
Abstract
For much of the past 20 years, MDM2 has been pursued as a cancer therapeutic target. Small molecule inhibitors that block the MDM2-p53 protein-protein interaction (MDM2 inhibitors) have been developed and a number of them have been evaluated in clinical trials for cancer treatment. Notwithstanding various setbacks, several MDM2 inhibitors have now progressed into late-stage clinical development. New strategies have also been developed to enhance the efficacy of MDM2 inhibitors and to mitigate their on-target toxicity. In this review, we summarize the progress and challenges in the development of a MDM2 targeted therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
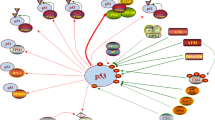
The MDM2 protein is the primary cellular inhibitor and regulator of the tumor suppressor, p53 (Fig. 1) [1,2,3,4,5,6,7]. MDM2 binds directly to p53 and inhibits p53 functions via several distinct mechanisms all of which are mediated by their direct binding (Fig. 1). By binding to the transactivation domain of p53, MDM2 blocks the binding of p53 to its target DNAs, thus rendering p53 ineffective as a transcriptional factor for gene transcription. Functioning as an E3 ligase, MDM2 induces ubiquitination and subsequent degradation of the p53 protein. Additionally, the binding of MDM2 to p53 promotes translocation of p53 from the nucleus to the cytoplasm, making p53 inaccessible to its targeted DNAs. MDM2 is also directly regulated by p53 and upon the activation of p53, p53 induces transcription of MDM2 by functioning as a transcriptional factor. Thus, through the auto-regulatory loop, p53 and MDM2 proteins are maintained at low levels in normal cells under unstressed conditions. In cancer cells harboring wild-type p53 but with overexpression of MDM2 caused by either amplification of the MDM2 gene or by other mechanisms, MDM2 severely suppresses the activity of p53, and this allows cancer cells to evade the p53 surveillance and the powerful tumor suppressor activity of p53. Based on this rationale, reactivation of p53 by inhibition of MDM2 has been pursued as a cancer therapeutic strategy [1,2,3,4,5,6,7].

The interplay between p53 tumor suppressor and MDM2 oncoprotein
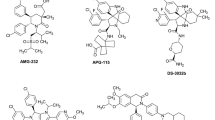
The co-crystal structure of MDM2 in a complex with a p53 peptide has revealed that their interaction is mediated by Phe19, Trp23 and Leu26 residues of p53 and a well-defined binding pocket in MDM2 [8]. This co-crystal structure suggested the possibility for the development of a non-peptide small molecule that could block the MDM2-p53 interaction. In 2004, scientists from Roche reported the discovery of Nutlins, compounds that comprise the first-in-class potent and selective MDM2 inhibitors with strong in vivo antitumor activity [9]. Subsequently, Wang et al. at the University of Michigan published a number of de novo designed spiro-oxindoles as a new class of MDM2 inhibitors [10]. These compounds have inspired the development of several classes of highly potent and druglike MDM2 inhibitors suitable for clinical development. Since 2007, a number of non-peptide small molecule MDM2 inhibitors have been advanced into clinical development [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26] and they are summarized in Fig. 2.

Non-peptide small molecule MDM2 inhibitors that have progressed into clinical development
Data from early clinical trials and lessons learned
RG7112 from Roche was the first MDM2 inhibitor advanced into human clinical development (Fig. 2). This compound was discovered by optimization of Nutlin-3 with the aim of developing compounds with higher binding affinities to MDM2 and suitable pharmacokinetics for clinical development [11]. RG7112 was evaluated against a wide range of cancers, including sarcoma, myelogenous leukemia and hematologic neoplasms [27], and objective clinical activities, including stable disease and partial responses, were observed with RG7112. Serious adverse events including neutropenia and thrombocytopenia in patients treated with RG7112 were noted, and are probably due to the persistent p53 reactivation in bone marrow caused by daily administration of RG7112. Based upon the initial clinical data, Roche decided not to progress RG7112 further, and advanced a second MDM2 inhibitor RG7388 (idasanutlin) (Fig. 2) into clinical development.
