondrial membrane potential (ΔΨm) is an essential component in the process of energy storage during oxidative phosphorylation (OXPHOS) [32]. We hypothesized that GSK-3α modulates mitochondrial membrane potential in I/R. To address this, GSK-3α overexpressing AC16 cardiomyocytes and control cells were challenged with H/R and ΔΨm was assessed through tetramethyl rhodamine ethyl ester (TMRE) staining. FACS analysis showed a significantly lower ΔΨm in the cells overexpressing GSK-3α compared to the control group (Fig. 3C, D).
We next assessed the expression levels of GSK-3 isoforms in cytosolic and mitochondrial fractions and explored whether their levels change under H/R. We observed significantly higher levels of both GSK-3α and GSK-3β isoforms in the cytosolic fraction under H/R compared to normoxia (Fig. 4A–C). Interestingly, GSK-3 isoform levels were low in mitochondria under normoxia but profoundly elevated upon H/R (Fig. 4A–C) though the ratio of GSK-3α vs. GSK-3β was comparable both in cytoplasm as well as mitochondria (Fig. 4D). These observations indicate that H/R promotes expression of GSK-3 isoforms in mitochondria, which directly facilitates the suppression of ΔΨm. These results strongly suggest that GSK-3α plays critical roles in regulating mitochondrial function post-I/R.

H/R induces the expression of GSK-3 isoforms in mitochondria. Representative Western blot images (A) and quantification show higher levels of B GSK-3α and C GSK-3β in cytosolic (including nuclear) and mitochondrial fractions of AC16 cells post-H/R vs. normoxia. D The relative expression of GSK-3α vs. GSK-3β in both cellular compartments was comparable. The levels of GSK-3 isoforms in cytosolic fraction were normalized with β-actin and mitochondrial fraction with mitochondrial marker COX IV. n = 3, **p < 0.005, ***p < 0.0005, ****p < 0.0001
Transcriptomic profiling of GSK-3α overexpressing human cardiomyocyte identifies several novel potential targets
To investigate the molecular mechanism underscoring the identified role of GSK-3α in I/R-induced cardiac injury, RNA transcriptomic studies were performed in cardiomyocytes subjected to normoxia, hypoxia, or H/R. mRNA sequencing and transcriptomic analysis revealed differentially expressed genes and pathways associated with GSK-3α overexpression compared to control cells. We performed differential gene expression analysis in each cluster to identify GSK-3α-dependent changes (Fig. 5A). The analysis revealed that 549 genes were upregulated while 330 genes were downregulated under normoxia conditions (Fig. 5A). Moreover, increased expression of GSK-3α differentially regulated 2664 and 2263 genes during hypoxia and HR, respectively, compared to Flag-transfected controls. Specifically, CCND1 and IL24 were the most upregulated genes post-H/R. These observations indicate that the GSK-3α overexpression dysregulates a significant number of genes under stress conditions. The top differentially expressed genes in each treatment condition are listed in Table 1 and Suppl. Tables 1–3.

Transcriptomic analysis of GSK-3α overexpressing AC16 cardiomyocytes. A Differential gene expression (DGE) analysis in GSK-3α overexpressing and Flag control cardiomyocytes under different conditions shows the number of upregulated and downregulated genes. B Venn diagrams show the number of overlapping and differentially expressed genes between GSK-3α overexpressing and Flag control cardiomyocytes under normoxia, hypoxia, and hypoxia/reoxygenation (H/R). C Volcano plots show upregulated (red dots), downregulated genes (green dots), and non-significantly different genes within the threshold line (blue dots)
Further analysis revealed several uniquely and differentially expressed genes in these stress conditions. GSK-3α overexpression in cardiomyocytes was associated with induction of 382 genes while 251 genes were repressed in the absence of stressor. This number of dysregulated genes profoundly increased in cardiomyocytes challenged with hypoxia where 550 genes were upregulated while 523 genes were downregulated in GSK-3α overexpressing AC16 cardiomyocytes vs. control. Reoxygenation after hypoxia further increases the number of upregulated genes to 579; however, the number of downregulated genes was less (346) in H/R vs. hypoxia (Fig. 5B). Volcano plot analysis revealed statistically significant differentially expressed genes in GSK-3α overexpressing cardiomyocytes relative to control cells under different conditions (Fig. 5C). The total numbers of upregulated genes were 1484 in cells challenged with H/R, 1488 genes in hypoxia, and 549 genes in normoxia group while 979 genes were repressed in H/R, 1176 genes in hypoxia, and 330 genes in normoxia groups, respectively. These results show the gain-of-GSK-3α function impacts significant numbers of genes in addition to uniquely expressed genes in all three conditions.
