
Abstract
The target new hybrid molecule types pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines phosphonates 4 and 2-(coumarin-3’’-yl)-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines 5 were prepared via Michaelis–Arbuzov rearrangement (Arbuzov reaction) of pyrazolotriazolopyrimidines chloride 3a–c, with trialkyl phosphate and Knoevenagel reaction of 2-cyanomethyl derivatives 3d–f with salicylic aldehyde, respectively. The precursors 3 were obtained in two steps starting from aminopyrazole 1. Target compounds 4 and 5 were completely characterized by 1H NMR, 13C NMR, 31P NMR, IR and HRMS. The anti-acetylcholinesterase activity of compounds 4 and 5 was evaluated, and results found indicated that they have possessed significant activities (IC50 = 1.73–39.86 µM), and the preliminary SAR of these compounds was investigated.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
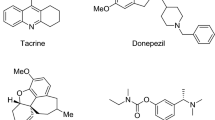
The study of heterocyclic compounds such as five- and six-membered rings containing nitrogen has a known considerable development due to the revealing of their varied effects in diverse domains. In this framework, pyrazoles, triazoles and pyrimidines, as well as their condensed derivatives, are very attractive targets from both a theoretical and a synthetic point of view. Moreover, they have been the subject of many chemical and biological studies on account of their pharmacological activity, such as anti-inflammatory, analgesic (Amin et al., 2009), cytotoxicity, antitumor (Rashad et al., 2010) and adenosine receptor antagonists (Baraldi et al., 2012). In addition, some of them have shown anti-acetylcholinesterase activity (Fig. 1a–c) (Zhi et al., 2008).

Previously reported AChEI compounds
On the other hand, diverse biological activity (Weiqin et al., 2006; Mado et al., 2014) of heterocyclic phosphonates containing the P–C bond has for a long time attracted considerable synthetic and pharmacological interest (Ali et al., 2001). Some of them exert their biological action on arthropods by attacking the system of neural transmission and inhibiting the function of acetylcholinesterase (Fig. 1d) (Fest and Schmidt 1973; Troev 2006). Thus, a large number of new phosphonate derivatives have been prepared hitherto with special attention to nitrogen heterocyclic compounds (Francesca et al., 2001; Yanchang et al., 2002; Francisco et al., 2003; Lise et al., 2007).
Furthermore, coumarins are a class of compounds, which occupies a special role in nature. They have attracted intense interest in recent years because of their diverse pharmacological properties like anti-HIV (Ma et al., 2008), anticoagulant (Kidane et al., 2004), antibacterial (Appendino et al., 2004), antioxidant (Kontogiorgis and Hadjipavlou-Litina 2004), cytotoxic (Musa et al., 2008) and particularly acetylcholinesterase inhibitors (Fig. 1e) (Razavi et al., 2013; Nam et al., 2014).
In addition, the synthesis of new hybrid molecules is a new concept in the field of drug development based on the association between pharmacophoric fragments of different bioactive substances to produce new hybrid compounds with improved efficiency and affinity. In this context and as a continuation of our previous work on the synthesis of new fused pyrimidine scaffolds (Rahmouni et al., 2014a, b), we report here the synthesis of some new hybrid molecules 4 and 5 bearing in their structures fragments described as anti-acetylcholinesterase agents, as indicated above, such as pyrazole, pyrimidine, triazole and phosphoric or coumarinic systems. Compounds 4 and 5 were evaluated for their anti-acetylcholinesterase activity, and preliminary SAR of these compounds was investigated (Table 1).
Results and discussion
Chemistry
Our key intermediate was the α-functionalized iminoethers type 2, which were synthesized from aminopyrazole 1 (Scheme 1), since such system constitutes a commonly used building block for the construction of a variety of polyheterocycles (Zaki 1998; El-Agordy et al., 2001).

Synthetic route of compounds 2
For this purpose and as described in schemes 2, 4 and 5, our approach to the target systems 4 and 5 was firstly started by the construction of the pyrazolotriazolopyrimidine skeleton type 3 via the intramolecular cyclocondensation reaction of 2. In fact, these intermediates, which possess two reactive sites: a cyano group and an imidic carbon, were made to react with appropriate acid hydrazide under ethanol reflux to give desired fused tricyclic triazolopyrimidines. Plausible pathway involves two successive nucleophilic additions of −NH2 group on the imidic carbon and on the cyano function followed by dehydrocyclization to give triazolopyrazolopyrimidines 3a–f (Scheme 2).

Synthetic route of compounds 3a–f
We note that triazolopyrimidine derivatives, such compounds 3, have generally been prepared, in two steps, via reaction of α-functionalized iminoethers with hydrazine followed by cyclocondensation reaction with one-carbon cyclizing reagents, namely acids, acid chlorides, orthoesters, CSI and DMFDMA (Romdhane et al., 2003, 2008; Scheme 3).

Synthesis of triazolopyrimidine derivatives, in two steps
To access new pyrazolotriazolopyrimidine phosphonates containing the P–C bond 4a–f, we thought about Michaelis–Arbuzov rearrangement (Arbuzov reaction) which is known as one of the most extensively investigated methods and is widely used to prepare phosphonates, by formation of P–C bond through reaction of an aryl/alkyl halide and trialkyl phosphite (Kosolapov et al., 1950). In fact, formation of new phosphonated compounds 4a–f in good yields (Table 2) was carried out via the reaction of pyrazolotriazolopyrimidines chloride 3a–c, with trialkyl phosphite.
In order to determine the best conditions for the preparation of pyrazolotriazolopyrimidines phosphonates 4, we examined the reaction under various conditions by changing the solvent (toluene, xylene), and we found that the best yields (Table 2) were obtained when compounds 3a–c were reacted under reflux with an excess of trialkyl phosphite used at the same time as a solvent. The reaction was conducted until TLC indicated that the starting materials have been completely converted to products 4.
The structures of compounds 4 have been assigned from their analytical data, IR, 1H NMR, 13C NMR, 31P NMR and mass spectrometry (ES-HRMS). In fact, the 1H NMR spectra of compounds 4 showed, in addition to the signals corresponding to the protons introduced by compounds 3, a new doublet (J = 21.3–21.6 Hz) at 3.58–3.66 ppm attributable to the methylene group (H-10) bearing the phosphoryl moiety. The large splitting was a result of a 2 J coupling of being directly bonded to the phosphorus. We also detected the presence of a doublet at 3.82–3.86 ppm (J = 12.3–12.4 Hz), when R2 = Me, as a result of protons of the methyl group coupled to the phosphorus atom and a multiplet at 4.07–4.11 ppm, when R2 = Et, due to the coupling of protons of the ethyl group to the phosphorus atom.
