
Abstract
Metabolic oncology is an exciting new field in cancer research, offering a new window to cancer’s molecular plasticity and promise for the development of effective, cancer-selective therapies and novel biomarkers. It is based on the realization that cancer’s unique metabolism (known since Warburg’s report in 1923) with suppression of mitochondrial glucose oxidation and upregulation of cytoplasmic glycolysis is not a secondary but a primary event, offering many growth advantages to cancer cells. Many mechanisms have been revealed, including growth factors, oncogenes, and mutations, all contributing to a suppression of mitochondria, similar to what takes place in hypoxia. This suppression leads to inhibition of mitochondria-driven apoptosis, promotes proliferation, and enhances angiogenesis and metastatic potential. A number of molecular tools and small molecules targeting metabolic enzymes, including pyruvate kinase, pyruvate dehydrogenase kinase, isocitrate dehydrogenase, and lactate dehydrogenase, have been developed, inhibiting cancer growth in vitro and in vivo in several cancer types. Several have already entered early-phase trials, a great translational success considering the young age of the field (less than 10 years). Here we review the mechanisms and effects of these metabolic modulators and the rationale for further development. This rapidly accumulating knowledge allows some optimism that this may prove to be a paradigm shift in the way we understand and treat cancer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The majority of chemotherapeutic drugs for cancer inhibit pathways fundamental to the life of all cells, leading to adverse effects on healthy tissues [1]. In addition, traditional drug development in cancer has been focusing on a single molecular pathway, following the “one gene - one drug” approach. It became apparent however that most tumors are characterized by multiple molecular abnormalities so that when the therapies are offered even in combination of 2-3 drugs, the tumor eventually relapses. It is a rather rare example to find therapies that are effective and not toxic. This requires the identification of molecular abnormalities that are not only critical for the life of cancer—but not normal cells—but are also the dominant or the only molecular abnormality within the tumor. For example, this can be seen in certain leukemias or tumors where a mutation in the majority of cancer cells not only dominates their molecular phenotype but is also critical for their survival, explaining the great success of agents such as Gleevec for chronic myelogenous leukemia [2] or Herceptin for certain breast cancers [3]. Yet, most cancers, like glioblastoma for example, are characterized by several cellular phenotypes within each tumor and yet, in each of these cell types, there are several genetic and molecular abnormalities [4], obviously not susceptible to a single or even a combination of two to three drugs. Is it possible to identify a common denominator across all of these abnormalities that is not only critical for the survival of cancer cells but is also not present in normal cells? And to push the envelope even further, is it possible that this common denominator is also present in cancer stem cells, so that if targeted, tumor relapse would be limited as well [5]? In other words, is there an Achilles’ heel for such complex and molecularly plastic tumors? Recent work over the past 10 years suggests that such a common denominator, unique to cancer cells, may exist and be no other than the unique metabolism of cancer cells, first identified by Otto Warburg, more than 90 years ago [6]. Unfortunately, the work of Warburg, who was awarded the Nobel Prize for his work on metabolism, did not translate in cancer therapies for many decades as it was wrongly assumed that the unique cancer metabolism was a consequence, not a cause or contributing factor in cancer. We now know that if not causal, like in certain examples of mutations in key metabolic enzymes, this metabolic remodeling is a strong contributing or promoting factor as it promotes proliferation, resistance to apoptosis, and tumor angiogenesis, facilitating tumor growth and metastasis. Even better, tumor metabolism is typically different than that of normal cells. The development of “metabolic modulators” over the past 10 years has already entered early-phase trials. Although it sounds “too good to be true” that metabolism is the long sought Achilles’ heel for cancer, a new field has already opened. Metabolic oncology is an exciting frontier in the battle against cancer, and this review focuses on the rationale for the development of metabolic modulators, concentrating on those that hold promise or already have entered human trials.
Cancer’s abnormal metabolism and its molecular consequences
The cornerstone of metabolism in the cell is the mitochondrion. Traditional biochemistry taught us that mitochondria’s main job is to generate heat and energy (ATP). We now know that although the most efficient means for ATP production lie within mitochondria, ATP can be adequately produced outside of mitochondria, through cytoplasmic glycolysis, at amounts enough to support the energy-hungry cancer cells. We also now know that mitochondria do much more: They produce diffusible mediators that can regulate multiple molecular pathways in the cell and even the nucleus. They can also induce apoptosis, an intriguing fact since the “provider of life” (i.e., ATP) can also be the provider of death for the cell. It was perhaps this “paradox” that did not allow mitochondria to be seen as targets for pro-apoptotic strategies until recently. Therefore, suppression of mitochondrial function can suppress apoptosis and alter cellular signaling toward a pro-proliferative phenotype as we discuss below. It is thus not a surprise that cancer cells have suppressed mitochondrial function.
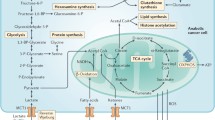
Normally, cells metabolize glucose to pyruvate in the cytoplasm by glycolysis; then, this is converted by pyruvate dehydrogenase (PDH) into acetyl-CoA, which enters the mitochondrial Krebs’ cycle, where it is oxidized to eventually produce the electron donors NADH and FADH2. These donate electrons down a redox gradient in the electron transport chain (ETC), while protons are pumped out of the mitochondria and mitochondria-derived reactive oxygen species (mROS; mostly the negatively charged superoxide) are generated, creating a membrane potential (ΔΨm) across the mitochondrial inner membrane (Fig. 1). This process uses oxygen as the final electron acceptor in Complex IV of the ETC (forming water) and uses the stored energy of the ΔΨm to produce and release ATP. As protons re-enter the inner membrane, they release energy used to pump ATP out in the cytoplasm and bring ADP in. A similar process is followed in the oxidation of fatty acids, forming acetyl-CoA, which is also oxidized in the Krebs’ cycle producing the same electron donors. Thus, mitochondria process fuel (carbohydrates, lipids, oxygen) to produce energy and have evolved to be important fuel sensors. When fuel supply is ample, the growth and differentiation of tissues can be kept under control by the coordinated induction of apoptosis, producing effective cell population control. In contrast, when fuel is limited, mitochondria suppress apoptosis in an attempt to preserve life under stressed conditions. It is here that mitochondria can be “fooled” by the cancer cells.

Suppressed mitochondrial function in cancer cells under normoxia (Warburg Effect). While the mitochondrial glucose oxidation (GO) is suppressed, the cytoplasmic glycolysis (Gly) is enhanced in cancer. This is caused by a combination of inhibited mitochondrial enzymes, whether by upregulation of PDK (a PDH inhibitor), LDH5 and PKM2 (isozymes that confer altered enzymatic activity) or by mutations in IDH. All of these enzymes are induced by HIF1α. On the other hand, the effects of this remodeling include decreased production of αKG (as a result of inhibited Krebs’ cycle) and increased levels of 2-HG, the product of mutated IDH. Both of which result in HIF1α stabilization, closing a powerful feedback loop. In addition, this remodeling results in mitochondrial hyperpolarization (in part of a translocation of the glycolytic enzyme HKII to the VDAC, inhibiting the function of the mitochondrial transition pore, a mega channel that allows the release of pro-apoptotic factors like cytochrome c and apoptosis inducing factor, thus inhibiting mitochondria-dependent apoptosis. Pyruvate dehydrogenase kinase (PDK), pyruvate dehydrogenase (PDH), lactate dehydrogenase 5 (LDH5), pyruvate kinase M2 (PKM2), isocitrate dehydrogenase (IDH), hypoxia inducible factor 1α (HIF1α), alpha-ketoglutarate (αKG), 2-hydroxyglutarate (2-HG), hexokinase II (HKII), and voltage-dependent anion channel (VDAC)
Let us say, for example, that oxygen is limited. This de facto inhibits oxidative phosphorylation in the mitochondria. Immediately, the mitochondria sense the lack of fuel and ignite a series of mechanisms for the cell to (a) seek alternate means of ATP production and (b) suppress apoptosis since stress may be imminent:
(a) The expression of glucose transporters is increased and more glucose enters the cell. This can be achieved by the activation of HIF1α directly by hypoxia but—remarkably—the mitochondrial suppression can also activate HIF1α. This is because alpha-ketoglutarate (αKG), a Krebs’ product that is a required cofactor for the prolyl-hydroxylases that de-stabilize HIF1α, is decreased [7]. Independent of its stabilization, the HIF1α transcription machinery, in part driven by the redox-sensitive p53, may also be activated when the diffusible mROS decrease (for example, H2O2 from superoxide’s dismutation from manganese superoxide dismutase) [8–11]. Activated HIF1α not only increases the expression of glucose transporters but also increases the transcription of almost all the glycolytic enzymes in the cytoplasm. Thus, with more glucose and activated glycolysis, ATP can be produced. Normally, the efficiency of glycolysis for ATP is less than that of mitochondria (each mole of glucose produces 36 mol of ATP in mitochondrial glucose oxidation but only 2 mol of ATP in glycolysis), but as glycolysis is enhanced, the cell may eventually compensate for the ATP that is missing from the inhibited mitochondria.