GSK-3α regulates genes associated with ECM organization, inflammatory pathways, and metabolic derangement in cardiomyocytes
The list of differentially expressed genes was analyzed for GO terms and KEGG pathway enrichment (p < 0.05) to gain insights into the molecular processes that are affected by GSK-3α in normoxia, hypoxia, and reoxygenation. The significant GO terms enriched in upregulated genes in normoxia conditions were ECM, response to IFNy and other immunology-related terms (Fig. 6A), while the most significant downregulated pathways were related to cell substrate junctions and cell substrate adhesion junctions (Fig. 6B). In cardiomyocytes challenged with hypoxia, the most significant upregulated term was inflammatory response; however, the most downregulated processes were related to ATPase activity, acyl-CoA, thioester, and other similar metabolic processes (Suppl. Fig. 1A, B). Upon reoxygenation, the immune-related genes were profoundly upregulated; however, the terms related to ECM, focal adhesion, and cell-substrate adherens junctions were downregulated (Fig. 6C, D).

Enrichment analysis to identify dysregulated cellular processes based on DEGs. Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEG) annotates the number of A upregulated and B downregulated genes categorized based on their involvement in cellular processes under normoxia and C, D hypoxia/reoxygenation (H/R)
KEGG pathway analysis revealed that genes associated with JAK-STAT and TNF signaling pathways and cytokine-cytokine receptor interaction were most enriched in the upregulated set in GSK-3α overexpressing AC16 cells under normoxia (Fig. 7A). However, the primary KEGG pathways enriched in the downregulated genes were mostly related to metabolism, including 2-oxo carboxylic acid and glutathione metabolism (Fig. 7B). In hypoxic conditions, the main KEGG pathways including NOD-like receptor, TNF, NF-κB, C-type lectin receptor (CLR), toll-like receptor (TLR), and JAK-STAT signaling pathways were enriched in the upregulated genes (Suppl. Fig. 1C). In contrast, metabolic pathways including fatty acid and pyruvate metabolism and OXPHOS were enriched in the downregulated gene set (Suppl. Fig. 1D). Consistent with hypoxia, NOD-like receptor (NLR), TNF, and NF-κB signaling pathways were most enriched in upregulated genes in cells challenged with H/R. Interestingly, mitogen-activated protein kinase (MAPK) and interleukin-17 (IL-17) signaling pathways were specifically enriched in the upregulated genes of AC16 cells challenged with H/R, and these were not observed in cells challenged with hypoxia (Fig. 7C, Table 2). Glutathione metabolism was one of the most enriched pathways in the downregulated genes of GSK-3α overexpressing cardiomyocytes post-H/R, which was not seen in the cells post-hypoxia (Fig. 7D). These observations suggest that GSK-3α activity modulates ECM organization, inflammation, and metabolism in cardiomyocytes in post-stress conditions.

KEGG pathway analysis identifies GSK-3α-mediated dysregulation of cellular pathways. Bar diagrams show significantly enriched pathways in A upregulated and B downregulated gene sets, respectively, in GSK-3α overexpressing cardiomyocytes under normoxia and C, D hypoxia/reoxygenation (H/R)
Validation of top dysregulated genes
Based on adjusted p-value and Log2 fold change, we randomly selected several of the highly differentially regulated genes (five most upregulated and downregulated) for validation. The overexpression of GSK-3α was first validated in the overexpressing cells; then, mRNA expression levels of PDCD1LG2, CCND1, IL24, ISG15, and RGS4 genes were assessed from the upregulated group (Fig. 8A) and genes LCNL1, OLFML2A, LRRC17, AKR1C3, and PCDH18, from the downregulated group (Fig. 8B). These data confirmed the trends of gene expression observed in transcriptome sequencing.