Structurally, RG7388 differs significantly from RG7112 and its design [13] was based upon the spiro-oxindole MDM2 inhibitors reported from the University of Michigan. In Phase 1 clinical trials, different dosing schedules were explored for RG7388 [28, 29] and a schedule of 500 mg administered daily, 5-days per week was recommended for Phase 2 clinical trials. Thrombocytopenia, neutropenia, febrile neutropenia and diarrhea were the dose-limiting toxicities [28]. Ultimately, RG7388 in combination with Cytarabine was evaluated in comparison with Cytarabine plus placebo in patients with relapsed or refractory acute myeloid leukemia (R/R AML) in a randomized Phase 3 trial [30]. While adding RG7388 to cytarabine to treat R/R AML improved the overall remission/response rate, it failed to improve the overall survival rate [30]. Consequently, Roche ended the clinical development of RG7388.
SAR405838 (MI-77301) was discovered by Wang et al. at the University of Michigan [12] and was advanced into clinical development by Sanofi. Approximately 90% of patients with de-differentiated liposarcoma (DDLPS) carry MDM2 gene amplification, and a Phase 1 study of SAR405838 was conducted in this patient population [31]. No partial or complete responses were observed [31], and the best responses were stable disease, which was observed in 71% of patients with DDLPS [31]. It was hypothesized that the absence of objective positive responses to SAR405838 may be due in part to emergence of TP53 gene mutations, which were observed in circulating tumor DNA [32]. As in RG7112 and RG7388, thrombocytopenia was the dose-limiting toxicity [31].
These early clinical data for RG7112, RG7388 and SAR405838 suggested the need to develop novel combinations to improve the efficacy and overcome resistance. In addition, mitigation of the on-target, thrombocytopenia dose-limiting toxicities associated with persistent MDM2 inhibition would be highly desirable.
Development of rational combination strategies
Currently, many of the ongoing clinical trials for MDM2 inhibitors is evaluation of their therapeutic performance in combination with a second agent, which are summarized in Table 1.
In preclinical studies, significant synergy was observed for an MDM2 inhibitor in combination with a Bcl-2 inhibitor. Overexpression of Mcl-1 is a major resistance mechanism for a selective Bcl-2 inhibitor. Activation of p53 by an MDM2 inhibitor induces up-regulation of PUMA and Noxa proteins, which are potent and effective cellular antagonists of Mcl-1. Mutations of TP53, the gene encoding p53 protein, can lead to resistance to MDM2 inhibitors, and Bcl-2 inhibitors are equally effective in leukemia cells carrying either wild-type or mutated p53 [33]. Consistently, combinations of MDM2 and Bcl-2 inhibitors can achieve impressive and often synergistic activity in preclinical models [33,34,35,36]. Currently, a number of clinical trials are being carried out to evaluate the combination of an MDM2 inhibitor such as RG7388, Siremadlin or APG-115 with a selective Bcl-2 inhibitor such as Venetoclax (ABT199) or APG-2575 (Table 1).