(b) The next thing that happens is that mitochondria hyperpolarize (increased ΔΨm) [12, 13]. The pro-apoptotic factors stored inside mitochondria (like cytochrome c or apoptosis-inducing factor) are unable to leak out, making cancer cells resistant to mitochondria-dependent apoptosis. This is because these factors leak through the mitochondrial transition pore (MTP), a mega-channel that, being voltage- and redox-sensitive, tends to close while membrane potential is high and mROS are low [14]. The mechanism for mitochondrial hyperpolarization is not clear although it has been known since the 1980s to be a feature of most cancers [15]. One potential mechanism is that glycolysis leads to a GSK-3β-driven translocation of the cytoplasmic hexokinase II (HK2) to the outer mitochondrial membrane, where it binds and inhibits the major channel that releases negatively charged ions, i.e., the voltage-dependent anion channel (VDAC; a critical component of the MTP), leading to a buildup of anions inside the mitochondria [16, 17] (Fig. 1). Another mechanism may be that the enhanced production of ATP in the cytoplasm due to the enhanced glycolysis causes a decrease in the ATP/ADP gradient in the microdomains around the outer mitochondrial membrane, decreasing the function of ATP synthase and thus preventing the re-uptake of the positively charged H+ ions back to the mitochondria [16]. Therefore, mitochondrial suppression supports a state of apoptosis resistance, while adequate production of ATP is maintained. Interestingly, HIF1α also induces pyruvate dehydrogenase kinase (PDK), inhibiting PDH, and thus decreasing the production of acetyl-CoA entering the Krebs’ cycle.
Let us now see what would happen if PDH were to be inhibited by other means, not by the hypoxia-mediated activation of HIF1α. Immediately, the mitochondria will sense a lack of fuel entering them since the supply of acetyl-CoA will decrease (despite the fact that the supply of glucose and oxygen to the cell remains normal). They will then ignite the same process described above, leading to resistance to apoptosis. One can follow the same logic and realize that inhibition of any critical enzyme used in glucose or fatty acid oxidation may have the same consequences, “fooling the mitochondria” and inhibiting oxidative phosphorylation, as if there was lack of fuel. This mechanism can be used by cancer cells to inhibit apoptosis, a sine qua non of cancer. At the same time, there are three additional advantages that the cancer cells gain by this “inappropriate” mitochondrial suppression:
First, the cancer cell can now use pyruvate that is not oxidized in the Krebs’ cycle, for biomass generation, as the tumor grows [18]. In other words, the unused pyruvate helps a dividing cell create the amino acids, nucleotides, and lipids needed to replicate. Specifically, unused pyruvate can be transaminated to produce amino acids. Similarly, unused pyruvate can be metabolized and shunted into the pentose phosphate pathway to produce both nucleotides and NADPH, which is required to synthesize lipids [18].
Second, as in the case of decreased production of αKG which contributes to HIF1α activation, there are other downstream signals from mitochondria, like decreased mROS, dysregulation of cytoplasmic calcium, or induction of chaperones that via the retrograde pathway (a series of mechanisms that mitochondria induce under stress to send signals to the nucleus) directly or indirectly activate other pro-proliferative master transcription factors (like NFAT for example) [12, 19–21]. Like HIF1α, NFAT can suppress several aspects of mitochondrial function, enhancing the proliferative potential of cancer cells and driving positively reinforcing feedback loops (proliferative signals → mitochondrial suppression → proliferative signals).
Third, this mitochondrial remodeling promotes angiogenesis, not surprising since this mitochondrial remodeling “mimics” hypoxia, a well-known driver of angiogenesis. While hypoxia and HIF1α activation induce mitochondrial suppression [22], the reverse is also true [10], establishing a powerful feedback loop that sustains angiogenesis even in the absence of hypoxia. The majority of solid tumors experience hypoxia during early tumor development as it occurs several cell layers away from capillaries [23]. This leads to an initial primary hypoxia-driven activation of HIF1α and angiogenic signaling to form new blood vessels to supply the growing tumor with nutrients. However, HIF1α stabilization is also regulated by several diffusible mitochondria-derived factors, like αKG and mROS as discussed above [23]. These lead to the inhibition of the degradation of HIF1α through VHL-dependent ubiquitination or activation of its transcriptional activity, respectively [24, 25]. Thus, as the newly formed blood vessels bring in more oxygen, limiting hypoxia, the mitochondrial suppression that takes place in the tumor now causes a primary normoxic activation of HIF1α, sustaining angiogenesis as the tumor continues to grow [10, 22]. Furthermore, by reducing pyruvate into lactic acid during glycolysis, cancer cells increase extracellular acidosis. This potentiates breakdown of the extracellular matrix and allows penetration of the cancer through the basement membrane, thereby driving metastasis [26].
Thus, mitochondrial suppression promotes suppressed apoptosis, increased proliferation, angiogenesis, and metastatic potential and so can be seen as a critical hub of cancer signaling. While hypoxia is a physiologic mechanism for global mitochondrial suppression, we will now discuss a variety of prominent cancer mechanisms that all lead to mitochondrial suppression, in a sense, mimicking hypoxia.
The cause of mitochondrial suppression in cancer
Mitochondria are well-known “integrators” of multiple signals [12]. Thus, mitochondrial suppression can occur through four overarching mechanisms in cancer: (1) growth and transcription factor signaling or (2) off-target effects of specific oncogenes that can suppress specific enzymes or ETC complexes, (3) suppression of factors regulating mitochondrial homeostasis, and (4) mutations in key metabolic enzymes. It is important to note that more than one of these factors may be present within any given tumor.
-
1.
Growth Factors: One of the pleiotropic effects of growth factors upregulated in cancer, such as epidermal growth factor (EGF) and fibroblast growth factor (FGF), is the regulation of the flux of pyruvate into the mitochondria by the gate-keeping enzyme PDH. EGF and FGF increase the activity of pyruvate dehydrogenase kinase (PDK) [27] which phosphorylates and inhibits PDH, thus preventing the entry of pyruvate into the Krebs’ cycle and suppressing glucose oxidation (GO) [28]. Furthermore, EGF signaling has recently been shown to also directly inhibit PDH function, through a PDK-independent tyrosine phosphorylation of its E1 subunit [29]. Furthermore, PDK is a target gene of HIF1α, which is upregulated in most solid tumors [23, 30]. HIF1α works at multiple levels to shift the balance of metabolism from GO toward glycolysis, including upregulation of glucose transporters and glycolytic enzymes. HIF1α also increases the translation of the EGF receptor, thereby suppressing mitochondrial function by both upregulating and activating PDK [31]. There are four PDK isoenzymes with variable tissue expression, and some, like PDK 4, are inducible in conditions of metabolic stress [32]. Thus, it is reasonable to assume that other, not yet identified, “fuel sensing” mechanisms exist in inducing the transcription of one or more PDKs in tumors, in addition to increasing PDK activity through tyrosine kinase signaling.
-
2.
Oncogenes: Mitochondrial function is also suppressed by many oncogenes, with p53 and c-MYC being prime examples. The Cancer Genome Atlas has identified p53 to be the most commonly mutated gene in cancer [33]. p53 inhibits glycolysis by inducing the expression of Tp53-induced glycolysis and apoptosis regulator (TIGAR) as well as by reducing the expression of glycolytic enzymes, like phosphoglycerate mutase [34, 35]. Furthermore, p53 enhances GO by increasing the expression of cytochrome c oxidase subunits of the ETC [36]. Loss of p53 function causes upregulation of both the glycolytic enzyme hexokinase II (HKII) and PDK [37, 38]. Overall, loss of p53 function suppresses mitochondria and shifts the cell to a more glycolytic phenotype. It is important to note that post-translational modifications like acetylation, a process intimately linked to mitochondrial function, are also known to regulate p53’s function in cancer and in the absence of mutations [39]. Like p53, c-MYC increases the expression of multiple glycolytic enzymes including HKII, phosphofructokinase, GAPDH, and enolase A, as well as glucose transporters and lactate dehydrogenase (LDH) [40–42]. On top of driving glycolysis, upregulation of LDH shifts the flux of pyruvate away from GO, thereby further suppressing mitochondrial function.