Validation of top differentially regulated genes. Relative mRNA expression of A top 5 upregulated and B top 5 downregulated genes in GSK-3α overexpressing vs. Flag control cardiomyocytes post-hypoxia/reoxygenation (H/R)
Discussion
Ischemic attack followed by reperfusion/stenting promotes myocardial cell death through apoptosis and/or necrosis and is associated with ROS production and mitochondrial dysfunction [16]. However, a better understanding of underlying molecular mechanisms of I/R-induced cardiac cell death would help design an effective therapeutic strategy to limit cardiac damage and remodeling post-I/R. Herein, we identified, for the first time, that cardiomyocyte-specific loss of GSK-3α significantly limits cardiac injury post-I/R. In vitro studies further attest the in vivo findings as increases in GSK-3α expression enhanced ROS production, mitochondrial dysfunction, and cardiomyocyte death post-H/R. The transcriptomic studies support the notion that GSK-3α acts to promote inflammatory pathways and metabolic derangement in the heart post-I/R.
The cell survival Akt/PKB pathway plays critical roles in cardiac pathogenesis [33, 34]. Akt is activated through upstream extracellular stimuli such as growth factors and cytokines [35]. It is well documented that Akt activation is critically important for the activity of downstream GSK-3α and its roles played in the cellular processes. Our in vivo studies show that the specific loss of GSK-3α limits I/R-induced cardiac injury. The related protein kinase GSK-3β has also been reported to limit I/R-induced cardiac injury by regulating mTOR and autophagy [36]. Myocardial I/R induces an enormous amount of ROS generation and mitochondrial dysfunction and decreases in ATP production which, ultimately, induces cell death [37]. Similarly, we observed that GSK-3α overexpression promotes ROS generation and cardiomyocyte death post-H/R. Here, we have shown that H/R induces the GSK-3α expression in mitochondria that essentially acts to suppress the mitochondrial membrane potential which is essential for the process of energy storage during OXPHOS [32]. Dysregulated OXPHOS can also alter cellular metabolic homeostasis, and cells are often forced to shift to less efficient, alternative energy production pathways. Our transcriptomic studies indicate that gain-of-GSK-3α function significantly downregulates the OXPHOS pathway in cardiomyocytes post-H/R, which is consistent with the observed detrimental role of GSK-3α post-I/R. These findings are in complete agreement with our previous ex vivo and in vivo studies where we reported that GSK-3α overexpression reduces the maximum respiration capacity of the mitochondria and OXPHOS ATP production in cardiomyocytes [20]. These observations suggest that GSK-3α induces cardiac cell death partly through regulating mitochondrial bioenergetics post-I/R.
Accumulating evidence suggests that inflammatory mediators play a crucial role in I/R-induced cardiac injury [16]. Our transcriptomic studies displayed profound upregulation of inflammatory genes IL24 and pro-inflammatory signaling pathways such as NOD-like receptor, TNF, NF-κB, CLR, TLR, and JAK-STAT in cardiomyocytes overexpressing GSK-3α under stressor. Normally, cytokines and growth factors play important roles in healing processes post-infarction [38]. However, the recruitment of excess amounts of inflammatory mediators further damages cardiac tissue. Though the relative role of IL-24 in acute cardiac injury is not well characterized, a recent study demonstrates that IL-24 promotes acute renal injury post-I/R [39], which is consistent with our finding of higher cardiac cell death in a model of acute cardiac I/R. It was previously unclear whether IL-24 is expressed by cardiomyocytes, but we observed a significant upregulation, at least at mRNA level, of IL-24 in AC16 cardiomyocytes post-H/R. IL-24-mediated signaling promotes secretion of inflammatory cytokines such as IFN-γ, IL-6, and TNF-α and increases inflammatory cell migration and recruitment [40]. Moreover, IL-24 is also known to activate the JAK/STAT pathway [41] which was identified as one of the most upregulated pathways in GSK-3α overexpressing cardiomyocytes post-hypoxia but not H/R. However, pro-inflammatory TNF, NF-κB, and NLR signaling pathways were found upregulated both under hypoxia and H/R conditions.