The 13C NMR spectra of these compounds were also in agreement with the proposed structures. In fact, in addition to the signals corresponding to the carbons introduced by the intermediate 3, we observed a doublet at 27.5–28.2 ppm with a very large coupling constant (161.3–161.5 Hz) as a result of being directly bonded of C-10 to the phosphorus atom and two doublets at 62.1–62.7 ppm (J = 24.3–24.4 Hz) and 161.1–162.2 ppm (J = 33.3–33.5 Hz) due to the coupling of the phosphorus atom with the methoxy carbon and C-2, respectively. For compounds 4b,d,e a new doublet at 16.5–16.8 ppm (J = 23.4–23.6 Hz) was observed resulting of the coupling between the methyl carbon of the ethoxy group and the phosphorus atom. The 31P NMR spectra of compounds 4a–f showed a signal corresponding to the phosphonyl group at 21.6–24.6 ppm. The infrared spectra also revealed the presence of an absorbance due to the P = O band at 1230–1238 cm−1 (Scheme 4).

Synthetic route of pyrazolotriazolopyrimidine phosphonates 4a–f
In the second part of this study, we have converted the intermediates 2 into pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine-2-acetonitriles 3d–f by reaction with cyanoacetic acid hydrazide, which was obtained by addition of hydrazine hydrate to ethyl cyanoacetate in ethanol with stirring at 0 °C, under reflux of toluene (Gorobets et al., 2004) (Scheme 2), in order to subsequently treat compounds 3d–f with salicylaldehyde via Knoevenagel condensation reaction, to access to the coumarin derivatives 5 (Scheme 5 ).

Synthetic route of coumarinic pyrazolotriazolopyrimidine derivatives 5a–c
The formed triazolopyrimidines 3d–f were characterized by their IR, 1H and 13C spectra. In fact, the IR spectra of compounds 3d–f showed an absorbance band at 2225–2230 cm−1 due to the cyano group. The 1H NMR spectra of compounds 3d–f showed, in addition to the signals of protons introduced by the intermediate 2, the appearance of a new signal due to the methylene group (–CH2–CN) at 4.08–4.10 ppm. The 13C NMR of 3d–f showed the appearance of new signals at 18.8–19.1, 116.8–117.5 and 164.5–165.1 ppm relative to –CH2–CN, –CN and C2, respectively.
The 2-cyanomethyl derivatives 3d–f were treated with salicylic aldehyde, via Knoevenagel reaction (Bylov et al., 1999; Kovalenko et al., 2000); under reflux of ethanol, an addition product formed 3′, from which reaction of an aqueous solution of hydrochloric acid gave the 2-(coumarin-3″-yl)-14(Aryl)-14H-naphto[2,1-b]pyrano[3,2-e][1,2,4]triazolo[1,5-c] pyrimidines 5a-c (scheme 5).
The reaction was conducted until TLC indicated that the starting materials have been completely converted to products 5. The IR spectra of these compounds revealed the presence of an absorbance band due to C = O at 1740–1747 cm−1 and the disappearance of any absorption frequency in the CN region. Further, the 1H NMR spectra of compounds 5 showed the disappearance of the singlet relative to the methylene protons (H-10) of compounds 3d–f (4,08–4,10 ppm) and the appearance of new signals, attributable to protons of the coumarin moiety, of which chemical shifts and multiplicities are in agreement with the proposed structure. Analysis of 13C NMR spectra of these compounds showed, in addition to the signals relative to the coumarin moiety carbons, the disappearance of two signals in the region of 18.8–19.1 ppm and 116.8–117.5 ppm attributable to – CH2–CN and –CN in compounds 3d–f, respectively.
The ES-HRMS showed essentially the correct protonated molecular peak [M + H]+ for all examined compounds 3–5.
Biological activity
Anti-acetylcholinesterase activity
Inhibition of acetylcholinesterase (AChE), the key enzyme in the breakdown of acetylcholine, is considered one of the treatment strategies against several neurological disorders such as Alzheimer’s disease, senile dementia, ataxia and myasthenia gravis (Orhan et al., 2006; Howes et al., 2003). Only compounds 4a-f and 5a-c were analysed on what concerns their acetylcholinesterase inhibition activity (Table 3) using an adaptation of the method described in the literature (Ferreira et al., 2006).
The results indicated in Table 3 showed that all the tested heterocycles gave significant activity. Compared to those given in the literature for crude pure products (Mata et al., 2007), we can say that the prepared derivatives 4 and 5 are considered good acetylcholinesterase inhibitors. Moreover, it has been found that the methylated phosphonate derivatives 4a,c,e were more active than their ethylated analogous 4b,d,f.
The greatest inhibitory activity was exhibited by the phosphonated compound 4a (R = H, R2 = Me, IC50 = 1.73 ± 0.05 µM), which was found almost twenty-three times more active than (E)-anethole (IC50 = 39.86 ± 0.1 µM) used as a standard compound, but it is about two times less active than tacrine (IC50 = 0.86 ± 0.02 µM), a drag used to treat Alzheimer.
On the other hand, it has been shown that the activity of these derivatives depends on the nature of R. In 4a,c,e (R2 = Me), the acetylcholinesterase inhibition decreases from R = H (4a) to R = Et (4e). The same phenomena has been observed with 4b,d,f (R2 = Et).
For coumarinic compounds, it has been shown also that the activity of these derivatives depends on the nature of R; in fact, the acetylcholinesterase inhibition decreases from R = H (5a) to R = Et (5c). Compound 5c (R = Et) was found to be the less inhibitor (IC50 = 36.54 ± 0.2 µM).
Inhibition of AChE by compounds 4a–f can be explained by forming covalent adducts with the reactive Ser200 hydroxyl group, which prevents ACh hydrolysis. In fact, the serine hydroxyl group, deactivated by the phosphorylated moiety of varying degrees that depends on the groups attached to the phosphorus atom (Ahmed et al., 2013; Gonçalves et al., 2011a; Bajqar 2004;), is no longer able to participate in the hydrolysis of ACh. The anti-AChE potential of phosphonate compounds 4a–f can be explained by the possible intervention of the serine from the AChE via its hydroxy group as indicated in Scheme 6.