-
3.
Regulation of factors that control mitochondrial homeostasis: Global mitochondrial function can be regulated through inhibition of mitochondria-specific factors like sirtuin 3 (Sirt3; the main de-acetylase in the mitochondria) and uncoupling protein-2 (UCP-2; a putative mitochondria calcium transporter). Inhibition of Sirt3 and UCP-2 suppresses mitochondrial function by increasing protein acetylation and decreasing mitochondrial calcium, respectively. When Sirt3 is deficient, the acetylation of the majority of proteins involved in oxidative metabolism, including Krebs’ enzymes and ETC complexes, increases; this post-translational modification typically leads to the inhibition of enzymatic function [43, 44]. Overall, loss of Sirt3 activity can cause up to 50 % reduction in mitochondria-derived ATP and respiration. Thus, it is not surprising that loss of Sirt3 function has been shown to promote the Warburg Effect and cancer. In fact, embryonic fibroblasts lacking Sirt3 require activation of only one other oncogene to transform into cancer [45], in contrast to wild-type cells that require at least two. Moreover, mice deficient in Sirt3 spontaneously develop breast cancer, and many human cancers are deficient in Sirt3 [45–47]. These observations have led investigators to propose that Sirt3 fulfills the criteria to be an oncogene [45].
Similarly, loss of UCP-2 function, which despite its name is a weak uncoupler but good mediator of calcium entry into the mitochondria, promotes proliferation [20] in part due to a reduction in mitochondrial calcium, which decreases activity of many calcium-dependent mitochondrial enzymes such as PDH, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase [48, 49]. Mice deficient in UCP-2 are more prone to develop colon cancer than wild-type UCP-2 littermates when exposed to a carcinogen, demonstrating a predisposition to oncogenic transformation in these glycolytic animals [50]. Similarly to Sirt3, UCP2 was recently proposed to be a tumor-suppressing factor [20].
Another way that mitochondrial function can be suppressed globally is disturbance of the way mitochondria form networks in the cell (mitochondrial fission and fusion) or the way these organelles are “recycled” (mitophagy) [51]. Since these mitochondrial properties are closely linked to cell cycle progression and cell division, it is not surprising that there is early evidence that they may be involved in carcinogenesis [52, 53]. Yet, the importance of these essential functions for many dividing healthy cells may limit their therapeutic potential.
-
4.
Enzymatic mutations: Mutations in Krebs’ cycle enzymes lead to suppressed mitochondrial function. Three examples of this are fumarase, succinate dehydrogenase (SDH), and isocitrate dehydrogenase (IDH). Mutations in fumarase have been identified in almost all cases of the tumor susceptibility syndrome, hereditary leiomyomatosis, and renal cell carcinoma [54]. Similarly, SDH mutations have been linked to the development of pheochromocytoma, paraganglioma, and renal cell carcinoma [55, 56]. Loss of fumarase or SDH function leads to an accumulation of intracellular fumarate or succinate, respectively. Each of these Krebs’ metabolites has been shown to stabilize HIF1α by inhibiting the prolyl-hydroxylases required for HIF1α degradation [57–59]. This leads to a high level of HIF1α activity even in the presence of oxygen. As a result, these tumors are very vascular. Similarly, a powerful pseudohypoxic environment is created in tumors housing IDH mutations due to gain-of-function activity. Two independent cancer genome sequencing projects found that missense mutations in patients with glioblastoma multiforme and acute myelogenous leukemia caused substitutions of a homologous arginine in the active site of the enzyme at R132 and R172 in IDH1 and IDH2, respectively [60, 61]. These mutations cause a gain-of-function, neomorphic activity, in which the mutant IDH-mediated reaction yields 2-hydroxyglutarate (2-HG) rather than the normal product, αKG [62, 63]. Nearly structurally identical to αKG, 2-HG may competitively inhibit the over 60 αKG-dependent dioxygenase enzymes in humans by binding to the αKG binding pocket in the enzyme’s active site [7]. These αKG-dependent enzymes (and thus 2-HG targets) are involved in a broad range of biological processes including HIF1α degradation, collagen synthesis, and histone methylation [64]. 2-HG also alters HIF1α degradation by inhibiting HIF prolyl-hydroxylases. As a result, cells producing 2-HG, due to an IDH mutation, are locked into a state of pseudohypoxia and mitochondrial suppression. Similar to renal cell carcinoma, glioblastomas are also particularly vascular tumors.
Like p53, many of the Krebs’ enzymes, including those discussed above, can be (at least partially) inhibited via post-translational modifications like acetylation, even in the absence of mutations, and this has also been shown to be present in several cancers [65, 66].
Mitochondria-targeting cancer therapies
We will now follow the sequential metabolism of glucose in cancer cells to highlight several cancer-specific metabolic targets that have been explored (Fig. 2), focusing on the translational potential of these discoveries.

Biomarkers and metabolic modulators arising from the metabolic theory of cancer (see text). Biomarkers (top) and metabolic modulators (bottom) that have been developed for target enzymes and have been or are in preclinical or clinical trials. Glucose transporter 1 (GLUT1), Hexokinase II (HKII), M2 isoform pyruvate kinase (PKM2), Lactate dehydrogenase 5 (LDH5), Pyruvate dehydrogenase (PDH), pyruvate dehydrogenase kinase (PDK), Tyrosine Kinase Inhibitors (TKIs) Isocitrate dehydrogenase (IDH), Phosphoenolpyruvate (PEP), Positron emission tomography (PET), Magnetic resonance (MR), 2-hydroxyglutarate (2-HG)
Glucose transport and phosphorylation
Since glycolysis is upregulated in many cancers, it may appear logical to attempt to inhibit it at its early stage. Glycolysis starts by glucose entry into the cell, through glucose transporters (GLUTs), followed immediately by phosphorylation by hexokinase (HK), required to “trap” glucose intracellularly. Logically, pharmacological inhibition of these two proteins may make sense as both are upregulated in many cancers in order to increase glucose uptake and glycolysis and compensate for the loss of GO. However, being so proximal in the metabolic pathway and ubiquitous in all cells, inhibition of GLUTs and HK, while effective in some pre-clinical studies, has suffered setbacks when tested in early phase human trials [67, 68]. For example, inhibition of GLUTs led to brain toxicity as neurons rely mostly on the use of carbohydrate metabolism [69]. Similarly, HK inhibition led to severe hepatic toxicity, an organ heavily involved in both catabolism and anabolism of glucose [69]. In fact, more than half of the clinical trials using the GLUT and HK inhibitors, 2-deoxyglucose and lonidamine, were terminated prematurely [70]. Nevertheless, the HK inhibitor, 3-bromopyruvate, which showed good effect in a xenotransplant model, was used in a patient with fibrolamellar hepatocellular carcinoma [71, 72]. While this patient survived the duration of therapy, with few reported serious systemic side effects, the drug was delivered directly to the tumor-related artery by transarterial chemoembolization (TACE) [73], perhaps limiting toxicity. Inhibiting glycolysis follows a more traditional “cytotoxic” pathway as inhibition of glycolysis causes non-specific necrosis; in fact, suppressing glycolysis will unavoidably further suppress mitochondrial function as it deprives mitochondria from a primary fuel in most tissues, limiting the therapeutic potential of this strategy. This is in contrast to the metabolic modulators discussed below that target the “coupling” of glycolysis to GO, actually enhancing mitochondrial function, allowing mitochondria to operate their intrinsic apoptotic machinery (an energy consuming function) or normalize their downstream signaling. While it is easy to criticize retrospectively, the investigators of these early clinical studies should be given credit as the first that attempted to target a metabolic process in human cancer, contributing to our re-examination of Warburg’s “forgotten” theory.