Interestingly, IL-17 and MAPK signaling pathways were upregulated in GSK-3α overexpressing cardiomyocytes specifically under H/R which suggests the specific implications of these pathways in GSK-3α-mediated H/R-induced cell deaths. There is evidence to suggest that IL-17–mediated signaling induces the expression of a variety of chemokines and cytokines in target cells [42, 43]. Though the precise role of IL-17 in I/R-induced cardiac injury is undefined, recent studies have reported the key roles of IL-17 in I/R-induced inflammatory processes and renal and intestinal injuries [44, 45]. Consistent with these reports, our transcriptomic studies revealed upregulation of IL-17 signaling pathways which suggests that GSK-3α may play a pivotal role in the induction of IL-17-mediated inflammatory reaction post-I/R. A study suggested that IL-17 induces I/R-mediated cell death and injury through MAPK pathway activation [44]. In agreement, MAPK pathway signaling was found to be profoundly upregulated in the GSK-3α overexpressing cardiomyocytes, particularly under H/R condition. Hence, GSK-3α promotes I/R-induced inflammatory reaction and cell death potentially through activation of MAPK.
Other than inflammatory pathways, we identified GSK-3α as an important regulator of cardiac metabolism under stressors. Several metabolic pathways, including glutathione and fatty acid metabolism, were among the top downregulated pathways in the GSK-3α overexpressing cells post-H/R. Glutathione is considered a potent endogenous antioxidant system that helps in detoxifying xenobiotics, scavenging ROS, and balancing cellular redox, which ultimately limits oxidative damage and cell death in the ischemic heart [46, 47]. GSK-3α-mediated downregulation of glutathione metabolism may partly contribute to elevated levels of ROS and oxidative stress in GSK-3α overexpressing cardiomyocytes. Studies have demonstrated that glutathione protects the heart from peroxynitrite-induced damage post-I/R [48, 49]. It is possible that GSK-3α induces peroxynitrite levels in cardiomyocytes through downregulation of the glutathione metabolism pathway post-I/R which may, in part, contribute to increased cell death.
In summary, we have identified a critical role of GSK-3α in promoting cell death under I/R conditions. Cardiomyocyte-specific loss of GSK-3α promotes cardiac injury post-I/R. GSK-3α overexpression significantly reduced cell viability and enhanced cardiomyocyte apoptosis post-H/R, which were accompanied by increased ROS generation and mitochondrial dysfunction. Transcriptomic analysis of GSK-3α overexpressing cardiomyocytes revealed that GSK-3α plays important roles in regulating inflammatory pathways and cellular metabolism under conditions that model ischemic stressors suffered by the heart. An increase in GSK-3α function upregulates MAPK, NLR, TNF, NF-κB, and IL-17 signaling pathways and downregulates glutathione and fatty acid metabolism indicating induction of inflammation and metabolic derangement in cardiomyocytes under ischemic stressors. These findings suggest that targeting specifically GSK-3α may limit cardiac injury and remodeling post-I/R. Small molecule inhibitors selective for GSK-3α may have therapeutic benefits in conditions associated with ischemic cardiac injury.
Data availability
Data is included in the manuscript and supplementary files. All the raw data is deposited in the NCBI database with the accession number PRJNA984601.