Blocking-up reaction of serine hydroxyl by the phosphorylated moiety
This proposed mechanism in addition to the probable steric hindrance due to the ethyl group (R2 = Et) in the phosphonate moiety, compared to that induced by the methyl group, may explain the relatively high activity of the compounds 4a,c,e (R2 = Me).
On the other hand, and according to some previous studies (Alipour et al., 2012; Razavi et al., 2013), we can explain the inhibition activity of coumarin derivatives 5a–c, by formation of an additional π-π interaction between the coumarin moiety and some actives sites of the AChE resulting more stability of the formed ligand which prevents more hydrolysis of ACh.
All these findings allowed to note that the covalent bond formation between the Ser200 hydroxyl group of AChE and the phosphorus atom in compounds 4a–f is more efficient than the π–π interaction between AChE and the coumarin system in derivatives 5a–c. However, the nature of coumarin attached to the triazole may also explain this difference in activity.
Conclusion
In conclusion, this work reports the synthesis of new hybrid compounds types pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines phosphonate 4 and coumarinic pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines 5 via Michaelis–Arbuzov rearrangement (Arbuzov reaction) of pyrazolotriazolopyrimidines chloride 3a–c, with trialkyl phosphate and Knoevenagel reaction of 2-cyanomethyl derivatives 3d–f with salicylic aldehyde, respectively. Precursors 3 were obtained in two steps starting from aminopyrazole 1. The anti-acetylcholinesterase activity of the synthesized hybrid compounds 4 and 5 was evaluated. It has been found that the methylated phosphonates 4a,c,e were the most active ones (IC50 = 1.73–6.50 µM).
Finally, we can say that the prepared derivatives 4 and 5 are considered good acetylcholinesterase inhibitors; this encouraged us to pursue this study in order to more explain the preliminary structure–activity relationship (SAR) of these compounds.
Experimental section
Chemistry
All reactions were monitored by TLC using aluminium sheets of SDS silica gel 60 F254, 0.2 mm. Melting temperatures were determined on an electrothermal 9002 apparatus and were reported uncorrected. NMR spectra were recorded on a Bruker AC-300 spectrometer at 300 MHz (1H), 75 MHz (13C) and 120 MHz (31P). All chemical shifts were reported as δ values (ppm) relative to residual non-deuterated solvent. IR spectra were recorded on FTS-6000 BIO-RAD apparatus. Mass spectra were obtained with ESI-TOF (LCT, Waters) using the reflectron mode in the positive ion mode. The starting materials 1 and 2 were prepared according to the literature (Rashad et al., 2005; Al-Afaleq et al., 2001).
General procedure for the synthesis of ethyl N-(4-cyano-1-phenyl-1H-pyrazol-5-yl) formimidate 2a–c
A mixture of compound 1 (10 mmol) and triethylorthoformate (3 mL) in acetic anhydride (25 mL) was refluxed for 1 h. After cooling, the precipitated product was filtered off and washed thoroughly with ethanol and recrystallized from ethanol to yield 2 as white solid.
Ethyl N-(4-cyano-1-phenyl-1H-pyrazol-5-yl)formimidate (2a)
White solid; Yield: 65 %; m.p.: 120–122 °C (ethanol); IR (KBr, cm−1) ν: 1620 (–C = N), 2228 (–CN); 1H NMR (CDCl3, 300 MHz): δ 1.34 (t, 3H, J = 7.6 Hz), 4.31 (q, 2H, J = 7.6 Hz),7.33–7.60 (m, 5H, Harom), 8.23 (s, 1H, H4), 8.72 (s, 1H, H7); 13C NMR (DMSO-d 6 , 75 MHz): δ 13.5 (C8), 64.3 (C9), 82.4 (C4), 115.3 (CN), 124.6 (C2′,6′), 127.3 (C4′), 129.7 (C3′,5′), 138.3 (C1′), 150.1 (C3), 151.4 (C5), 160.6 (C7).
Ethyl N-(4-cyano-3-methyl-1-phenyl-1H-pyrazol-5-yl)formimidate (2b)
White solid; Yield: 65 %; m.p.: 120–122 °C (ethanol); IR (KBr, cm−1) ν: 1625 (–C = N), 2225 (–CN); 1H NMR (CDCl3, 300 MHz): δ 1.35 (t, 3H, J = 7.5 Hz), 2.39 (s, 3H), 4.31 (q, 2H, J = 7.5 Hz),7.31–7.61 (m, 5H, Harom), 8.36 (s, 1H, H7); 13C NMR (DMSO-d 6 , 75 MHz): δ 13.1 (C8), 13.9, 64.2 (C9), 81.7 (C4), 114.7 (CN), 123.9 (C2′,6′), 127.6 (C4′), 128.8 (C3′,5′), 138.1 (C1′), 150.0 (C3), 151.7 (C5), 160.3 (C7).
Ethyl N-(4-cyano-3-ethyl-1-phenyl-1H-pyrazol-5-yl)formimidate (2c)
White solid; Yield: 65 %; m.p.: 120–122 °C (ethanol); IR (KBr, cm−1) ν: 1625 (–C = N), 2228 (–CN); 1H NMR (CDCl3, 300 MHz): δ 1.32 (t, 3H, J = 7.3 Hz), 1.34 (t, 3H, J = 7.6 Hz), 2.70 (q, 2H, J = 7.3 Hz), 4.45 (q, 2H, J = 7.6 Hz),7.35–7.63 (m, 5H, Harom), 8.46 (s, 1H, H7); 13C NMR (DMSO-d 6 , 75 MHz): δ 14.2 (C9), 14.8, 64.5, 65.1 (C8), 82.4 (C4), 116.3 (CN), 124.7 (C2′,6′), 127.6 (C4′), 129.5 (C3′,5′), 138.5 (C1′), 150.3 (C3), 151.1 (C5), 160.2 (C7).
General procedure for the synthesis of 2-chloromethyl-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidines 3a–c
To a solution of iminoethers 2 (1 mmol) in ethanol (30 mL), 2-chloroacetohydrazide hydrochloride (1.1 mmol) and triethylamine (1.1 mmol) were added. The reaction mixture was boiled at reflux for 4 h, and after cooling, it is poured into ice water (50 mL) to give a white solid which was crystallized from ethanol.