In recent years, immune-oncology (IO) therapies, either alone or in combinations have become frontline treatments for different types of human cancers. Since p53 plays a major role in the activation of innate antitumor immunity, preclinical studies have been conducted to evaluate the immune response to MDM2 inhibitors in cancer. A preclinical study of the MDM2 inhibitor APG-115 showed that MDM2 inhibition and activation of p53 supports immune regulation in the tumor microenvironment and achieves an antitumor immune response, regardless of the p53 mutation status of the tumors [37]. MDM2 inhibition was shown to modulate the immune response in several ways including shift of macrophage polarization from M2 to M1, enhancement of activation of CD4 + T-cells and upregulation of PD-L1 expression in the tumor cells. Combination of APG-115 with an anti-PD-1 antibody also improved the efficacy when compared to treatment with only the anti-PD-1 antibody [37]. Similarly, preclinical combination studies have shown that combination of HDM201 or BI-907828 with an anti-PD-1 antibody achieved synergistic antitumor activity, but interestingly, only in wild type (wt) TP53 tumors [21, 37, 38]. A recent study showed that MDM2 plays a key role in survival and function of tumor-infiltrating CD8 + T cells [39]. By competing with c-Cbl, an E3 ligase, for binding with STAT5, a transcriptional factor critical for T-cell function, MDM2 reduces c-Cbl-mediated STAT5 degradation and enhances STAT5 stability in tumor-infiltrating CD8 + T-cells. Blocking the MDM2-p53 interaction by an MDM2 inhibitor activates p53 and increases the levels of MDM2 in T-cells, boosting T-cell immunity through stabilization of STAT5 [39]. MDM2 inhibition and cancer immunotherapy are synergistic, regardless of the tumor’s p53 status [39]. These preclinical studies provide a rationale for the clinical combination of MDM2 inhibitors with immune check-point inhibitors such as PD-1 and PD-L1 antibodies.
Currently, a number of clinical trials have been conducted to evaluate MDM2 inhibitors in combination with immune check-point blockades and these are also included in Table 1. In combination with Pembrolizumab (Keytruda), an anti-PD-L1 antibody, APG-115 was evaluated in Phase 2 in various solid tumors in adults and children by Ascentage Pharma. It was reported that the combination of APG-115 with Pembrolizumab was tolerated well and showed antitumor activity in several of the tumor types including those of patients with IO drug-resistant or recurrent melanoma. The results included two complete responses (CR), an 11% overall-response rate (ORR) and a disease-control rate (DCR) of 57% [40]. The same group reported a DCR of 50% in malignant peripheral nerve sheath tumor (MPNST), a rare pediatric sarcoma, that currently has no effective treatment options [40]. BI-907828 is also being evaluated in combination with a PD1 blockade and a LAG-3 blockade, a new type of immune checkpoint inhibitor (NCT03964233 in Table 1). AMG-232 is also being evaluated in combination with anti-PD1 and anti-PD-L1 antibodies (NCT05705466, NCT03787602 in Table 1).
In addition to the combinations with Bcl-2 inhibitors and immune checkpoint blockades, MDM2 inhibitors are also being evaluated in combination with traditional chemotherapeutic agents, DNA-damaging agents and targeted kinase inhibitors [41].
Intermittent high dosing strategy to mitigate on-target toxicity
Thrombocytopenia is a dose-limiting toxicity of MDM2 inhibitors in clinical trials, and is attributed to persistent, continuous activation of p53 in the bone marrow. It has been proposed that the use of an intermittent high dose administration regimen for an MDM2 inhibitor would allow time between doses for hematologic recovery, therefore reducing the toxicity of on-target thrombocytopenia. Using HDM201, preclinically in mouse and rat models, it has been shown that continuous daily dosing versus pulsed/intermittent high dosing regimens produces different p53 responses [18]. Intermittent administration of high doses of HDM201 induces higher levels of activated p53 and leads to a faster and more persistent antitumor response resulting from the high extent of induction of PUMA. Low continuous administration on the other hand, causes lower levels of activated p53 and initially induces cell cycle arrest and subsequent onset of apoptosis after multiple doses. Although continuous administration caused tumor regression after several doses, the tumors rebounded more rapidly. The use of weaker, less optimized MDM2 inhibitors such as CGM097 failed, revealing a potency threshold beyond which MDM2 inhibitors produce robust antitumor activity with intermittent dosing regimens. In a phase 1 clinical trial (NCT02143635, Table 1) of HDM201, in which the continuous administration of low doses and high dose intermittent administration regimens was conducted, the intermittent high dosing regimen showed higher levels of GDF-15, a protein secreted in response to activated p53, which served as a p53 activation biomarker in patient serum [18]. Such intermittent administration of MDM2 inhibitors thus appears to provide an opportunity to allow hematological recovery between doses and improved tolerability for patients, while achieving robust antitumor activity.
