
Abstract
c-Jun N-terminal kinase is an important regulator, activating several transcription factors in response to proinflammatory cytokines, ultraviolet radiations, environmental stress, hypoxia and osmotic shock and is known to be reported a cause for many diseases, such as diabetes, cancer, inflammation, stroke, etc. In the present study, we aim to predict novel therapeutic leads against c-Jun N-terminal kinase-3 by employing structure based virtual screening in combination with various in silico toxicity filters. We screened ZINC database virtually using a known potent c-Jun N-terminal kinase inhibitor, SP600125, as reference molecule. We obtained 128 molecules sharing ≥70% structure identity with SP600125. These 128 compounds were subjected to virtual screening and various toxicity filters. Finally, three molecules were identified as novel c-Jun N-terminal kinase inhibitors. Further binding mode analysis suggested that these molecules inhibit c-Jun N-terminal kinase activity through binding the adenosine triphosphate binding pocket.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
c-Jun N-terminal kinase (JNK) is a hepatic MAP-2 protein kinase known to be activated after the intraperitonial injection of cycloheximide in rodents (Kyriakis and Avruch 1990; Zhou et al. 2015). The name indicates its kinase activity on the c-Jun, an oncoprotein, at the amino terminal trans-activation domain (Hibi et al. 1993). JNKs are encoded by three genes of which jnk1, jnk2 are ubiquitously expressed and jnk3 expression is restricted to brain, heart, and testis. Alternative splicing of these gene transcripts create ten JNK isoforms (Gupta et al. 1996). JNKs are activated by proinflammatory cytokines, ultraviolet (UV) radiations, environmental stress, hypoxia and osmotic shock (Xie et al. 1998) by triggering the activation of MAP kinase cascade through sequential phosphorylation of MAP3K, MAP2K, MAPK, and JNK. Complete activation of JNK requires dual phosphorylation of threonine and tyrosine residues within a threonine-proline-tyrosine motif located in the kinase domain. Upon activation, JNKs regulate several transcription factors such as c-Jun (a component of activator protein-1), Elk1, P53, insulin receptor substrate 1, and several members of the Bcl-2 family of apoptosis-related proteins etc. (Bode and Dong 2007).
JNK3 also plays an important role in the brain to mediate neurodegeneration in Alzheimer’s disease, neurotoxicity in Parkinson’s disease. It is reported that JNKs are activated in many diseases, such as diabetes, cancer, inflammation, stroke, etc. Therefore, JNK inhibitors are expected to be effective therapeutic agents against these diseases (Asano et al. 2008). Determination of the X-ray structure of JNK3 has provided an opportunity to design selective inhibitors. The present study is an attempt to predict novel JNK3 inhibitors from SP600125, an anthrapyrazolone inhibitor of c-Jun N-terminal kinase (Bennett et al. 2001).
Materials and methods
Virtual screening
ZINC database screening and ligand dataset preparation
SP600125 is a reversible adenosine triphosphate (ATP) competitive inhibitor of JNK. Using this known potent inhibitor as reference molecule, we screened ZINC database (Irwin and Shoichet 2005) for compounds with at least 70% structure similarity. All the small molecules obtained from structure similarity screening were downloaded in structure data format and subjected to ligand preparation in chimera 1.10.2. (Pettersen et al. 2004).
Target structure preparation
The X-ray crystal structures of several JNK3-ligand complexes have been previously reported. The structure with the best resolution amongst the available crystal structures of human JNK3 bound with inhibitor at 1.70 Å resolution (PDB: 3OY1) (Gary D. Probst et al. 2011) was chosen for our study. As an initial step, the crystallographic water molecules and ligand were removed from the co-crystal structure. Hydrogen atoms and Gesteiger partial charges were added to the receptor using UCSF Chimera1.10.2.