2-chloromethyl-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (3a)
White solid; Yield: 76 %; m.p.: 184–186 °C (ethanol); IR (KBr, cm−1) ν: 1620 (–C = N); 1H NMR (CDCl3, 300 MHz): δ 4.85 (s, 2H, –CH2Cl), 7.45–8.15 (m, 5H, Harom), 8.58 (s, 1H, H9), 9.15 (s, 1H, H5); 13C NMR (DMSO-d 6 , 75 MHz): δ 37.8 (–CH2Cl), 104.4 (C9a), 122.4 (C2′,6′), 127.4 (C4′), 129.5 (C3′,5′), 138.1 (C1′), 138.5 (C5), 146.5 (C9b), 149.0 (C6a), 149.3 (C9),164.6 (C2), ES-HRMS [M + H]+ calcd for (C13H10ClN6)+: 285.0577, found: 285.0569.
2-chloromethyl-9-methyl-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (3b)
White solid; Yield: 82 %; m.p.: 192-194 °C (ethanol); IR (KBr, cm−1) ν: 1615 (–C = N); 1H NMR (CDCl3, 300 MHz): δ 2.91 (s, 3H, –CH3), 4.83 (s, 2H, –CH2Cl), 7.47–8.21 (m, 5H, Harom), 9.20 (s, 1H, H5); 13C NMR (DMSO-d 6 , 75 MHz): δ 14.1 (–CH3), 37.6 (–CH2Cl), 103.9 (C9a), 121.9 (C2′,6′), 126.8 (C3′), 128.9 (C3′,5′), 138.2 (C1′), 138.8 (C5), 145.9 (C9b), 148.9 (C6a), 149.1 (C9), 165.1 (C2), ES-HRMS [M + H]+ calcd for (C14H12ClN6)+: 299.0734, found: 299.0740.
2-chloromethyl-9-ethyl-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (3c)
White solid; Yield: 74 %; m.p.: 196–198 °C (ethanol); IR (KBr, cm−1) ν: 1617 (–C = N); 1H NMR (CDCl3, 300 MHz): δ 1.54 (t, 3H, J = 7.5 Hz), 3.24 (q, 2H, J = 7.5 Hz), 4.87 (s, 2H, –CH2Cl), 7.36–8.10 (m, 5H, Harom), 9.14 (s, 1H, H5); 13C NMR (DMSO-d 6 , 75 MHz): δ 12.9, 22.0, 37.4 (–CH2Cl), 102.5 (C9a), 122.3 (C2′,6′), 127.5 (C4′), 129.3 (C3′,5′), 138.2 (C1′), 138.4 (C5), 146.4 (C9b), 149.1 (C6a), 149.2 (C9),164.5 (C2), ES-HRMS [M + H]+ calcd for (C15H14ClN6)+: 313.0890, found: 313.0899.
General procedure for the synthesis of 7-phenylpyrazolo[3,2-e][1,2,4]triazolo[1,5-c]pyrimidine-2-acetonitriles 3d–f
A mixture of 2 (1 mmol) and cyanoacetic hydrazide (1.2 mmol) in absolute ethanol (20 mL) was refluxed for 10 h; after cooling, the precipitated product was filtered off and crystallized from ethanol to give 3d–f.
7-Phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine-2-acetonitrile (3d)
White solid; Yield: 68 %; m.p.: 172–174 °C (ethanol); IR (KBr, cm−1) ν: 2225 (–CN); 1H NMR (CDCl3, 300 MHz): δ 4.08 (s, 2H, -CH2-CN), 7.42–8.13 (m, 5H, Harom), 8.60 (s, 1H, H9), 9.13 (s, 1H, H5); 13C NMR (DMSO-d 6 , 75 MHz): δ 18.8 (–CH2–CN), 104.4 (C9a), 117.3 (–CN), 122.2 (C2′,6′), 127.4 (C4′), 129.5 (C3′,5′), 138.1 (C1′), 138.5 (C5), 146.5 (C9b), 149.0 (C6a), 149.3 (C9), 164.6 (C2), ES-HRMS [M + H]+ calcd for (C14H10N7)+: 276.0916, found: 276.0908.
9-Methyl-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine-2-acetonitrile (3e)
White solid; Yield: 80 %; m.p.: 176–178 °C (ethanol); IR (KBr, cm−1) ν: 2228 (-CN); 1H NMR (CDCl3, 300 MHz): δ 2.91 (s, 3H, –CH3), 4.10 (s, 2H, –CH2–CN), 7.47–8.21 (m, 5H, Harom), 9.20 (s, 1H, H5); 13C NMR (DMSO-d 6 , 75 MHz): δ 14.3 (–CH3), 18.8 (–CH2–CN), 104.2 (C9a), 117.5 (–CN), 122.3 (C2′,6′), 126.5 (C4′), 129.2 (C3′,5′), 138.4 (C1′), 139.3 (C5), 145.3 (C9b), 148.3 (C6a), 149.7 (C9), 164.4 (C2), ES-HRMS [M + H]+ calcd for (C15H12N7)+: 290.1074, found: 290.1066.
9-Ethyl-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine-2-acetonitrile (3f)
White solid; Yield: 82 %; m.p.: 180–182 °C (ethanol); IR (KBr, cm−1) ν: 2230 (–CN); 1H NMR (CDCl3, 300 MHz): δ 1.54 (t, 3H, J = 7.5 Hz), 3.24 (q, 2H, J = 7.5 Hz), 4.10 (s, 2H, –CH2–CN), 7.17-8.21 (m, 5H, Harom), 9.17 (s, 1H, H5); 13C NMR (DMSO-d 6 , 75 MHz): δ 12.9 (–CH2CH3), 18.8 (–CH2–CN), 22.3 (–CH2CH3), 102.3 (C9a), 116.8 (–CN), 122.6 (C2′,6′), 128.1 (C4′), 129.8 (C3′,5′), 136.5 (C1′), 137.3 (C5), 146.7 (C9b), 148.7 (C6a), 150.7 (C9), 165.7(C2), ES-HRMS [M + H]+ calcd for (C16H14N7)+: 304.1563, found: 304.1557.