Pyruvate kinase M2 (PKM2) activation
Pyruvate stands at a crossroads of metabolic fates: It is the product of cytoplasmic glycolysis, the product of cytosolic malate oxidation (to make anabolic NADPH), a precursor for amino acid production through transamination, the substrate of PDH versus LDH to either make acetyl-CoA which drives the mitochondrial Krebs’ cycle, or lactate and complete glycolysis, respectively [69, 74, 75]. Pyruvate kinase (PK) is the last enzymatic step in glycolysis, catalyzing the reaction of phosphoenolpyruvate to pyruvate, and consists of four isoforms [76]. Two of these isoforms, M1 (PKM1) and M2 (PKM2), are encoded by alternative splicing of the PKM2 (15q23) gene [77, 78]. Enzymatically, PKM1 is the highly active version and is found in normal tissues requiring large amounts of glucose-derived ATP, like skeletal muscle or brain [79, 80]. In contrast, the less active PKM2 is expressed in most tissues during development and has been found in many cancer cell lines [80–82]. Indeed, preferential expression of PKM2 over PKM1 is associated with mitochondrial suppression and enhanced tumorigenesis [80, 83, 84]. The enzymatic activity of PKM2 is regulated by several factors including glycolytic intermediates, tyrosine phosphorylation, and acetylation [85–87]. Therefore, even though PKM2 expression is increased in cancer, its overall activity may be decreased [80]. Furthermore, it is dynamic since in nutrient-abundant states, PKM2 forms active tetramers that function similarly to the more active PKM1 [80, 88]. The importance of this dynamic regulation of PKM2 has recently been explored in cancer [89]. Israelson et al. found that mice conditionally deficient in PKM2 had a more accelerated tumor growth and mortality than their wild-type littermates. Furthermore, the ratio of the inactive over active form of PKM2 was found to tip the balance toward cancer growth. In contrast to the earlier belief that it was the switch from one isozyme to the other that promotes cancer, this work showed that it is the overall suppression of PKM1/2 activity as a whole (and thus the suppressed GO) that promotes cancer (Fig. 3a). Indeed, overexpressing PKM1 in cancer cell lines lead to reduced tumor growth in xenotransplant models [83]. Thus, while the early discovery of an isozyme “specific for cancer” would have triggered efforts to inhibit it, it appears that it is PKM2 activators that may hold promise as cancer therapies. Several small molecules have been developed, such as TEPP-46, DASA-58, and ML265, which bind PKM2 at the subunit interaction interface to promote formation of enzymatically active tetramers [90]. This leads to a constitutively active enzyme with over 200 % enhanced activity [83]. In vitro, treatment with these small-molecule activators decreased the intermediates necessary for biomass generation, reduced lactate production and lipid synthesis, and lead to smaller and slower growing tumors in vivo [83, 91].

Ratio of Glycolysis to Glucose Oxidation increases in cancer due to changes in several key metabolic enzymes.(a) Cells that express a low amount of Pyruvate kinase M1 (PKM1) and an abundance of low-activity Pyruvate kinase M2 (PKM2; which results in an overall decrease in pyruvate kinase activity) prevent the entry of pyruvate into the mitochondria, inhibiting glucose oxidation (GO). (b) Pyruvate dehydrogenase kinase (PDK) also inhibits the entry of pyruvate into the mitochondria by phosphorylating and inhibiting pyruvate dehydrogenase (PDH). (c) The Lactate dehydrogenase (LDH) enzyme is made from four subunits comprised of H (Heart) or M (Muscle) isoforms. Overexpression of the hypoxia-inducible factor 1α (HIF1α) inducible LDH-M creates an enzyme comprised of four M subunits resulting in increased activity, favouring the reduction of pyruvate to lactate, thereby shunting pyruvate away from GO. (d) Mutation in isocitrate dehydrogenase (IDH) leads to the production of the oncometabolite, 2-hydroxyglutarate (2-HG), which antagonizes the normal product, alpha-ketoglutarate (αKG), leading to suppressed GO via the stabilization and accumulation HIF1α
Despite these promising results, pharmacologic activation of PKM2 may not induce cytotoxic changes in cancer. While populations of cancer cells expressing PKM2 do not proliferate, they continue to persist in a non-proliferative, perhaps senescent state [89]. Therefore, once a tumor has been detected, treatment with PKM2 activators may only limit further growth and would need to be combined with another agent or treatment modality to debulk the tumor by increasing cytotoxicity or by inducing apoptosis.
Pyruvate dehydrogenase kinase (PDK) inhibition
PDK phosphorylates and inhibits the E1a subunit of PDH [28]. The net result of inhibited PDH is an increase in the ratio of glycolysis over GO (Fig. 3b), with all the subsequent downstream signaling events that were discussed earlier. It is possible that increased expression and activity of PDK (via HIF1α or tyrosine kinase signaling as discussed earlier) is enough to induce the Warburg Effect and be the dominant mechanism in certain cancers [13, 92] or other proliferative diseases, like pulmonary arterial hypertension, characterized by a proliferative vascular remodeling and mitochondrial suppression [12, 93]. For example, PDK is significantly more increased in glioblastoma tumors compared to that in healthy brain tissues from the same patient [94], although this has not been systematically studied in cancer yet. There is strong evidence that PDK inhibition decreases cancer growth in vitro and in vivo in a variety of tumors as discussed below.
Dichloroacetate is an orally available small-molecule inhibitor of PDK (structurally resembling pyruvate) that can reach most tissues and cross the blood brain barrier. Dichloroacetate (DCA) inhibits PDK at concentrations of 10–250 μM, while it is more active against some of the four PDK isoforms (i.e., the ubiquitously expressed PDK2) compared to others [95, 96]. DCA’s mechanism of action is quite specific as it is mimicked by molecular PDK knockdown; in addition, DCA has no additional effects in cells with effective PDK knockdown [94, 97].
Originally, DCA was pioneered by Dr. Stacpoole’s group at the University of Florida to limit lactic acidosis in children with congenital mitochondrial diseases (for example, deficiencies of PDH or other mitochondrial enzymes), and over the past 40 years, it has been explored in a number of disease states associated with lactic acidosis or with a primary mitochondrial suppression, including diabetes, malaria, pulmonary arterial hypertension, lactic acidosis, heart failure, and exercise tolerance in chronic respiratory disease [98–106]. In 2007, we described DCA’s pro-apoptotic and antiproliferative effects due to normalization of mitochondrial function in a variety of cancers (non-small-cell lung cancer, breast cancer, glioblastoma) in vitro and in xenotransplant models in vivo (Fig. 4a) [97]. DCA, a generic drug, cannot be patented, creating the potential for financial barriers in its development as a cancer treatment. Yet, a number of investigators have shown interest since and have described similar effects in a variety of tumors. Some examples include prostate cancer [107], colon cancer [108–110], gastric cancer [111], endometrial cancer [112], glioblastoma [113], neuroblastoma [114], T cell lymphoma [115], non-Hodgin’s lymphoma [116], fibrosarcoma [117], and metastatic breast cancer [118] (Table 1). In this last study, DCA reduced lung metastases by 58 % in a highly metastatic breast cancer model [118].

Translation of DCA from animal studies to an early phase human trial. DCA decreased tumor size (a), vascularity and FDG uptake (measured by microPET-CT (b) in a xenograft rat model with non-small cell lung cancer. In a small human glioblastoma trial, DCA normalized mitochondrial metabolism, increased apoptosis, suppressed angiogenesis and reduced tumor growth after debulking surgery for at least 18 months, in a patient that had failed all approved therapies and was otherwise destined for hospice care (c)
DCA appears to not have significant effects in normal cells, perhaps because of low levels of PDK activity in healthy tissues. Generally speaking, normal cells need active mitochondria with active PDH and keep the levels of its inhibitor (PDK) low. For example, DCA normalized the high ΔΨm of non-small-cell lung cancer, glioblastoma, and breast cancer cell lines without altering the ΔΨm of each cancer’s non-malignant tissue analog, i.e., small airway epithelial cells, mammary epithelial cells, or healthy brain tissues [10, 94, 97].
In addition to the induction of apoptosis and inhibition of proliferation, DCA can exhibit effects apparent only in vivo, as it suppresses angiogenesis by reversing the pseudohypoxic state caused by activated PDK. By promoting the decarboxylation of pyruvate into acetyl-CoA, DCA drives the Krebs’ cycle to produce αKG, as well as NADH and FADH2 to be used in the ETC [10]. This leads to increased mROS, which increases activity of redox sensitive tumor suppressors like p53 [10, 13, 94, 115]. Together, increased p53 activity and increased levels of αKG prevent the stabilization of HIF1α as well as reduce HIF1α transcriptional activity and the expression of downstream HIF1α targets [10, 119]. DCA reduces the levels of angiogenic signaling molecules such as VEGF and SDF-1 and prevents neovascularization both in vitro and in xenotransplant tumor models [10]. In addition, inhibition of PDK using RNA interference has recently been shown to promote oncogene-induced senescence in melanoma in vitro and in vivo, providing another mechanism through which DCA may be exerting antitumor effects [120].