References
Whelan RS, Kaplinskiy V, Kitsis RN (2010) Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 72:19–44. https://doi.org/10.1146/annurev.physiol.010908.163111
Cormier KW, Woodgett JR (2017) Recent advances in understanding the cellular roles of GSK-3. F1000Res 6. https://doi.org/10.12688/f1000research.10557.1
Lal H, Ahmad F, Woodgett J, Force T (2015) The GSK-3 family as therapeutic target for myocardial diseases. Circ Res 116:138–149. https://doi.org/10.1161/CIRCRESAHA.116.303613
Ahmad F, Woodgett JR (2020) Emerging roles of GSK-3alpha in pathophysiology: emphasis on cardio-metabolic disorders. Biochim Biophys Acta Mol Cell Res 1867:118616. https://doi.org/10.1016/j.bbamcr.2019.118616
Gupte M, Tumuluru S, Sui JY, Singh AP, Umbarkar P, Parikh SS, Ahmad F, Zhang Q, Force T, Lal H (2018) Cardiomyocyte-specific deletion of GSK-3beta leads to cardiac dysfunction in a diet induced obesity model. Int J Cardiol 259:145–152. https://doi.org/10.1016/j.ijcard.2018.01.013
Zhang N, Tian YN, Zhou LN, Li MZ, Chen HD, Song SS, Huan XJ, Bao XB, Zhang A, Miao ZH et al (2021) Glycogen synthase kinase 3beta inhibition synergizes with PARP inhibitors through the induction of homologous recombination deficiency in colorectal cancer. Cell Death Dis 12:183. https://doi.org/10.1038/s41419-021-03475-4
Griebel G, Stemmelin J, Lopez-Grancha M, Boulay D, Boquet G, Slowinski F, Pichat P, Beeske S, Tanaka S, Mori A et al (2019) The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci Rep 9:18045. https://doi.org/10.1038/s41598-019-54557-5
Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, Greytak S, Woulfe K, Trivedi CM, Woodgett JR et al (2008) Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest 118:3609–3618
Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, Farber J, Wang Y, Ma XL, Woodgett J et al (2013) GSK-3alpha is a central regulator of age-related pathologies in mice. J Clin Invest 123: 1821–1832. https://doi.org/10.1172/JCI64398
Lal H, Zhou J, Ahmad F, Zaka R, Vagnozzi RJ, Decaul M, Woodgett J, Gao E, Force T (2012) Glycogen synthase kinase-3alpha limits ischemic injury, cardiac rupture, post-myocardial infarction remodeling and death. Circulation 125:65–75. https://doi.org/10.1161/CIRCULATIONAHA.111.050666
Embi N, Rylatt DB, Cohen P (1980) Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur J Biochem 107:519–527
Woodgett JR (1991) cDNA cloning and properties of glycogen synthase kinase-3. Methods Enzymol 200:564–577
MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, Nagy A, Woodgett JR (2007) Glycogen synthase kinase 3alpha-specific regulation of murine hepatic glycogen metabolism. Cell Metab 6:329–337. https://doi.org/10.1016/j.cmet.2007.08.013
Cohen P (2002) Protein kinases- the major drug targets of the twenty-first century? Nat Rev Drug Disc 1:309–315
Linkermann A, Brasen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H et al (2013) Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 110:12024–12029. https://doi.org/10.1073/pnas.1305538110
Eltzschig HK, Eckle T (2011) Ischemia and reperfusion–from mechanism to translation. Nat Med 17:1391–1401. https://doi.org/10.1038/nm.2507
Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto K, Takagi H, Tamamori-Adachi M, Kitajima S et al (2008) Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci USA 105:20900–20905
Ahmad F, Lal H, Zhou J, Vagnozzi RJ, Yu JE, Shang X, Woodgett JR, Gao E, Force T (2014) Cardiomyocyte-specific deletion of gsk3alpha mitigates post-myocardial infarction remodeling, contractile dysfunction, and heart failure. J Am Coll Cardiol 64:696–706. https://doi.org/10.1016/j.jacc.2014.04.068
Umbarkar P, Tousif S, Singh AP, Anderson JC, Zhang Q, Tallquist MD, Woodgett J, Lal H (2022) Fibroblast GSK-3alpha promotes fibrosis via RAF-MEK-ERK pathway in the injured heart. Circ Res 131:620–636. https://doi.org/10.1161/CIRCRESAHA.122.321431