BI-907828 is a potent MDM2 inhibitor developed by Boehringer Ingelheim (BI) (Fig. 2). Early clinical data suggested that BI-907828 has a T1/2 in patients of 27.9–59.4 h, making it suitable for intermittent administration [42]. Preliminary results of a Phase 1 trial (NCT03449381, Table 1) of treatment with intermittent administration of BI-907828 (once every 3 weeks, or Q3W) in patients with advanced solid tumors were reported at the 2022 American Society of Clinical Oncology Annual Meeting (ASCO 2022). This trial showed partial responses (PR) or stable disease (SD) in 88.9% of patients with DDLPS and in 92.9% of patients with well-differentiated LPS. The progression-free survival for patients with LPS was over 10.5 months and all patients who achieved an objective response had MDM2 amplifications. Based upon these encouraging Phase 1 clinical data, a Phase 2/3 clinical trial (Brightline-1, NCT05218499, Table 1) is being conducted to determine whether intermittently administered BI-907828 at 45 or 60 mg (Q3W) is better than doxorubicin as first-line systemic treatment for advanced or metastatic DDLPS based on progression-free survival (PFS).
Milademetan (DS-3032) is an MDM2 inhibitor developed by Daiichi-Sankyo and acquired by Rain therapeutics. In its first-in-human phase 1 study in patients with advanced liposarcoma, solid tumors, or lymphomas it was found that extended or continuous dose schedules led to unfavorable thrombocytopenia and other hematologic events, similar to those observed with other MDM2 inhibitors. Furthermore, they reported that dose reduction and prolonged dose interruptions were often needed because of the delayed onset of adverse events [43]. As a consequence the dose escalation part of the same study was expanded to explore different intermittent dosing schedules. They determined that an intermittent dosing of 260 mg on days 1-3 and 15-17 every 28 days significantly reduced thrombocytopenia and other hematological events, allowed time for bone marrow recovery, and displayed an elevated serum GDF15 level which is a biomarker of p53 reactivation. As a single-agent, milademetan showed a disease control rate (DCR) of 46% in the overall population and was better in the DDLPS subpopulation with a DCR of 59% with prolonged partial responses (PR) in two patients and a median PFS of 7.2 months [43]. Based upon these encouraging Phase 1 clinical data, a phase 3 clinical trial (MANTRA; RAIN-3201, NCT04979442) is ongoing to compare milademetan versus trabectedin in unresectable or metastatic DDLPS patients with disease progression on prior therapies.
Navtemadlin (AMG-232/KRT-232) is a potent MDM2 inhibitor developed by Amgen and now acquired by Kartos therapeutics. In a first clinical proof-of-concept study of KRT-232 as a monotherapy it was found that an intermittent dose (240 mg on Day 1-7 of 28 day cycle) showed promising efficacy and tolerability in patients with myelofibrosis relapsed or refractory (R/R) to prior JAK inhibitor treatment. Rationale for this new indication came from the observation that MDM2 was overexpressed in circulating CD34+ cells of myelofibrosis patients and 96% of these patients retained wild-type p53 [44]. In the clinic, it was found that the established intermittent dosing yielded a best spleen volume reduction (SVR) ≥ 35% in 16% of patients, best total symptom score (TSS) response >50% in 30% of patients, an 87% reduction of CD34+ cells in peripheral blood at Week 24, and a tolerable safety profile by including prophylaxis for nausea/vomiting [44]. Based on these encouraging results, a Phase 3 clinical trials (BOREAS, NCT03662126) is being conducted evaluating KRT-232 for treatment of myelofibrosis patients who no longer benefit from JAK inhibitor treatment and determine if KRT-232 (240 mg on Day 1-7/28-day cycle) monotherapy is better than best available treatment.