General procedure for the synthesis of pyrazolotriazolopyrimidine dialkyl phosphonates 4a–f
In a typical procedure, the solution of pyrazolotriazolopyrimidine 3a–c (1 mmol) and an excess of trialkylphosphite (10 mL) was refluxed for 6 h. The reaction evolution was checked by TLC. When all the starting material was consumed, the mixture was cooled to room temperature, and then, the precipitate formed was filtered, dried and purified by silica gel column chromatography eluted with petroleum ether–ethyl acetate (7:3).
Dimethyl ((7-phenyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)methyl)phosphonate (4a)
White solid; Yield: 88 %; mp 248–250 °C; IR (KBr, cm−1) ν: 1625 (C = N), 1235 (P = O), 975 (P–O–C); 1H NMR (300 MHz, DMSO-d 6): δ 3.62 (d, 2 J P–H = 21.3 Hz, 2H, H10), 3.86 (d, J P–H = 12.3 Hz, 6H, 2 P–O–CH 3 ), 7.48–8.09 (m, 5Harom.), 8.72 (s, 1H, H9), 9.65 (s, 1H, H5); 13C NMR (75 MHz, DMSO-d 6): δ 27.6 (d, 1 J P–C = 161.3 Hz, C10), 62.4 (d, 2 J P–C = 24.3 Hz, P–O–CH 3 ), 103.5 (C9a), 122.6 (C2′,6′), 128.1 (C4′), 129.7 (C3′,5′), 133.7 (C1′), 138.4 (C5), 140.6 (C9b), 146.2 (C6a), 148.1 (C9), 161.2 (d, 2 J P–C = 33.3 Hz, C2); 31P NMR (121 MHz, CDCl3): δ 24,6; ES-HRMS [M + H]+ calcd. For (C15H16N6O3P)+: 359.0943; found: 359.0937.
Diethyl ((7-phenyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)methyl)phosphonate (4b)
White solid; Yield: 90 %; mp 253–255 °C; IR (KBr, cm−1) ν: 1620 (C = N), 1238 (P = O), 978 (P–O–C); 1H NMR (300 MHz, DMSO-d 6): δ 1.23 (t, J H–H = 7.2 Hz, 6H, 2 P–O–CH2–CH 3 ), 3.64 (d, 2 J P–H = 21.3 Hz, 2H, H10), 4.07 (m, 4H, 2 P–O–CH 2 –CH3), 7.41–8.11 (m, 5Harom.), 8.73 (s, 1H, H9), 9.70 (s, 1H, H5); 13C NMR (75 MHz, DMSO-d 6): δ 16.7 (d, 3 J P–C = 23.4 Hz,P–O–CH2–C H 3), 28.1 (d, 1 J P–C = 161.5 Hz, C10), 62.8 (d, 2 J P–C = 24.4 Hz, P–O–CH 2 –CH3), 103.9 (C9a), 123.5 (C2′,6′), 128.0 (C4′), 130.1 (C3′,5′), 134.7 (C1′), 138.8 (C5), 141.3 (C9b), 146.4 (C6a), 149.3 (C9), 161.4 (d, 2 J P–C = 33.3 Hz, C2); 31P NMR (121 MHz, CDCl3): δ 21,8; ES-HRMS [M + H]+ calcd. For (C17H20N6O3P)+: 387.1256; found: 387.1248.
Dimethyl ((9-methyl-7-phenyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)methyl)phosphonate (4c)
White solid; Yield: 92 %; mp 260-262 °C; IR (KBr, cm−1) ν: 1618 (C = N), 1234 (P = O), 973 (P–O–C); 1H NMR (300 MHz, DMSO-d 6): δ 2.87 (s, 3H, –CH3), 3.61 (d, 2 J P–H = 21.4 Hz, 2H, H10), 3.84 (d, J P–H = 12.4 Hz, 6H, 2 P–O–CH 3 ), 7.43-8.12 (m, 5Harom.), 9.63 (s, 1H, H5); 13C NMR (75 MHz, DMSO-d 6): δ 14.3 (-CH3), 28.1 (d, 1 J P–C = 161.4 Hz, C10), 62.2 (d, 2 J P–C = 24.5 Hz, P-O-CH 3 ), 103.8 (C9a), 122.8 (C2′,6′), 128.3 (C4′), 129.5 (C3′,5′), 133.4 (C1′), 138.6 (C5), 141.1 (C9b), 146.3 (C6a), 149.2 (C9), 162.1 (d, 2 J P–C = 33.4 Hz, C2); 31P NMR (121 MHz, CDCl3): δ 24,5; ES-HRMS [M + H]+ calcd. For (C16H18N6O3P)+: 373.1100; found: 373.1108.
Diethyl ((9-methyl-7-phenyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)methyl)phosphonate (4d)
White solid; Yield: 98 %; mp 258-260 °C; IR (KBr, cm−1) ν: 1622 (C = N), 1235 (P = O), 975 (P–O–C); 1H NMR (300 MHz, DMSO-d 6): δ 1.24 (t, J H–H = 7.3 Hz, 6H, 2 P–O–CH2–CH 3 ), 2.91 (s, 3H, –CH3), 3.66 (d, 2 J P–H = 21.6 Hz, 2H, H10), 4.11 (m, 4H, 2 P–O–CH 2 –CH3), 7.44–8.09 (m, 5Harom.), 9.68 (s, 1H, H5); 13C NMR (75 MHz, DMSO-d 6): δ 14.7 (–CH3), 16.5 (d, 3 J P–C = 23.6 Hz, P–O–CH2–C H 3), 27.5 (d, 1 J P–C = 161.5 Hz, C10), 62.7 (d, 2 J P–C = 24.4 Hz, P–O–CH 2 –CH3), 104.1 (C9a), 122.3 (C2′,6′), 128.1 (C4′), 129.7 (C3′,5′), 133.6 (C1′), 138.7 (C5), 141.0 (C9b), 146.2 (C6a), 148.3 (C9), 161.6 (d, 2 J P–C = 33.5 Hz, C2); 31P NMR (121 MHz, CDCl3): δ 22,0; ES-HRMS [M + H]+ calcd. For (C18H22N6O3P)+: 401.1413; found: 401.1406.