DCA has had success in early-phase small clinical trials for glioblastoma (GBM) and recurrent brain tumors [94, 121]. Compared to healthy brain tissue removed during surgery for epilepsy, tumors from 49 patients with GBM exhibited significantly higher levels of PDK and hyperpolarized ΔΨm. Treatment of five patients with DCA (for which brain tissue was removed at the time of debulking surgery at baseline as well as after DCA treatment, allowing direct pre-post comparisons) caused mitochondrial depolarization, increased rates of apoptosis, activated p53, reduced proliferation, and inhibited HIF1α activity and tumor vascularity [94]. Despite the very advanced stage of their disease, some patients showed evidence of tumor regression whereas others remained clinically stable for >18 months (Fig. 4b). No patient developed hematologic, hepatic, renal, or cardiac toxicity. Peripheral neuropathy developed in some patients but reversed at a lower dose of DCA. Similar results, supporting the safety of the drug and the need for phase II trials in glioblastoma, were confirmed by another phase I trial performed by an independent group, demonstrating clinically stable disease with no significant side effects beyond peripheral neuropathy, which when it occurs is dose-dependent and reversible [121]. Recently, the University of Florida group published their experience with DCA in children that were treated with DCA in their group continuously from 9.7 to 16.5 years at a dose of 12.5 mg/kg two times a day (i.e., higher or equal to the doses used in the glioblastoma trials in humans). They reported no hematological, electrolyte, renal, or hepatic toxicity, with the only toxicity being a reversible and dose-dependent peripheral neuropathy (treated with dose reduction or only temporary discontinuation of DCA) [122].
Although the initial half-life of DCA is very short (i.e., approximately 2 h) [96], the drug inhibits its own metabolism until it reaches a plateau, and thus therapeutic concentrations can be achieved in plasma with time (for example, at a dosing regimen of 6.25 mg/kg two times a day for 3 months) [94]. Nevertheless, this may take more than 3 months. Thus, DCA may not be a good choice as a monotherapy in advanced and rapidly proliferating tumors. Like the PKM2 targeting drugs, it is not cytotoxic and could be seen as a drug that “sensitizes” the tumors to apoptosis, perhaps best used as a part of a combination therapy with a more cytotoxic drug at the early stages of treatment.
Another fact that may support DCA’s sustained effects in the long term is its ability to inhibit HIF1α. For example, the early effectiveness of VEGF inhibitors is limited by the eventual escape of the tumor, which, having sustained HIF1α activity, can continue to generate alternate pro-angiogenic factors. Thus, metabolic modulators like DCA that have the ability to inhibit the normoxic activation of HIF1α may offer much more sustained and effective inhibition of angiogenesis [10]. Indeed, synergy between DCA and VEGF inhibitors was recently shown in glioblastoma cancer models [113].
The anticancer efficacy of DCA increases when combined with other agents. Similar to the synergy between DCA and VEGF inhibitors or DCA and temozolomide, a combination agent, called mitaplatin, combines DCA with cisplatin [123]. This unique drug is synthesized by adding two DCA moieties (one onto each end) of a cisplatin core. Once this drug enters a cancer cell, it is reduced to release one molecule of free cisplatin and two molecules of free DCA. The result is the combined effect of DCA on mitochondria (increasing cytochrome c release and apoptosis) as well as cisplatin-mediated DNA crosslinking. Mitaplatin exceeded the anticancer efficacy of cisplatin alone in a variety of cancer cell lines [123]. DCA has also been used in combination with 5-fluorouracil (5-FU) to re-sensitize hypoxic gastric cancer cells that developed resistance to 5-FU monotherapy [111]. Similarly, the combination of DCA with tamoxifen and omeprazole exhibited more potent antitumor activity than those agents alone [117]. Furthermore, other therapeutic modalities, such as external beam radiation, have been found to be efficacious in combination with metabolic modulators. DCA sensitized prostate cancer cells (which were previously resistant due to overexpression of BCL-2), to radiation therapy [107]. Furthermore, in combination with etoposide or irradiation, DCA decreases the apoptosis resistance seen in gliomas compared to treatment with either of these agents alone [124]. These examples suggest that mitochondrial activation may be effective in combination strategies for several tumor types.
DCA’s proven ability to increase the GO/glycolysis ratio in the treated tumors and its ability to decrease HIF1α activity and thus reverse the upregulation of glucose transporters suggest that metabolic imaging, like FDG-PET, may be used to track its effects in vivo, a very desirable tool in drug development (Fig. 4c). Tumor cells, expressing a relative abundance of glucose transporters and glycolytic enzymes take up much more fluorinated deoxyglucose (a metabolite that once uptaken remains trapped intra-cellularly allowing its imaging) than surrounding non-cancerous tissue. In theory, one of the first signs of DCA’s effectiveness in vivo maybe its ability to decrease the FDG uptake under FDG-PET imaging, a possibility that although has been documented in animal models [10], should be systematically pursued in future clinical trials.
Lactate dehydrogenase A (LDHA) inhibition
Suppressed mitochondria in cancer cells force pyruvate to be reduced to lactate in order to allow glycolysis to continue. This is achieved by lactate dehydrogenase (LDH), a tetrameric enzyme that facilitates the recycling of NAD+ from NADH by reducing pyruvate to lactate in the cytoplasm. There are five isoforms of LDH made from differing subunit combinations of the products from two genes, LDHB and LDHA: LDHB expresses a constitutively active form, LDH-H (heart); LDHA is a HIF1α responsive gene that transcribes a more efficient enzyme, LDH-M (muscle) [74, 125] (Fig. 3c). In highly glycolytic tumors, the isoform made exclusively from four subunits of LDH-M, known as LDH5, predominates [126]. Similarly, tumors epigenetically silence the LDHB gene through hypermethylation of its promoter region, thereby further shifting the ratio of LDH toward LDH5 [127]. Indeed, the increased expression of this highly active tetramer is a marker of poor prognosis in multiple malignancies [126, 128, 129]. In tissues where LDH activity is enhanced, its inhibition will facilitate pyruvate’s entry into the mitochondria (assuming that PDH is active), increasing GO and preventing the shift of pyruvate’s metabolism into anabolic precursors.
Indeed, inhibition of LDH5 with short hairpin RNA enhanced respiration and reduced ΔΨm. LDHA knockdown reduced cancer growth rates in vitro and in vivo in animal models [130]. This work led to the development of a small-molecule inhibitor of LDH, FX11, which was shown to be effective in animal models of lymphoma and pancreatic cancer [131]. Another class of LDH5 competitive inhibitors, N-hydroxy-2-carboxy-substituted indoles, called NHI-1 and NHI-2, has been developed [132, 133]. These more specific and efficient LDH5 inhibitors decrease lactate production and reduce proliferation in multiple cancer cells lines.
Recently, 13C-labeled magnetic resonance spectroscopy has been adapted to follow dynamic metabolic conversions in vivo [134]. This imaging biomarker assesses real-time changes in intracellular metabolism such as decreased reduction of pyruvate to lactate in response to drugs like LDH inhibitors or DCA [135, 136].
Mutant isocitrate dehydrogenase (IDH) inhibition
As discussed above, mutant IDH leads to pseudo-hypoxic signaling, due to the production of 2-HG (Fig. 3d). Recently, pharmacological inhibition of IDH has been explored. Several small-molecule inhibitors that specifically inhibit the mutant form of IDH have been developed [137, 138]. Discovered through high-throughput screening, AGI-5198 inhibits the production of 2-HG by mutant IDH1 while AGI-6780 inhibits 2-HG production by mutant IDH2. This inhibition appears highly specific for the mutant isoform as AGI-5198 impairs only the growth of IDH1 mutant but not IDH1 wild-type glioma xenotransplant tumors [138].
2-HG-producing tumors cause exponentially higher levels of this metabolite in the patient’s circulation [63]. Fathi et al. have taken advantage of this unique cancer metabolite, to non-invasively diagnose and subsequently follow response to treatment in AML by detecting the levels of 2-HG in patients’ serum and urine samples before and during therapy [139]. While IDH inhibitors have yet to be tested in humans, this powerful biomarker will facilitate IDH inhibitor development in clinical trials.