Molecular docking
Molecular docking method is widely used to predict the binding mode of the ligand with protein, which further helps in the calculation of binding affinities (Meng et al. 2011). Molecular docking was performed using AutoDock Vina in PyRx program, an open-source software with an intuitive user interface that runs on all major operating systems (Dallakyan and Olson 2015). The grid box was generated around the active site residues with grid center X-axis −18.33 A°, Y-axis 7.61 A°, Z-axis −30.44 A° and grid dimensions x-24.41 A°, Y-23.16 A°, Z-23.71 A°.
Analysis of drug-likeness and prediction of adverse effects
Lipinski “Rule of five” is widely used as a filter for drug-like properties (Lajiness et al. 2004). The rule states that most molecules with good membrane permeability have logP ≤ 5, molecular weight ≤500, the number of hydrogen bond acceptors ≤10, and the number of hydrogen bond donors ≤5 (Lipinski et al. 2012). Lipinski “Rule of five”, adverse effects of the chemicals i.e., skin sensitization, carcinogenicity, mutagenicity, and developmental toxicity, the in silico pharmacokinetic properties and ADME (absorption, distribution, metabolism and elimination) analysis were predicted using DataWarrior (http://www.openmolecules.org/datawarrior.html). DataWarrior is a universal data analysis and visualization program whose embedded cheminformatics algorithms make it a versatile tool to explore large data sets of chemical structures with alphanumerical properties (Sander et al. 2015). DataWarrior tries to assess the toxicity risk by finding substructures within the chemical structure being indicative of a toxicity risk within one of aforementioned four major toxicity classes.
Results and discussion
Virtual screening
Docking protocol validation
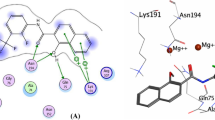
The docking protocol was validated using redocking experiment by removing ligand from JNK and docked back into the same binding pocket using Autodock Vina in PyRx with default parameters. It showed root mean square deviation value of 0.843 A°, obtained from all atom coordinates between redocked and experimental confirmations. Moreover, as shown in Fig. 1b, the docked native ligand is bound tightly to the ATP binding site by a hydrogen-bond with oxygen atom of the carboxyl group (C=O) on Met149 in addition to hydrophobic interactions with the residues Asp150, Ile70, Val196, Val78, Leu206, Ala91, Met146, Lys93, Leu144, Ile124 as in co-crystallized structure (Fig. 1a). It indicates that these parameters are adequate in reproducing experimental structure and can be extended to search for novel JNK inhibitors in the present study.

PoseView (Stierand and Rarey 2010) representation of c-Jun N-terminal kinase-3 and native ligand interactions. (a) Interactions between 3OY1 receptor and ligand taken from Protein Data Bank (b)
Results from molecular docking of selected inhibitor analogs
Results from the Zinc database search showed 128 compounds sharing greater than 70% structural similarity with SP600125. In order to study the molecular basis of interaction and binding affinity of SP600125 and its analogs, all these compounds were docked with the target protein. Docking of SP600125 to the receptor structure showed a binding energy of −9.2 kcal/mol. Among the 128 structural analogs, 30 compounds were found to contain binding energy greater −10 kcal/mol i.e., better than that of SP600125 to JNK3 (Table 1).
Assessment of pharmacological properties
All the compounds were tested for Lipinski “Rule of 5”. DataWarrior results showed that among 30 compounds, 7 compounds i.e., ZINC02054325, ZINC13743542, ZINC08277424, ZINC03196840, ZINC03206809, ZINC22007326, ZINC34253221 did not satisfy Lipinski rule.