Development of PROTAC® MDM2 degraders
Since 2015, induced target protein degradation by the PROTAC® technology for the discovery and development of new therapies has gained tremendous momentum [45,46,47]. PROTAC® degraders are heterobifunctional small molecules, consisting of a ligand for a protein of interest such as MDM2, a ligand for an E3 ligase or an E3 ligase complex, and a linker to join these two ligands together. Distinct from traditional small-molecule inhibitors, which are occupancy-driven, a PROTAC® degrader achieves protein degradation by an event-driven mechanism. By depleting the POI through degradation, a PROTAC® degrader can also achieve more complete inhibition of all the functions associated with the POI, allowing a degrader molecule to achieve better efficacy than the corresponding inhibitors. Blocking the MDM2-p53 interactions by the traditional small-molecule inhibitors increases the levels of MDM2 protein and attenuates the activation of p53. In contrast, MDM2 degradation induced by a PROTAC® degrader can achieve stronger p53 activation than traditional MDM2 inhibitors. As a result, induced MDM2 degradation by a PROTAC® mechanism represents a new therapeutic strategy with which to target MDM2.
MDM2 PROTAC® degraders have been reported and have demonstrated impressive preclinical activity and efficacy [48,49,50,51,52]. The first MDM2 degraders were reported by the Wang et al. laboratory at the University of Michigan, as exemplified by MD-224 (Fig. 3) [53].

Mechanism of MDM2 degradation induced by a PROTAC® MDM2 degrader
In preclinical studies, by depletion of MDM2, MD-224 induces stronger p53 activation at much lower concentrations than its corresponding MDM2 inhibitor and is >100-times more potent than its corresponding MDM2 inhibitor in cell growth inhibition. MD-224 achieves a more sustained tumor regression at a much lower dose in vivo, even with less frequent administration than its corresponding MDM2 inhibitor [53]. MD-224 is well-tolerated in mice [53].
Additional PROTAC® MDM2 degraders, including WB156, WB214 and XY-27, were subsequently reported (Fig. 4) [54,55,56]. More recently, Kymera Therapeutics has disclosed the development of KT-253 (Table 2) as a PROTAC® MDM2 degrader [57]. In preclinical studies, KT-253 induces MDM2 degradation with DC50 = 0.4 nM and in cell growth inhibition is >200-times more potent than DS-3032 (Fig. 1), an MDM2 inhibitor. In vivo, KT-253, administered Q3W at 1 mg/kg, induces rapid apoptosis in the tumor tissues and achieves sustained tumor regression in MV;411 and RS4;11 mouse xenograft models. It was demonstrated that a single dose of KT-253 at 1 mg/kg in combination with daily administration of Venetoclax achieves complete and sustained tumor regression in the MOLM-13 xenograft model and is superior to KT-253 or Venetoclax alone or a combination of Venetoclax and cytarabine [58]. These preclinical data obtained for KT-253 suggest that intermittent dosing schedules may be suitable for clinical development to achieve strong efficacy with acceptable toxicity profiles for an MDM2 degrader. KT-253 is the first MDM2 degrader, that has entered into clinical development.

Reported PROTAC® MDM2 degraders
Summary
In the last 20 years, targeting of MDM2 has been pursued as a cancer therapeutic strategy and several highly potent and orally bioavailable small-molecule inhibitors of the MDM2-p53 interaction have progressed into clinical development. Data from early clinical trials indicate that on-target toxicities due to persistent p53 activation and emergence of p53 mutations are two of the main factors limiting the clinical benefits of MDM2 inhibitors. This has led to the development of rational combination strategies and an exploration of intermittent dosing regimens. Preclinical studies supported the combinations of MDM2 inhibitors with selective Bcl-2 inhibitors or with immune checkpoint blockades in clinical trials. Initial but encouraging clinical results for the combination of one such MDM2 inhibitor (APG-115) with Pembrolizumab (Keytruda), an anti-PD-L1 antibody have been reported. Intermittent administration of BI-907828, a MDM2 inhibitor with a long half-life, demonstrated highly promising single-agent activity in DDLPS, a sarcoma with 90% of MDM2 gene amplification. This has led to a Phase 2/3 clinical trial, currently in progress, whose goal is evaluation of BI-907828 as a frontline treatment for DDLPS. In addition, by taking advantage of the recent advancement in the PROTAC® technology, highly potent and efficacious MDM2 degraders have been developed and one such compound (KT-253) has entered clinical development.