Diethyl ((9-methyl-7-phenyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)methyl)phosphonate (4e)
White solid; Yield: 96 %; mp 268-270 °C; IR (KBr, cm−1) ν: 1620 (C = N), 1232 (P = O), 977 (P–O–C); 1H NMR (300 MHz, DMSO-d 6): δ 1.51 (t, 3H, J = 7.5 Hz), 3.18 (q, 2H, J = 7.5 Hz), 3.64 (d, 2 J P–H = 21.6 Hz, 2H, H10), 3.82 (d, J P–H = 12.3 Hz, 6H, 2 P–O–CH 3 ), 7.52–8.11 (m, 5Harom.), 9.62 (s, 1H, H5); 13C NMR (75 MHz, DMSO-d 6): δ 11.7, 21.7, 28.2 (d, 1 J P–C = 161.3 Hz, C10), 62.2 (d, 2 J P–C = 24.3 Hz, P–O–CH 3 ), 103.4 (C9a), 122.8 (C2′,6′), 127.9 (C4′), 130.1 (C3′,5′), 133.2 (C1′), 137.9 (C5), 141.1 (C9b), 146.4 (C6a), 147.9 (C9), 161.4 (d, 2 J P–C = 33.3 Hz, C2); 31P NMR (121 MHz, CDCl3): δ 24,3; ES-HRMS [M + H]+ calcd. For (C17H20N6O3P)+: 387.1256; found: 3871248.
Diethyl ((9-ethyl-7-phenyl-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)methyl)phosphonate (4f)
White solid; Yield: 94 %; mp 266–268 °C; IR (KBr, cm−1) ν: 1625 (C = N), 1230 (P = O), 975 (P–O–C); 1H NMR (300 MHz, DMSO-d 6): δ 1.25 (t, J H–H = 7.3 Hz, 6H, 2 P–O–CH2–CH 3 ), 1.46 (t, 3H, J = 7.5 Hz), 3.10 (q, 2H, J = 7.5 Hz), 3.63 (d, 2 J P–H = 21.6 Hz, 2H, H10), 4.10 (m, 4H, 2 P–O–CH 2 –CH3), 7.30–8.10 (m, 5Harom.), 9.61 (s, 1H, H5); 13C NMR (75 MHz, DMSO-d 6): δ 13.8, 16.6 (d, 3 J P–C = 23.6 Hz, P–O–CH2–C H 3), 26.3, 28.1 (d, 1 J P–C = 161.3 Hz, C10), 62.5 (d, 2 J P–C = 24.3 Hz, P–O–CH 2 –CH3), 103.1 (C9a), 122.0 (C2′,6′), 127.5 (C4′), 129.7 (C3′,5′), 138.5 (C1′), 140.4 (C5), 142.9 (C9b), 146.5 (C6a), 148.5 (C9), 161.4 (d, 2 J P–C = 33.3 Hz, C2); 31P NMR (121 MHz, CDCl3): δ 23,3; ES-HRMS [M + H]+ calcd. For (C19H24N6O3P)+: 415.1569; found: 415.1563.
General procedure for the synthesis of 2-(coumarin-3″-yl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine 5a-c
A mixture of 2-cyanomethyl derivatives 3d–f (1 mmol) and salicylic aldehyde (1 mmol) in ethanol (20 mL) was refluxed for 1 h in the presence of few drops of piperidine. The intermediate 3′d–f obtained after filtration was reacted with a mixture of ethanol/water/concentrated hydrochloric acid (20:2:2, v:v:v) at reflux for 3 h, and after cooling to room temperature, the precipitated product was filtered off and recrystallized from ethanol to give 5a-c.
2-(Coumarin-3″-yl)-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine (5a)
White solid; Yield: 78 %; m.p.: 272–274 °C (ethanol); IR (KBr, cm−1) ν: 1740 (CO); 1H NMR (CDCl3, 300 MHz): δ 7.40–8.10 (m, 8H, Harom), 8.21 (d, 1H, Harom., J = 8.1 Hz), 8.62 (s, 1H, H9), 9.11 (s, 1H, H5), 9.21 (s, 1H, H4″); 13C NMR (DMSO-d 6 , 75 MHz): δ 103.7 (C9a), 121.7 (C8″), 121.8 (C2′,6′), 122.7 (C4a’’), 125.7 (C6″), 126.8 (C5″), 127.1 (C4′), 128.4 (C7″), 129.3 (C3″), 130.3 (C3′,5′), 137.8 (C1′), 139.2 (C5), 146.1 (C4″), 147.2 (C9b), 149.2 (C6a), 149.5 (C9), 150.1 (C8a’), 164.2 (C2), 165.3 (CO). ES-HRMS [M + H]+ calcd for (C21H13N6O2)+: 381.1022, found: 381.1015.
2-(Coumarin-3″-yl)-9-methyl-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c] pyrimidine (5b)
White solid; Yield: 86 %; m.p.: 280-282 °C (ethanol); IR (KBr, cm−1) ν: 1745 (CO); 1H NMR (CDCl3, 300 MHz): δ 2.90 (s, 3H, –CH3), 7.51–8.12 (m, 8H, Harom), 8.22 (d, 1H, Harom., J = 8.1 Hz), 8.63 (s, 1H, H9), 9.15 (s, 1H, H5), 9.24 (s, 1H, H4′); 13C NMR (DMSO-d 6 , 75 MHz): δ 14.2 (–CH3), 104.5 (C9a), 121.8 (C8″), 122.8 (C2′,6′), 123.3 (C4a″), 124.8 (C6″), 126.1 (C5″), 127.6 (C4′), 128.7 (C7″), 129.1 (C3″), 129.7 (C3′,5′), 138.2 (C1′), 139.1 (C5), 146.7 (C4″), 147.2 (C9b), 149.3 (C6a), 150.1 (C9), 150.7 (C8a′), 163.2 (C2), 164.2 (CO). ES-HRMS [M + H]+ calcd for (C22H15N6O2)+: 395.1172, found: 395.1178.