Cancer stem cells
Mitochondrial metabolism also determines stem cell fate. Temporally, a switch to glycolysis precedes expression of stem cell markers and subsequent entry into a pluripotent state [140]. Conversely, an increase in mitochondrial glucose oxidation is necessary for initiating stem cell differentiation [141]. Normal stem cells, similar to cancer cells, exhibit increased LDHA, HKII, PDK, and phosphorylated PDH, compared to differentiated progeny, leading to mitochondrial suppression [142]. This mitochondrial suppression is further exacerbated in hypoxic tumor cells and cancer stem cells. For example, when lung cancer stem cells (LCSCs) are directly compared to differentiated lung cancer cells, LCSCs demonstrate lower oxygen consumption, mROS, mitochondrial numbers, and ATP levels, as well as higher mitochondrial membrane potential [143]. Similarly, when studied in parallel, healthy brain tissue, glioblastoma cancer cells, and glioblastoma putative cancer stem cells exhibit a graded increase in mitochondrial suppression, with the highest levels of ΔΨm seen in the cancer stem cells [94]. Mechanistically, overwhelming mitochondrial suppression in cancer stem cells provides significant resistance to apoptosis, potentially contributing to the cancer stem cells’ resistance to conventional chemotherapeutics.
Activating mitochondria unlocks apoptosis resistance in cancer stem cells. For example, tumor biopsies from GBM patients treated with DCA found induction of apoptosis in glioblastoma putative cancer stem cells, particularly when used in combination with temozolomide in vivo and in vitro [94]. Similarly, DCA reduces cancer stem cell viability in embryonal cancer stem cells when the substrate for PDH, pyruvate, is available [144].
Despite the induction of apoptosis in cancer stem cells, there is evidence that DCA exhibits specificity for cancer stem cells and not healthy stem cells. For example, patients treated with long-term (up to 16 years) DCA did not suffer any hematological side effects, suggesting lack of effects on bone marrow stem cells [122]. Yet, it is possible that more potent mitochondrial activators may have this problem, an important issue that has to be addressed in the future with long-term studies. On the other hand, metabolic disturbances may affect the ability of stem cells to differentiate and, in the case of IDH mutations in leukemic cells, there is evidence that they may impair the ability of hematopoietic stem cells to differentiate, resulting in leukemias that mimic a difficult to treat, hematopoietic stem cell phenotype [145].
Glucose oxidation and histone acetylation
Histone acetylation has received a lot of attention in cancer research [146]. As the source of the acetyl group is acetyl-CoA (a prime mitochondrial product), it is possible that the mitochondria suppression discussed herein may actually also impact epigenetic mechanisms. Isolated nuclei exposed to acetyl-CoA exhibit increased histone acetylation [147]. Intriguingly, the acetyl-CoA molecule is extremely unstable and has to be used in the organelle that is produced. In other words, acetyl-CoA cannot simply leak out of mitochondria and enter the nucleus. Recent work, however, has shown two mechanisms by which mitochondria can regulate histone acetylation:
-
(a)
PDH activity promotes citrate production in the Krebs’ cycle, which can diffuse out of the mitochondria and into the nucleus to be used as a substrate to acetylate histones by the enzyme ATP-citrate lyase, which is present both in the cytoplasm and the nucleus [148]. Thus, a primary inhibition of PDH will also result eventually in a suppression of citrate production and histone acetylation, unless citrate can be replenished by an alternate pathway like the reductive glutamine pathway, which can produce citrate in the cytoplasm from the amino acid glutamine [149].
-
(b)
We recently described an alternate way of nuclear production of acetyl-CoA by showing that PDH can actually translocate into the nucleus in a cell-cycle-dependent manner [150]. Interestingly, several subunits of the PDH complex, E1a and E2, had previously been shown to be present in the nucleus of leukemic T cells, although at the time their presence was not linked to the main function of PDH, i.e., production of acetyl-CoA [151]. We found that nuclear PDH (which included all subunits of the complex), although in small amounts, is functional, providing a source of acetyl-CoA to be used to acetylate specific histone residues involved in cell cycle progression [150]. Intriguingly, PDK does not follow PDH in the nucleus (potentially being displaced from its binding site on the E2 subunit by HSP70, which then transports a “PDK-free” HSP70-PDH complex to the nucleus), suggesting that nuclear PDH will be immune to DCA and perhaps represent a potential “escape” mechanism to DCA treatment (Fig. 5) [150].
Fig. 5 
Nuclear translocation of the PDH Complex (PDC) provides Acetyl-CoA for histone acetylation. PDC dynamically translocates from the mitochondria to the nucleus in response to growth factors, like epidermal growth factor (EGF), to provide acetyl-CoA for histone acetylation and cell cycle progression

The fact that translocation of such a large enzyme like PDH takes place between mitochondria and the nucleus is not as surprising as one may first think. For example, while mitochondrial PDH has been classically thought to be localized in the mitochondrial matrix, it has also been shown to move through the mitochondrial inner membranes and remain functional in the outer mitochondrial membrane [27]. This is in keeping with our finding that the chaperone HSP70 may be involved in its nuclear translocation since it may easily reach PDH on the outer mitochondrial membrane [150]. The role of nuclear PDH is not entirely clear but suggests that it may provide a critical regulatory mechanism for gene expression by mediating a shift from heterochromatin to euchromatin thereby facilitating transcription factor binding. Identifying a mechanism by which this translocation is blocked may represent a new means of cancer therapy, merging the fields of metabolism and epigenetic regulation therapeutics.
Conclusion
Suppressed mitochondrial function and in particular GO appears to be a universal feature of cancer, giving cancer cells a proliferative advantage, while simultaneously repressing apoptosis. A multitude of mechanisms underlie this mitochondrial remodeling, exposing a number of novel therapeutic targets. Although not cytotoxic, limiting their use as monotherapies in rapidly growing tumors, these metabolic modulators appear to be selective to the tumor. Metabolic oncology also has the potential to utilize a number of unique metabolism-based biomarkers. Multiple small molecules targeting both cytoplasmic and mitochondrial enzymes involved in cancer metabolism, such as PKM2, PDK, IDH, and LDH5, are currently in use in preclinical and early phase human clinical trials, a remarkable achievement of translational oncology that was essentially born less than 10 years ago.