Therefore, while challenges remain in the continuous development of MDM2-targeted therapies for the treatment of human cancer, it is very possible that an MDM2 target therapy will emerge as a new treatment for human cancer.
References
Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–32.
Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45.
Freedman DA, Wu L, Levine AJ. Functions of the MDM2 oncoprotein. Cell Mol Life Sci. 1999;55:96–107.
Juven-Gershon T, Oren M. Mdm2: the ups and downs. Mol Med. 1999;5:71–83.
Momand J, Wu HH, Dasgupta G. MDM2 - master regulator of the p53 tumor suppressor protein. Gene. 2000;242:15–29.
Bond GL, Hu WW, Levine AJ. MDM2 is a central node in the p53 pathway: 12 years and counting. Curr Cancer Drug Tar. 2005;5:3–8.
Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol. 2007;8:275–83.
Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–53.
Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8.
Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, et al. Structure-based design of potent non-peptide MDM2 inhibitors. J Am Chem Soc. 2005;127:10130–1.
Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med Chem Lett. 2013;4:466–9.
Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014;74:5855–65.
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem. 2013;56:5979–83.
Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem. 2014;57:1454–72.
Rew Y, Sun D. Discovery of a small molecule MDM2 inhibitor (AMG 232) for treating cancer. J Med Chem. 2014;57:6332–41.
Holzer P, Masuya K, Furet P, Kallen J, Valat-Stachyra T, Ferretti S, et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J Med Chem. 2015;58:6348–58.
Holzer P. Discovery of Potent and Selective p53-MDM2 Protein-Protein Interaction Inhibitors as Anticancer Drugs. Chimia (Aarau). 2017;71:716–21.
Jeay S, Ferretti S, Holzer P, Fuchs J, Chapeau EA, Wartmann M, et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the p53-MDM2 Inhibitor HDM201. Cancer Res. 2018;78:6257–67.
Arnhold V, Schmelz K, Proba J, Winkler A, Wunschel J, Toedling J, et al. Reactivating TP53 signaling by the novel MDM2 inhibitor DS-3032b as a therapeutic option for high-risk neuroblastoma. Oncotarget. 2018;9:2304–19.
Aguilar A, Lu J, Liu L, Du D, Bernard D, McEachern D, et al. Discovery of 4-((3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-1’-ethyl-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamido)bicyclo[2.2.2]octane -1-carboxylic Acid (AA-115/APG-115): A Potent and Orally Active Murine Double Minute 2 (MDM2) Inhibitor in Clinical Development. J Med Chem. 2017;60:2819–39.
Rudolph D, Reschke M, Blake S, Rinnenthal J, Wernitznig A, Weyer-Czernilofsky U, et al. BI 907828: A novel, potent MDM2 inhibitor that induces antitumor immunologic memory and acts synergistically with an anti-PD-1 antibody in syngeneic mouse models of cancer. Cancer Res. 2018;78:13.
Cornillie J, Wozniak A, Li H, Gebreyohannes YK, Wellens J, Hompes D, et al. Anti-tumor activity of the MDM2-TP53 inhibitor BI-907828 in dedifferentiated liposarcoma patient-derived xenograft models harboring MDM2 amplification. Clin Transl Oncol. 2020;22:546–54.
Rinnenthal J, Rudolph D, Blake S, Gollner A, Wernitznig A, Weyer-Czernilofsky U, et al. BI 907828: A highly potent MDM2 inhibitor with low human dose estimation, designed for high-dose intermittent schedules in the clinic. Cancer Res. 2018;78:13.
Kang MH, Reynolds CP, Kolb EA, Gorlick R, Carol H, Lock R, et al. Initial Testing (Stage 1) of MK-8242-A Novel MDM2 Inhibitor-by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2016;63:1744–52.
Ravandi F, Gojo I, Patnaik MM, Minden MD, Kantarjian H, Johnson-Levonas AO, et al. A phase I trial of the human double minute 2 inhibitor (MK-8242) in patients with refractory/recurrent acute myelogenous leukemia (AML). Leuk Res. 2016;48:92–100.