2-(Coumarin-3″-yl)-9-ethyl-7-phenylpyrazolo[4,3-e]-1,2,4-triazolo[1,5-c] pyrimidine (5c)
White solid; Yield: 82 %; m.p.: 284–286 °C (ethanol); IR (KBr, cm−1) ν: 1747 (CO); 1H NMR (CDCl3, 300 MHz): δ 1.52 (t, 3H, J = 7.5 Hz), 3.22 (q, 2H, J = 7.5 Hz), 7.43–8.13 (m, 8H, Harom), 8.41 (d, 1H, Harom., J = 8.1 Hz), 8.71 (s, 1H, H9), 9.09 (s, 1H, H5), 9.31 (s, 1H, H4′); 13C NMR (DMSO-d 6 , 75 MHz): δ 12.4, 22.3, 104.8 (C9a), 121.1 (C8″), 122.5 (C2′,6′), 123.1 (C4a″), 124.7 (C6″), 127.1 (C5″), 127.8 (C4′), 128.5 (C7″), 129.8 (C3″), 130.3 (C3′,5′), 138.6 (C1′), 139.3 (C5), 146.3 (C4″), 146.6 (C9b), 150.2 (C6a), 151.2 (C9), 152.8 (C8a′), 165.3 (C2), 166.8 (CO). ES-HRMS [M + H]+ calcd for (C23H17N6O2)+: 409.1327, found: 409.1321.
Biological activity
Determination of AChE inhibitory activity
Cholinesterase (ChE) inhibitory activity was measured using Ellman’s method (Ellman et al., 1961), with modifications (Moyo et al., 2010). In this study, 50 μL of 0.1 M sodium phosphate buffer (pH 8.0), 25 μL of AChE solution, 25 μL of each compound, and 125 μL of DTNB [5,50—dithiobis (2-nitrobenzoic acid)] were added in a 96-well microplate and incubated for 15 min at 25 C. All compounds were re-suspended in the DMSO followed by dilution in the buffer so that the DMSO does not exceed 1 %. 25 μL of a solution of acetylthiocholine iodide was added, and the final mixture incubated, for 15 min, at 25 C. The hydrolysis of acetylthiocholine iodide was monitored by the formation of the yellow 5-thio-2-nitrobenzoate anion as a result of the reaction of DTNB with thiocholines, catalysed by enzymes at a wavelength of 412 nm. The concentration of the compounds which caused 50 % inhibition of the AChE activity (IC50) was calculated by nonlinear regression analysis. The percentage of inhibition was calculated from (1 to S/E) × 100, where E and S were the respective enzyme activity without and with the test samples, respectively.
References
Ahmed SK, Belabassi Y, Sankaranarayanan L, Chao CK, Gerdes JM, Thompson CM (2013) Synthesis and anti-acetylcholinesterase properties of novel β-and γ-substituted alkoxy organophosphonates. Bioorg Med Chem Lett 23(7):2048–2051
Al-Afaleq EI, Abubshait SA (2001) Heterocyclic o-aminonitriles: preparation of pyrazolo[3,4-d]pyrimidine with modification of the susbstituents at the 1-position. Molecules 6(7):621–638
Ali MM, Tasneem Rajanna KC, Prakash PKS (2001) An efficient and facile synthesis of 2-chloro-3-formyl quinolines from acetanilides in micellar media by Vilsmeier-Haack cyclisation. Synlett 251–253
Alipour M, Khobbi M, Foroumadi A, Nadri H, Moradi A, Sakhteman A, Ghandi M, Shafiee A (2012) Novel coumarin derivatives bearing N-benzyl pyridinium moiety: potent and dual binding site acetylcholinesterase inhibitors. Bioorg Med Chem 20:7214–7222
Amin KM, Hanna MM, Abo-Youssef HE, George RF (2009) Synthesis, analgesic and anti-inflammatory activities evaluation of some bi-, tri- and tetracyclic condensed pyrimidines. Eur J Med Chem 44(11):4572–4584
Appendino G, Mercalli E, Fuzzati N, Arnoldi L, Stavri M, Gibbons S, Ballero M, Maxia A (2004) Antimycobacterial coumarins from the Sardinian giant fennel (Ferula communis). J Nat Prod 67:2108–2110
Bajqar J (2004) Organophosphates/nerve agent poisoning: mechanism of action, diagnosis, prophylaxis, and treatment. Adv Clin Chem 38:151–216
Baraldi PG, Saponaro G, Tabrizi MA, Baraldi S, Romagnoli R, Moorman AR, Varani K, Borea PA, Preti (2012) Pyrrolo- and pyrazolo-[3,4-e][1,2,4]triazolo[1,5-c]pyrimidines as adenosine receptor antagonists. Bioorg Med Chem 20:1046–1059
Bylov IE, Vasylyev MV, Bilokin YV (1999) Synthesis and anti-inflammatory activity of N-substituted 2-oxo-2H-1-benzopyran-3-carboxamides and their 2-iminoanalogues. Eur J Med Chem 11:997–1001
El-Agordy AM, Abd El-Latif MS, El-Hady NA, Fakery AH, Bedair AH (2001) Heteroaromatization with 4- Hydroxycoumarin Part II: synthesis of Some New Pyrano[2,3-d]pyrimidines[1,2,4]triazolo[1,5-c]pyrimidines and Pyrimido[1,6-b][1,2,4]triazine Derivatives. Molecules 6:519–527
Ellman GL, Courtney KD, Andres VJ, Featherstone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95
Ferreira A, Proença C, Serralheiro MLM, Araujo MEM (2006) The in vitro screening for acetylcholinesterase inhibition and antioxidant activity of medicinal plants from Portugal J Eth Pharmacol 108:31–37
Fest C, Schmidt KJ (1973) Chemistry of Organophosphorus Compounds. Springer-Verlag, Berlin, pp 122–124
Francesca C, Maria LG, Elena P, Marinella V (2001) Isothiazoles. Part 12: isothiazolylphosphonates, a new class of isothiazole dioxides. Tetrahedron 57:5455–5459
Francisco P, Ana Maria OR, Eduardo MDM, Marta R, Jaione P (2003) Aza-Wittig reaction of N-phosphazenes with carbonyl compounds and phenylisocyanate. Synthesis of 4-amino-3-phosphoryl-2-azadienes and pyrazine-phosphonates. Tetrahedron 59:2617–2623
Gonçalves ADS, França TCC, Figueroa-Villarb JD, Pascutti PG (2011a) Molecular Dynamics Simulations and QM/MM Studies of the Reactivation by 2-PAM of Tabun Inhibited Human Acethylcolinesterase. J Braz Chem Soc 22(1):155–165
Gonçalves ADS, França TCC, Figueroa-Villarb JD, Pascutti PG (2011b) Molecular dynamics simulations and QM/MM studies of the reactivation by 2-PAM of tabun inhibited human acethylcolinesterase. J Braz Chem Soc 22(1):155–165