References
Le Tourneau C, Lee JJ, Siu LL (2009) Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst 101:708–720
Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S et al (2001) Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 344:1031–1037
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507
Loureiro R, Mesquita KA, Oliveira PJ, Vega-Naredo I (2013) Mitochondria in cancer stem cells: a target for therapy. Recent Pat Endocr Metab Immune Drug Discov 7:102–114
Warburg O (1923) Metabolism of tumours. Biochem Zeitschr 142:317–333
Loenarz C, Schofield CJ (2008) Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol 4:152–156
Schmid T, Zhou J, Kohl R, Brune B (2004) p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem J 380:289–295
Vousden KH, Ryan KM (2009) p53 and metabolism. Nat Rev Cancer 9:691–700
Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS, Michelakis ED (2012) Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 32:1638–1650
Maddocks OD, Vousden KH (2011) Metabolic regulation by p53. J Mol Med (Berl) 89:237–245
Dromparis P, Michelakis ED (2013) Mitochondria in vascular health and disease. Annu Rev Physiol 75:95–126
Sutendra G, Michelakis ED (2013) Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front Oncol 3:38
Zamzami N, Kroemer G (2001) The mitochondrion in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol 2:67–71
Chen LB (1988) Mitochondrial membrane potential in living cells. Annu Rev Cell Biol 4:155–181
Lemasters JJ, Holmuhamedov E (2006) Voltage-dependent anion channel (VDAC) as mitochondrial governator—thinking outside the box. Biochim Biophys Acta 1762:181–190
Pastorino JG, Hoek JB, Shulga N (2005) Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res 65:10545–10554
Vander Heiden MG, Cantley LC, Thompson CB (2009) Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033
Butow RA, Avadhani NG (2004) Mitochondrial signaling: the retrograde response. Mol Cell 14:1–15
Esteves P, Pecqueur C, Ransy C, Esnous C, Lenoir V, Bouillaud F, Bulteau AL, Lombes A, Prip-Buus C, Ricquier D et al (2014) Mitochondrial retrograde signaling mediated by UCP2 inhibits cancer cell proliferation and tumorigenesis. Cancer Res 74:3971–3982
Wallace DC (2012) Mitochondria and cancer. Nat Rev Cancer 12:685–698
Semenza GL (2010) HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20:51–56
Denko NC (2008) Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8:705–713
Yu F, White SB, Zhao Q, Lee FS (2001) HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc Natl Acad Sci U S A 98:9630–9635
Ke Q, Costa M (2006) Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol 70:1469–1480
Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4:891–899
Hitosugi T, Fan J, Chung TW, Lythgoe K, Wang X, Xie J, Ge Q, Gu TL, Polakiewicz RD, Roesel JL et al (2011) Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol Cell 44:864–877
Korotchkina LG, Patel MS (2001) Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J Biol Chem 276:5731–5738
Fan J, Kang HB, Shan C, Elf S, Lin R, Xie J, Gu TL, Aguiar M, Lonning S, Chung TW et al (2014) Tyr-301 phosphorylation inhibits pyruvate dehydrogenase by blocking substrate binding and promotes the Warburg effect. J Biol Chem 289:26533–26541
Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177–185
Franovic A, Gunaratnam L, Smith K, Robert I, Patten D, Lee S (2007) Translational up-regulation of the EGFR by tumor hypoxia provides a nonmutational explanation for its overexpression in human cancer. Proc Natl Acad Sci U S A 104:13092–13097
Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA (1999) Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 48:1593–1599
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA et al (2013) Mutational landscape and significance across 12 major cancer types. Nature 502:333–339
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126:107–120
Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D (2005) Glycolytic enzymes can modulate cellular life span. Cancer Res 65:177–185
Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM (2006) p53 regulates mitochondrial respiration. Science 312:1650–1653
Contractor T, Harris CR (2012) p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res 72:560–567
Mathupala SP, Heese C, Pedersen PL (1997) Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem 272:22776–22780
Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol 15:536–550
Dang CV, Semenza GL (1999) Oncogenic alterations of metabolism. Trends Biochem Sci 24:68–72
Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV (1997) c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A 94:6658–6663
Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV (2004) Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol Cell Biol 24:5923–5936
Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, Horvath TL, Sinclair DA, Pfluger PT, Tschop MH (2012) Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev 92:1479–1514
He W, Newman JC, Wang MZ, Ho L, Verdin E (2012) Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol Metab: TEM 23:467–476
Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM et al (2010) SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 17:41–52
Guarente L (2014) The many faces of Sirtuins: Sirtuins and the Warburg effect. Nat Med 20:24–25
Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB et al (2011) SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 19:416–428
Denton RM (2009) Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta 1787:1309–1316
Dromparis P, Paulin R, Sutendra G, Qi AC, Bonnet S, Michelakis ED (2013) Uncoupling protein 2 deficiency mimics the effects of hypoxia and endoplasmic reticulum stress on mitochondria and triggers pseudohypoxic pulmonary vascular remodeling and pulmonary hypertension. Circ Res 113:126–136
Derdak Z, Fulop P, Sabo E, Tavares R, Berthiaume EP, Resnick MB, Paragh G, Wands JR, Baffy G (2006) Enhanced colon tumor induction in uncoupling protein-2 deficient mice is associated with NF-kappaB activation and oxidative stress. Carcinogenesis 27:956–961
Archer SL (2013) Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med 369:2236–2251
Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J (2009) A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A 106:11960–11965
Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, Salgia R, Husain AN, Wietholt C, Archer SL (2012) Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J: Off Publ Fed Am Soc Exp Biol 26:2175–2186
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S et al (2002) Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30:406–410
Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA et al (2004) Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. Jama 292:943–951
Baysal BE (2003) On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab: TEM 14:453–459
Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, Merino M, Trepel J, Zbar B, Toro J et al (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8:143–153
King A, Selak MA, Gottlieb E (2006) Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25:4675–4682
Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E (2005) Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7:77–85
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL et al (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD et al (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361:1058–1066
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE et al (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17:225–234
Rose NR, McDonough MA, King ON, Kawamura A, Schofield CJ (2011) Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev 40:4364–4397
Guan KL, Xiong Y (2011) Regulation of intermediary metabolism by protein acetylation. Trends Biochem Sci 36:108–116
Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H et al (2010) Regulation of cellular metabolism by protein lysine acetylation. Science 327:1000–1004
Devi MA, Das NP (1993) In vitro effects of natural plant polyphenols on the proliferation of normal and abnormal human lymphocytes and their secretions of interleukin-2. Cancer Lett 69:191–196
Kobori M, Shinmoto H, Tsushida T, Shinohara K (1997) Phloretin-induced apoptosis in B16 melanoma 4A5 cells by inhibition of glucose transmembrane transport. Cancer Lett 119:207–212
Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P (2011) Anticancer targets in the glycolytic metabolism of tumors: a comprehensive review. Front Pharmacol 2:49
Tennant DA, Duran RV, Gottlieb E (2010) Targeting metabolic transformation for cancer therapy. Nat Rev Cancer 10:267–277
Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL (2012) A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J Bioenerg Biomembr 44:163–170
Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, Hullihen J, Pedersen PL (2004) Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun 324:269–275
Biolato M, Marrone G, Racco S, Di Stasi C, Miele L, Gasbarrini G, Landolfi R, Grieco A (2010) Transarterial chemoembolization (TACE) for unresectable HCC: a new life begins? Eur Rev Med Pharmacol Sci 14:356–362
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB (2007) Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 104:19345–19350
Hutson SM, Sweatt AJ, Lanoue KF (2005) Branched-chain [corrected] amino acid metabolism: implications for establishing safe intakes. J Nutr 135:1557S–1564S
Gupta V, Bamezai RN (2010) Human pyruvate kinase M2: a multifunctional protein. Protein Sci 19:2031–2044
Takenaka M, Noguchi T, Sadahiro S, Hirai H, Yamada K, Matsuda T, Imai E, Tanaka T (1991) Isolation and characterization of the human pyruvate kinase M gene. Eur J Biochem 198:101–106
Noguchi T, Inoue H, Tanaka T (1986) The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem 261:13807–13812
Munoz ME, Ponce E (2003) Pyruvate kinase: current status of regulatory and functional properties. Comp Biochem Physiol B Biochem Mol Biol 135:197–218
Wong N, De Melo J, Tang D (2013) PKM2, a central point of regulation in cancer metabolism. Int J Cell Biol 2013:242513
Imamura K, Tanaka T (1972) Multimolecular forms of pyruvate kinase from rat and other mammalian tissues. I electrophoretic studies. J Biochem 71:1043–1051
Mazurek S (2011) Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol 43:969–980
Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, Hong BS, Tempel W, Dimov S, Shen M, Jha A et al (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8:839–847
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452:230–233
Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y et al (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 42:719–730
Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, et al (2009) Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2: ra73
Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC (2008) Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452:181–186
Ashizawa K, Willingham MC, Liang CM, Cheng SY (1991) In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J Biol Chem 266:16842–16846