Ahmad F, Singh AP, Tomar D, Rahmani M, Zhang Q, Woodgett JR, Tilley DG, Lal H, Force T (2019) Cardiomyocyte-GSK-3alpha promotes mPTP opening and heart failure in mice with chronic pressure overload. J Mol Cell Cardiol 130:65–75. https://doi.org/10.1016/j.yjmcc.2019.03.020
Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, DeCaul M, Shang X, Patel S, Woodgett JR et al (2010) Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res 106:1635–1645. https://doi.org/10.1161/CIRCRESAHA.109.211482
Lal H, Ahmad F, Zhou J, Yu JE, Vagnozzi RJ, Guo Y, Yu D, Tsai EJ, Woodgett J, Gao E et al (2014) Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 130:419–430. https://doi.org/10.1161/CIRCULATIONAHA.113.008364
Zhou J, Ahmad F, Parikh S, Hoffman NE, Rajan S, Verma VK, Song J, Yuan A, Shanmughapriya S, Guo Y et al (2016) Loss of adult cardiac myocyte GSK-3 leads to mitotic catastrophe resulting in fatal dilated cardiomyopathy. Circ Res 118:1208–1222. https://doi.org/10.1161/CIRCRESAHA.116.308544
Zhou J, Ahmad F, Lal H, Force T (2016) Response by Zhou et al to letter regarding article, “Loss of adult cardiac myocyte GSK-3 leads to mitotic catastrophe resulting in fatal dilated cardiomyopathy.” Circ Res 119:e29–e30. https://doi.org/10.1161/CIRCRESAHA.116.309093
Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ (2010) A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res 107:1445–1453
Yusuf AM, Qaisar R, Al-Tamimi AO, Jayakumar MN, Woodgett JR, Koch WJ, Ahmad F (2022) Cardiomyocyte-GSK-3beta deficiency induces cardiac progenitor cell proliferation in the ischemic heart through paracrine mechanisms. J Cell Physiol 237:1804–1817. https://doi.org/10.1002/jcp.30644
Ahmad F, Tomar D, Aryal ACS, Elmoselhi AB, Thomas M, Elrod JW, Tilley DG, Force T (2020) Nicotinamide riboside kinase-2 alleviates ischemia-induced heart failure through P38 signaling. Biochim Biophys Acta Mol Basis Dis 1866:165609. https://doi.org/10.1016/j.bbadis.2019.165609
Shahzadi SK, Marzook H, Qaisar R, Ahmad F (2022) Nicotinamide riboside kinase-2 inhibits JNK pathway and limits dilated cardiomyopathy in mice with chronic pressure overload. Clin Sci (Lond) 136:181–196. https://doi.org/10.1042/CS20210964
Marzook H, Gupta A, Tomar D, Saleh MA, Patil K, Semreen MH, Hamoudi R, Soares NC, Qaisar R, Ahmad F (2023) Nicotinamide riboside kinase-2 regulates metabolic adaptation in the ischemic heart. J Mol Med (Berl) 101:311–326. https://doi.org/10.1007/s00109-023-02296-6
Khan AA, Gul MT, Karim A, Ranade A, Azeem M, Ibrahim Z, Ramachandran G, Nair VA, Ahmad F, Elmoselhi A et al (2022) Mitigating sarcoplasmic reticulum stress limits disuse-induced muscle loss in hindlimb unloaded mice. NPJ Microgravity 8:24. https://doi.org/10.1038/s41526-022-00211-w
Gupte M, Lal H, Ahmad F, Sawyer DB, Hill MF (2017) Chronic neuregulin-1beta treatment mitigates the progression of postmyocardial infarction heart failure in the setting of type 1 diabetes mellitus by suppressing myocardial apoptosis, Fibrosis, and Key Oxidant-Producing Enzymes. J Card Fail 23:887–899. https://doi.org/10.1016/j.cardfail.2017.08.456
Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M et al (2018) Mitochondrial membrane potential. Anal Biochem 552:50–59. https://doi.org/10.1016/j.ab.2017.07.009
Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard MH, Force T, Franke T, Hajjar R, Rosenzweig A (2001) Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104:330–335
Ghafouri-Fard S, Khanbabapour Sasi A, Hussen BM, Shoorei H, Siddiq A, Taheri M, Ayatollahi SA (2022) Interplay between PI3K/AKT pathway and heart disorders. Mol Biol Rep 49:9767–9781. https://doi.org/10.1007/s11033-022-07468-0
Ruderman NB, Kapeller R, White MF, Cantley LC (1990) Activation of phosphatidylinositol 3-kinase by insulin. Proc Natl Acad Sci USA 87:1411–1415. https://doi.org/10.1073/pnas.87.4.1411
Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J (2011) Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ Res 109:502–511. https://doi.org/10.1161/CIRCRESAHA.111.249532