Gorobets NY, Yousefi BH, Belaj F, Kappe CO (2004) Rapid microwave-assisted solution phase synthesis of substituted 2-pyridone libraries. Tetrahedron 60:8633–8644
Howes MR, Houghton P (2003) Plants used in Chinese and Indian traditional medicine for improvement of memory and cognitive function. J Pharmacol Biochem Behav 75:513–527
Kidane AG, Salacinski H, Tiwari A, Bruckdorfer KR, Seifalian AM (2004) Anticoagulant and antiplatelet agents: their clinical and device application(s) together with usages to engineer surfaces. Biomacromolecules 5:798–813
Kontogiorgis CA, Hadjipavlou-Litina DJ (2004) Synthesis and biological evaluation of novel coumarin derivatives with a 7-azomethine linkage. Bioorg Med Chem Lett 14:611–614
Kosolapov G (1950) Organophosphorus compounds. Wiley: New York, Chapter 7
Kovalenko SM, Bylov IE, Sytnik KM, Chernykh VP, Bilokhin YV (2000) A new pathway to 3-hertarly-2-oxo-2H-chromenes: on the proposed mechanisms for the reaction of 3-carbamoyl-2-iminochromenes with dinucleophiles. Molecules 5(10):1146–1165
Lise DB, Didier V, Jean-Francois L, Jane S, Paul-Alin J (2007) Synthesis of triazolylalkylphosphonate starting from ω-azidoalkylphosphonates or ω-alkynylphosphonates. Tetrahedron 63:9677–9684
Ma T, Liu L, Xue H, Li L, Han C, Wang L, Chen Z, Liu G (2008) The chemical library and structure-activity relationship of Calanolide-A analogues against HIV-1. J Med Chem 51:1432–1446
Mado N, Bunta W, Liyou H, Bun-ichi S, Kei W, Keiichi F, Hideyuki S, Jun H (2014) Glutathione-analogous peptidyl phosphorus esters as mechanism-based inhibitors of c-glutamyl transpeptidase for probing cysteinyl-glycine binding site. Bioorg Med Chem 22:1176–1194
Mata AT, Proença C, Ferreira AR, Serralheiro MLM, Nogueira JMF, Araújo MEM (2007) Antioxidant and antiacetylcholinesterase activities of five plants used as Portuguese food spices. Food Chem 103:778–786
Meth-Cohn O, Narine B (1987) Versatile new synthesis of quinolines, thienopyridines and related fused pyridines. Tetrahedron Lett 19:2045–2048
Moyo M, Ndhlala AR, Finnie JF, Staden JV (2010) Phenolic Composition, antioxidant and acetylcholinesterase inhibitory activities of Sclerocarya birrea and Harpephyllum caffrum (Anacardiaceae) extracts. Food Chem 123:69–76
Musa MA, Cooperwood JS, Khan MO (2008) A review of coumarin derivatives in pharmacotherapy of breast cancer. Curr Med Chem 15(26):2664–2679
Nam SO, Park DH, Lee YH, Ryu JH, Lee YS (2014) Synthesis of aminoalkylsubstituted coumarin derivatives as acetylcholinesterase inhibitors. Bioorg Med Chem 22:1262–1267
Orhan G, Orhan I, Sener B (2006) Recent developments in natural and synthetic drug research for Alzheimer’s disease. Lett Drug Des Discov 3(4):268–274
Rahmouni A, Romdhane A, Ben Saïd A, Majouli K, Ben Jannet H (2014a) Synthesis of new pyrazole and antibacterial pyrazolopyrimidine derivatives. Turk J Chem 38:210–221
Rahmouni A, Romdhane A, Besbes M, Elie N, Touboul D, Ben Jannet H (2014b) Synthesis of novel pyrazolo[3, 4d]pyrimidinone derivatives as cytotoxic inhibitors. Mediterr J Chem 2:679–690
Rashad AE, Shamroukh AH, Hegab MI, Awad HM (2005) Synthesis of some biologically active pyrazoles and C-nucleosides. Acta Chim Slov 52:429–434
Rashad AE, Hegab MI, Abdel-Megeid RE, Ali MM, Abdel-Megeid FME (2010) Synthesis and antitumor evaluation of some newly synthesized pyrazolopyrimidine and pyrazolotriazolopyrimidine derivatives. Phosphor Sulfur Silicon 185:74–83
Razavi SF, Khoobi M, Nadri H, Sakhteman A, Moradi A, Emami S, Foroumadi A, Shafiee A (2013) Synthesis and evaluation of 4-substituted coumarins as novel acetylcholinesterase inhibitors. Eur J Med Chem 64:252–259
Romdhane A, Gharbi R, Mighri Z (2003) Condensation of iminoethers with diarylnitrilimines. Novel synthesis of 1,2,4-triazoles. J Soc Chem Tunisie. 5:189–192
Romdhane A, Hammami S, Trimeche B, Gharbi R, Hamza MA, Mighri Z (2008) Novel synthesis of 1,2,4-oxadiazoles by condensation of arylamidoximes with N-substituted iminoethers. Heterocycl Commun 14:45–49
Troev KD (2006) Chemistry and applications of H-phosphonate. Elsevier, Amsterdam, pp 253–261
Weiqin J, George A, James JF, Olivia L, Pamela T, Jun X, Peifang Z, Joseph G, Keith D, Scott L, Zhihua S (2006) New progesterone receptor antagonists: phosphorus-containing 11b-aryl-substituted steroids. Bioorg Med Chem 14:6726–6736
Yanchang S, Yuming Z, Jei S (2002) Regiospecific synthesis of perfluoroalkylated pyrrolo[2,1-a]isoquinolinyl phosphonates. J Fluor Chem 116:157–161
Zaki MEA (1998) Synthesis of novel fused heterocycles based on pyrono [2,3-c]pyrazole. Molecules 3:71–79
Zhi H, Chen L, Zhang L, Liu S, Wan DCC, Lin H, Hu C (2008) 5H-thiazolo[3,2a]pyrimidine derivatives as a new type of acetylcholinesterase inhibitors. Arkivoc, xiii: 266–227
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Romdhane, A., Said, A.B., Cherif, M. et al. Design, synthesis and anti-acetylcholinesterase evaluation of some new pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine derivatives. Med Chem Res 25, 1358–1368 (2016). https://doi.org/10.1007/s00044-016-1576-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-016-1576-0