Konopleva M, Martinelli G, Daver N, Papayannidis C, Wei A, Higgins B, et al. MDM2 inhibition: an important step forward in cancer therapy. Leukemia. 2020;34:2858–74.
Andreeff M, Kelly KR, Yee K, Assouline S, Strair R, Popplewell L, et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin Cancer Res. 2016;22:868–76.
Yee K, Papayannidis C, Vey N, Dickinson MJ, Kelly KR, Assouline S, et al. Murine double minute 2 inhibition alone or with cytarabine in acute myeloid leukemia: Results from an idasanutlin phase 1/1b study small star, filled. Leuk Res. 2021;100:106489.
Italiano A, Miller WH Jr., Blay JY, Gietema JA, Bang YJ, Mileshkin LR, et al. Phase I study of daily and weekly regimens of the orally administered MDM2 antagonist idasanutlin in patients with advanced tumors. Investig New Drugs. 2021;39:1587–97.
Konopleva MY, Rollig C, Cavenagh J, Deeren D, Girshova L, Krauter J, et al. Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv. 2022;6:4147–56.
de Weger VA, de Jonge M, Langenberg MHG, Schellens JHM, Lolkema M, Varga A, et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br J Cancer. 2019;120:286–93.
Jung J, Lee JS, Dickson MA, Schwartz GK, Le Cesne A, Varga A, et al. TP53 mutations emerge with HDM2 inhibitor SAR405838 treatment in de-differentiated liposarcoma. Nat Commun. 2016;7:12609.
Hoffman-Luca CG, Ziazadeh D, McEachern D, Zhao Y, Sun W, Debussche L, et al. Elucidation of Acquired Resistance to Bcl-2 and MDM2 Inhibitors in Acute Leukemia In Vitro and In Vivo. Clin Cancer Res. 2015;21:2558–68.
Van Goethem A, Yigit N, Moreno-Smith M, Vasudevan SA, Barbieri E, Speleman F, et al. Dual targeting of MDM2 and BCL2 as a therapeutic strategy in neuroblastoma. Oncotarget. 2017;8:57047–57057.
Pan R, Ruvolo V, Mu H, Leverson JD, Nichols G, Reed JC, et al. Synthetic Lethality of Combined Bcl-2 Inhibition and p53 Activation in AML: Mechanisms and Superior Antileukemic Efficacy. Cancer Cell. 2017;32:748–760.e6.
Luo Q, Pan W, Zhou S, Wang G, Yi H, Zhang L, et al. A Novel BCL-2 Inhibitor APG-2575 Exerts Synthetic Lethality With BTK or MDM2-p53 Inhibitor in Diffuse Large B-Cell Lymphoma. Oncol Res. 2020;28:331–44.
Fang DD, Tang Q, Kong Y, Wang Q, Gu J, Fang X, et al. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J Immunother Cancer. 2019;7:327.
Wang HQ, Liang J, Mulford I, Sharp F, Gaulis S, Chen Y, et al. Abstract 5560: PD-1/PD-L1 blockade enhances MDM2 inhibitor activity in p53 wild-type cancers. Cancer Res. 2018;78:5560.
Zhou J, Kryczek I, Li S, Li X, Aguilar A, Wei S, et al. The ubiquitin ligase MDM2 sustains STAT5 stability to control T cell-mediated antitumor immunity. Nat Immunol. 2021;22:460–70.
ASCO 2022: Ascentage Pharma Releases Updated Results Demonstrating the Therapeutic Potential of Alrizomadlin (APG-115) plus Pembrolizumab in Patients with Solid Tumors who Progressed on Immunotherapies. https://www.prnewswire.com/news-releases/asco-2022--ascentage-pharma-releases-updated-results-demonstrating-the-therapeutic-potential-of-alrizomadlin-apg-115-plus-pembrolizumab-in-patients-with-solid-tumors-who-progressed-on-immunotherapies-301562392.html.