Toxicity prediction
Although Lipinski “Rule of 5” describes the molecular properties important for a drug’s pharmacokinetics in the human body, including its ADME, it does not predict the pharmacological activity of the compounds. Therefore, pharmacokinetic properties and toxicities were predicted for all the 30 compounds using DataWarrior. Results of pharmacokinetic properties and toxicity analysis were shown in Table 2. Solubility and logP values were calculated for pharmacokinetic properties, whereas mutagenicity, tumorigenicity, irritation effect, and risk of reproductive effect were predicted for toxicity study. To determine the hydrophilicity we have predicted logP value, which is the logarithm of its partition coefficient between n-octanol and water (log(c octanol/c water)), is a well-established measure of the compound’s hydrophilicity. Low hydrophilicity and therefore high logP values cause poor absorption or permeation. It is suggestive for compounds to have a reasonable probability of being well absorbed in their logP value which must be less than 5 (Vyas et al. 2013). Results showed that among 30 compounds 23 compounds present within this limit. In general, a poor solubility is associated with bad absorption and the aqueous solubility (Log S) of a compound significantly affects its absorption and distribution characteristics. Results showed that ZINC13743541, ZINC28917266, ZINC28917268, ZINC26510110, ZINC28917178, ZINC28917176 have better LogS values than others. Among the 30 compounds, two compounds i.e., ZINC59468290, ZINC34253221 have high mutagenic activity, one compound i.e., ZINC27304443 has tumorigenic activity, two compounds i.e., ZINC59468290 and ZINC02054325 showed the low reproductive effect and one compound i.e., ZINC34253221 showed high reproductive effect. Among these 30 compounds except six compounds i.e., ZINC13743541, ZINC13743549, ZINC27304443, ZINC00062970, ZINC13743542, ZINC34253221, all are predicted to be skin sensitizers. Among these 30 compounds, based on the results from the DataWarrior, it is predicted to have less LogP, better LogS, good drug score and less toxicity risk parameters as shown in the Table 2 ZINC compounds, ZINC13743541, ZINC28917266, ZINC28917268 might be chosen as the new inhibitor for JNK with 0.393, 0.336, and 0.336 drug scores, respectively.
Binding mode analysis
The binding mode of these three compounds was shown in Fig. 2. Binding mode analysis revealed that these molecules inhibit JNK activity through binding the ATP binding pocket by forming hydrogen bonds, hydrophobic interactions and π–π stacking interactions. Among them, compound ZINC28917268 is the most potent with a binding energy of −10.8 kcal/mol (Table 1). As shown in Fig. 2a this compound is bound tightly to the ATP binding site by two hydrogen-bonding interactions with a carboxyl group present on Ala91 and amino group of Asn152. The structural analog of aforementioned compound ZINC28917268, ZINC28917266 also showed good binding affinity of −10.4 kcal/mol (Table 1). This compound is bound to JNK ATP binding site with a hydrogen bond. Hydrogen is donated by an amino group of lys93 to the oxygen atom of hydroxyl group presented on 28917266 (Fig. 2b). The third compound, ZINC13743541, also showed the better binding energy of −10.2, than that of SP600125. It is bound to the JNK by forming a hydrogen bond. A hydrogen bond donor is an amino group of lysine to the nitrogen atom present on ZINC13743541 (Fig. 2c). Figure 2 shows that all these three compounds can form hydrophobic interactions with the same residues that are involved in the formation of hydrophobic interactions with SP600125.

3D and 2D representations of binding mode of proposed ligands with c-Jun N-terminal kinase-3 (a) ZINC28917268 (b) ZINC28917266 (c) ZINC13743541
Conclusion
JNKs are being considered as potential therapeutic targets in number of human diseases. In this study, we proposed novel JNK inhibitors using an integrated computational approach by combining structural based virtual screening, molecular docking, in silico pharmacokinetic and toxicity studies. Redocking experiment was successful in reproducing the experimental structure. The docking results showed that each compound presented in dataset is able to bind to JNK with reasonably good binding affinity. Results from pharmacokinetic and toxicity calculated in DataWarrior, showed that many of these compounds are not suitable as drug candidates. Only three compounds i.e., 4-benzylλ-methyl-indeno[3,2-c]pyridazin-5-one (ZINC13743541), [(2R)-2-hydroxy-2-phenyl-ethyl]BLAHone (ZINC28917266), and its isomer [(2S)-2-hydroxy-2-phenyl-ethyl]BLAHone (ZINC28917268), have good pharmacokinetic and drug-likeness properties. Further binding mode analysis has suggested these molecules inhibit JNK activity through binding the ATP binding pocket. Hence, it can be concluded that combination of the above in silico methods is able to identify new hits that may act as a good lead against JNK. Further pharmacological studies are needed to be performed for considering these three small molecules as effective.