Israelsen WJ, Dayton TL, Davidson SM, Fiske BP, Hosios AM, Bellinger G, Li J, Yu Y, Sasaki M, Horner JW et al (2013) PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155:397–409
Walsh MJ, Brimacombe KR, Anastasiou D, Yu Y, Israelsen WJ, Hong BS, Tempel W, Dimov S, Veith H, Yang H, et al (2010) ML265: A potent PKM2 activator induces tetramerization and reduces tumor formation and size in a mouse xenograft modelProbe Reports from the NIH Molecular Libraries Program, Bethesda (MD)
Parnell KM, Foulks JM, Nix RN, Clifford A, Bullough J, Luo B, Senina A, Vollmer D, Liu J, McCarthy V et al (2013) Pharmacologic activation of PKM2 slows lung tumor xenograft growth. Mol Cancer Ther 12:1453–1460
Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99:989–994
Sutendra G, Michelakis ED (2014) The metabolic basis of pulmonary arterial hypertension. Cell Metab 19:558–573
Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2: 31ra34
Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 329(Pt 1):191–196
Stacpoole PW (1989) The pharmacology of dichloroacetate. Metabolism 38:1124–1144
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G et al (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11:37–51
Stacpoole PW, Moore GW, Kornhauser DM (1978) Metabolic effects of dichloroacetate in patients with diabetes mellitus and hyperlipoproteinemia. N Engl J Med 298:526–530
Stacpoole PW, Wright EC, Baumgartner TG, Bersin RM, Buchalter S, Curry SH, Duncan CA, Harman EM, Henderson GN, Jenkinson S et al (1992) A controlled clinical trial of dichloroacetate for treatment of lactic acidosis in adults. The Dichloroacetate-Lactic Acidosis Study Group. N Engl J Med 327:1564–1569
Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN et al (2006) Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117:1519–1531
McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED (2004) Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 95:830–840
Krishna S, Supanaranond W, Pukrittayakamee S, Kuile FT, Ruprah M, White NJ (1996) The disposition and effects of two doses of dichloroacetate in adults with severe falciparum malaria. Br J Clin Pharmacol 41:29–34
Bersin RM, Wolfe C, Kwasman M, Lau D, Klinski C, Tanaka K, Khorrami P, Henderson GN, de Marco T, Chatterjee K (1994) Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J Am Coll Cardiol 23:1617–1624
Calvert LD, Shelley R, Singh SJ, Greenhaff PL, Bankart J, Morgan MD, Steiner MC (2008) Dichloroacetate enhances performance and reduces blood lactate during maximal cycle exercise in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 177:1090–1094
Stacpoole PW (1969) Review of the pharmacologic and therapeutic effects of diisopropylammonium dichloroacetate (DIPA). J Clin Pharmacol J New Drugs 9:282–291
Holloway PA, Knox K, Bajaj N, Chapman D, White NJ, O'Brien R, Stacpoole PW, Krishna S (1995) Plasmodium berghei infection: dichloroacetate improves survival in rats with lactic acidosis. Exp Parasitol 80:624–632
Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 68:1223–1231
Sanchez-Arago M, Chamorro M, Cuezva JM (2010) Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 31:567–576
Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG (2010) Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer 102:1746–1752
Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D et al (2012) The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 151:1185–1199
Xuan Y, Hur H, Ham IH, Yun J, Lee JY, Shim W, Bae Kim Y, Lee G, Han SU, Kwan Cho Y (2013) Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res 321(2):219–230
Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 109:394–402
Kumar K, Wigfield S, Gee HE, Devlin CM, Singleton D, Li JL, Buffa F, Huffman M, Sinn AL, Silver J et al (2013) Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J Mol Med (Berl) 91:749–758
Vella S, Conti M, Tasso R, Cancedda R, Pagano A (2012) Dichloroacetate inhibits neuroblastoma growth by specifically acting against malignant undifferentiated cells. Int J Cancer 130:1484–1493
Kumar A, Kant S, Singh SM (2012) Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma: Implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem Biol Interact 199:29–37
Flavin DF (2010) Non-Hodgkin’s lymphoma reversal with dichloroacetate. J Oncol 2010
Ishiguro T, Ishiguro R, Ishiguro M, Iwai S (2012) Co-treatment of dichloroacetate, omeprazole and tamoxifen exhibited synergistically antiproliferative effect on malignant tumors: in vivo experiments and a case report. Hepatogastroenterology 59:994–996
Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2010) Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 120:253–260
Kaluzova M, Kaluz S, Lerman MI, Stanbridge EJ (2004) DNA damage is a prerequisite for p53-mediated proteasomal degradation of HIF-1alpha in hypoxic cells and downregulation of the hypoxia marker carbonic anhydrase IX. Mol Cell Biol 24:5757–5766
Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T et al (2013) A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498:109–112
Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW (2013) Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs 32(2):452–464
Abdelmalak M, Lew A, Ramezani R, Shroads AL, Coats BS, Langaee T, Shankar MN, Neiberger RE, Subramony SH, Stacpoole PW (2013) Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab 109:139–143
Dhar S, Lippard SJ (2009) Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc Natl Acad Sci U S A 106:22199–22204
Morfouace M, Lalier L, Bahut M, Bonnamain V, Naveilhan P, Guette C, Oliver L, Gueguen N, Reynier P, Vallette FM (2012) Comparison of spheroids formed by rat glioma stem cells and neural stem cells reveals differences in glucose metabolism and promising therapeutic applications. J Biol Chem 287:33664–33674
Markert CL, Shaklee JB, Whitt GS (1975) Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 189:102–114
Koukourakis MI, Giatromanolaki A, Sivridis E, Bougioukas G, Didilis V, Gatter KC, Harris AL (2003) Lactate dehydrogenase-5 (LDH-5) overexpression in non-small-cell lung cancer tissues is linked to tumour hypoxia, angiogenic factor production and poor prognosis. Br J Cancer 89:877–885
Leiblich A, Cross SS, Catto JW, Phillips JT, Leung HY, Hamdy FC, Rehman I (2006) Lactate dehydrogenase-B is silenced by promoter hypermethylation in human prostate cancer. Oncogene 25:2953–2960
Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Trarbach T, Folprecht G, Shi MM, Lebwohl D, Jalava T, Laurent D et al (2011) Prognostic and predictive role of lactate dehydrogenase 5 expression in colorectal cancer patients treated with PTK787/ZK 222584 (vatalanib) antiangiogenic therapy. Clin Cancer Res 17:4892–4900
Koukourakis MI, Giatromanolaki A, Winter S, Leek R, Sivridis E, Harris AL (2009) Lactate dehydrogenase 5 expression in squamous cell head and neck cancer relates to prognosis following radical or postoperative radiotherapy. Oncology 77:285–292
Fantin VR, St-Pierre J, Leder P (2006) Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9:425–434
Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV (2010) Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A 107:2037–2042
Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, Lanza M, Betti L, Giannaccini G, Lucacchini A et al (2011) Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J Med Chem 54:1599–1612
Maftouh M, Avan A, Sciarrillo R, Granchi C, Leon LG, Rani R, Funel N, Smid K, Honeywell R, Boggi U et al (2014) Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br J Cancer 110(1):172–182
Golman K, Zandt RI, Lerche M, Pehrson R, Ardenkjaer-Larsen JH (2006) Metabolic imaging by hyperpolarized 13C magnetic resonance imaging for in vivo tumor diagnosis. Cancer Res 66:10855–10860
Dutta P, Le A, Vander Jagt DL, Tsukamoto T, Martinez GV, Dang CV, Gillies RJ (2013) Evaluation of LDH-A and glutaminase inhibition in vivo by hyperpolarized 13C-pyruvate magnetic resonance spectroscopy of tumors. Cancer Res 73:4190–4195
Hill DK, Orton MR, Mariotti E, Boult JK, Panek R, Jafar M, Parkes HG, Jamin Y, Miniotis MF, Al-Saffar NM et al (2013) Model free approach to kinetic analysis of real-time hyperpolarized 13C magnetic resonance spectroscopy data. PLoS One 8:e71996
Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C et al (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340:622–626
Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E et al (2013) An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 340:626–630
Fathi AT, Sadrzadeh H, Borger DR, Ballen KK, Amrein PC, Attar EC, Foster J, Burke M, Lopez HU, Matulis CR et al (2012) Prospective serial evaluation of 2-hydroxyglutarate, during treatment of newly diagnosed acute myeloid leukemia, to assess disease activity and therapeutic response. Blood 120:4649–4652
Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A (2011) Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14:264–271
Xu X, Duan S, Yi F, Ocampo A, Liu GH, Izpisua Belmonte JC (2013) Mitochondrial regulation in pluripotent stem cells. Cell Metab 18:325–332
Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CA, Ramalho-Santos J, Van Houten B, Schatten G (2011) Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 6:e20914
Ye XQ, Li Q, Wang GH, Sun FF, Huang GJ, Bian XW, Yu SC, Qian GS (2011) Mitochondrial and energy metabolism-related properties as novel indicators of lung cancer stem cells. Int J Cancer 129:820–831
Vega-Naredo I, Loureiro R, Mesquita KA, Barbosa IA, Tavares LC, Branco AF, Erickson JR, Holy J, Perkins EL, Carvalho RA et al (2014) Mitochondrial metabolism directs stemness and differentiation in P19 embryonal carcinoma stem cells. Cell Death Differ 21:1560–1574
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18:553–567
Minucci S, Pelicci PG (2006) Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6:38–51
Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S et al (2014) Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 20(2):306–319
Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324:1076–1080
Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ (2012) Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481:385–388
Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED (2014) A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 158:84–97
Chueh FY, Leong KF, Cronk RJ, Venkitachalam S, Pabich S, Yu CL (2011) Nuclear localization of pyruvate dehydrogenase complex-E2 (PDC-E2), a mitochondrial enzyme, and its role in signal transducer and activator of transcription 5 (STAT5)-dependent gene transcription. Cell Signal 23:1170–1178
Funding
Adam Kinnaird is a Vanier Scholar supported by the Canadian Institutes of Health Research (CIHR). Evangelos Michelakis is supported by CIHR and the Canada Research Chair Program.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kinnaird, A., Michelakis, E.D. Metabolic modulation of cancer: a new frontier with great translational potential. J Mol Med 93, 127–142 (2015). https://doi.org/10.1007/s00109-014-1250-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-014-1250-2