Mullard A. p53 programmes plough on. Nat Rev Drug Discov. 2020;19:497–500.
Schoffski P, Lahmar M, Lucarelli A, Maki RG. Brightline-1: phase II/III trial of the MDM2-p53 antagonist BI 907828 versus doxorubicin in patients with advanced DDLPS. Future Oncol.2023;19:621–29.
Gounder MM, Bauer TM, Schwartz GK, Weise AM, LoRusso P, Kumar P, et al. A First-in-Human Phase I Study of Milademetan, an MDM2 Inhibitor, in Patients With Advanced Liposarcoma, Solid Tumors, or Lymphomas. J Clin Oncol. 2023;41:1714–24.
Verstovsek S, Al-Ali HK, Mascarenhas J, Perkins A, Vannucchi AM, Mohan SR, et al. BOREAS: a global, phase III study of the MDM2 inhibitor navtemadlin (KRT-232) in relapsed/refractory myelofibrosis. Future Oncol.2022;18:4059–69.
Chan KH, Zengerle M, Testa A, Ciulli A. Impact of Target Warhead and Linkage Vector on Inducing Protein Degradation: Comparison of Bromodomain and Extra-Terminal (BET) Degraders Derived from Triazolodiazepine (JQ1) and Tetrahydroquinoline (I-BET726) BET Inhibitor Scaffolds. J Med Chem. 2018;61:504–13.
Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13:514–21.
Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol. 2015;11:611–7.
Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. P Natl Acad Sci USA. 2001;98:8554–9.
Schneekloth JS Jr, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748–54.
Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16:101–14.
Crews CM, Georg G, Wang SM. Inducing Protein Degradation as a Therapeutic Strategy. J Med Chem. 2016;59:5129–30.
Bekes M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21:181–200.
Li Y, Yang J, Aguilar A, McEachern D, Przybranowski S, Liu L, et al. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J Med Chem. 2019;62:448–66.
Wang B, Wu S, Liu J, Yang K, Xie H, Tang W. Development of selective small molecule MDM2 degraders based on nutlin. Eur J Med Chem. 2019;176:476–91.
Wang B, Liu J, Tandon I, Wu S, Teng P, Liao J, et al. Development of MDM2 degraders based on ligands derived from Ugi reactions: Lessons and discoveries. Eur J Med Chem. 2021;219:113425.
Marcellino B, Yang XB, Chen H, Chen K, Brady C, Clementelli C, et al. Development of an MDM2 Degrader for Treatment of Acute Leukemias. Blood. 2021;138:1866–+.
Chutake Y, Mayo M, Chen D, Enerson B, Cho P, Filiatrault J, et al. Abstract 3934: KT-253, a highly potent and selective heterobifunctional MDM2 degrader for the treatment of wildtype p53 tumors with superior potency and differentiated biological activity compared to small molecule inhibitors (SMI). Cancer Res. 2022;82:3934.
Mayo M, Karnik YC, McDonald A, Cho P, Filiatrault J, Chen D, et al. Development of KT-253, a Highly Potent and Selective Heterobifunctional MDM2 Degrader for the treatment of Acute Myeloid Leukemia. 64th Annual Meeting of the American Society of Hematology (ASH), December 10–13, 2022, New Orleans, LA. https://www.kymeratx.com/wp-content/uploads/2022/12/POSTER-Kymera_ASH_2022_Mayo-1.pdf.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SW and AA are co-inventors of an MDM2 inhibitor APG-115 and MDM2 degraders, which have been licensed by Ascentage Pharma Group for clinical development and they receive royalty from the University of Michigan. SW is a co-founder of Ascentage, owns stock in Ascentage and is a paid consultant of Ascentage. The University of Michigan also has a research contract with Ascentage.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Aguilar, A., Thomas, J.E. & Wang, S. Targeting MDM2 for the development of a new cancer therapy: progress and challenges. Med Chem Res 32, 1334–1344 (2023). https://doi.org/10.1007/s00044-023-03102-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-023-03102-1