References
Asano Y, Kitamura S, Ohra T, Itoh F, Kajino M, Tamura T, Kaneko M, Ikeda S, Igata H, Kawamoto T, Sogabe S (2008) Discovery, synthesis and biological evaluation of isoquinolones as novel and highly selective JNK inhibitors (2). Bioorg Med Chem 16:4699–4714
Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS (2001) SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci 98:13681–13686
Bode AM, Dong Z (2007) The functional contrariety of JNK. Mol Carcinog 46:591–598
Dallakyan S, Olson AJ (2015) Small-molecule library screening by docking with PyRx. Chem Biol 1263:243–250
Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ (1996) Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J 15:2760
Hibi M, Lin AN, Smeal T, Minden A, Karin M (1993) Identification of an oncoprotein-and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 7:2135–2148
Irwin JJ, Shoichet BK (2005) ZINC-a free database of commercially available compounds for virtual screening. J Chem Inf model 45:177–182
Kyriakis JM, Avruch J (1990) pp54 microtubule-associated protein 2 kinase. A novel serine/threonine protein kinase regulated by phosphorylation and stimulated by poly-l-lysine. J Biol Chem 265:17355–17363
Lajiness MS, Vieth M, Erickson J (2004) Molecular properties that influence oral drug-like behavior. Curr Opin Drug Discov Dev 7:470–477
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2012) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 64:4–17
Meng XY, Zhang HX, Mezei M, Cui M (2011) Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des 7:146–157
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Probst GD, Bowers S, Sealy JM, Truong AP, Hom RK, Galemmo RA, Konradi AW, Sham HL, Quincy DA, Pan H, Yao N (2011) Highly selective c-Jun N-terminal kinase (JNK) 2 and 3 inhibitors with in vitro CNS-like pharmacokinetic properties prevent neurodegeneration. Bioorg Med Chem Lett 21:315–319
Sander T, Freyss J, von Korff M, Rufener C (2015) Data warrior: an open-source program for chemistry aware data visualization and analysis. J Chem Inf Model 55:460–473
Stierand K, Rarey M (2010) Poseview–molecular interaction patterns at a glance. J Chem Inform 2:1
Vyas VK, Ghate M, Goel A (2013) Pharmacophore modeling, virtual screening, docking and in silico ADMET analysis of protein kinase B (PKB β) inhibitors. J Mol Graph Model 42:17–25
Xie X, Gu Y, Fox T, Coll JT, Fleming MA, Markland W, Caron PR, Wilson KP, Su MS (1998) Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure 6:983–991
Zhou YY, Li Y, Jiang WQ, Zhou LF (2015) MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci Rep 35:e00199
Acknowledgements
DT is thankful to University Grants Commission, New Delhi for financial support in the form of UGC—BSR (RFSMS) Senior Research Fellowship. Y.S. is thankful to Human Resource Development for Health Research, New Delhi (F.No.V.25011/542-HRD/2016-HR). We are very grateful to the Journal Editor, anonymous reviewers, and Prof. P. Sreedhara Reddy, Department of Physics, Sri Venkateswara University, Tirupati for their constructive and useful comments which improve the scientific content of the original paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Daggupati, T., Chitrala, K., Pamanji, R. et al. Molecular screening and analysis of novel therapeutic inhibitors against c-Jun N-terminal kinase. Med Chem Res 26, 2112–2118 (2017). https://doi.org/10.1007/s00044-017-1919-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-017-1919-5