Cadenas S (2018) ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med 117:76–89. https://doi.org/10.1016/j.freeradbiomed.2018.01.024
Sharma HS, Das DK (1997) Role of cytokines in myocardial ischemia and reperfusion. Mediators Inflamm 6:175–183. https://doi.org/10.1080/09629359791668
Schutte-Nutgen K, Edeling M, Kentrup D, Heitplatz B, Van Marck V, Zarbock A, Meersch-Dini M, Pavenstadt H, Reuter S (2022) Interleukin 24 promotes cell death in renal epithelial cells and is associated with acute renal injury. Am J Transplant 22:2548–2559. https://doi.org/10.1111/ajt.17143
Buzas K, Oppenheim JJ, Zack Howard OM (2011) Myeloid cells migrate in response to IL-24. Cytokine 55:429–434. https://doi.org/10.1016/j.cyto.2011.05.018
Menezes ME, Bhoopathi P, Pradhan AK, Emdad L, Das SK, Guo C, Wang XY, Sarkar D, Fisher PB (2018) Role of MDA-7/IL-24 a Multifunction protein in human diseases. Adv Cancer Res 138:143–182. https://doi.org/10.1016/bs.acr.2018.02.005
Hartupee J, Liu C, Novotny M, Li X, Hamilton T (2007) IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol 179:4135–4141. https://doi.org/10.4049/jimmunol.179.6.4135
Xu S, Cao X (2010) Interleukin-17 and its expanding biological functions. Cell Mol Immunol 7:164–174. https://doi.org/10.1038/cmi.2010.21
Xiao L, Zhang WH, Huang Y, Huang P (2022) Intestinal ischemia-reperfusion induces the release of IL-17A to regulate cell inflammation, apoptosis and barrier damage. Exp Ther Med 23:158. https://doi.org/10.3892/etm.2021.11081
Zhang J, Li Q, Zou YR, Wu SK, Lu XH, Li GS, Wang J (2021) HMGB1-TLR4-IL-23-IL-17A axis accelerates renal ischemia-reperfusion injury via the recruitment and migration of neutrophils. Int Immunopharmacol 94:107433. https://doi.org/10.1016/j.intimp.2021.107433
Matuz-Mares D, Riveros-Rosas H, Vilchis-Landeros MM, Vazquez-Meza H (2021) Glutathione participation in the prevention of cardiovascular diseases. Antioxidants (Basel) 10. https://doi.org/10.3390/antiox10081220
Tan M, Yin Y, Ma X, Zhang J, Pan W, Tan M, Zhao Y, Yang T, Jiang T, Li H (2023) Glutathione system enhancement for cardiac protection: pharmacological options against oxidative stress and ferroptosis. Cell Death Dis 14:131. https://doi.org/10.1038/s41419-023-05645-y
Fan Q, Yang XC, Cao XB, Wang SY, Yang SL, Liu XL, Gao F (2006) Glutathione reverses peroxynitrite-mediated deleterious effects of nitroglycerin on ischemic rat hearts. J Cardiovasc Pharmacol 47:405–412. https://doi.org/10.1097/01.fjc.0000210073.48991.bf
Cheung PY, Wang W, Schulz R (2000) Glutathione protects against myocardial ischemia-reperfusion injury by detoxifying peroxynitrite. J Mol Cell Cardiol 32:1669–1678. https://doi.org/10.1006/jmcc.2000.1203
Funding
The work was supported by collaborative (22010901112) research grants from the University of Sharjah to Firdos Ahmad.
Author information
Authors and Affiliations
Contributions
FA designed the study, performed experiments, acquired funding, supervised the project, performed data analysis and interpretation, and wrote the manuscript. HM performed experiments, collected data, and helped with manuscript writing. AA and AG performed experiments and collected data. AAK and MAS helped with data analysis, interpretation, and manuscript writing. WJK and JRW helped with reagents, data analysis, and manuscript editing. RQ helped with data analysis, interpretation, and supervision.
Corresponding author
Ethics declarations
Ethics approval
The Institutional Animal Care and Use Committee (IACUC) of Vanderbilt University Medical Center approved all animal procedures and treatments.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ahmad, F., Marzook, H., Gupta, A. et al. GSK-3α aggravates inflammation, metabolic derangement, and cardiac injury post-ischemia/reperfusion. J Mol Med 101, 1379–1396 (2023). https://doi.org/10.1007/s00109-023-02373-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-023-02373-w