
Abstract
A series of amide-linked 1,4-disubstituted 1,2,3-bistriazoles was tested for antimicrobial activity against Gram-positive bacteria Staphylococcus aureus and Bacillus subtilis, Gram-negative bacteria Escherichia coli and two fungal strains Aspergillus niger and Candida albicans. The antimicrobial evaluation data indicated that most of the compounds exhibited potential activity. To describe activity on the structural basis, QSAR studies were performed and statistically significant models were developed. Further, binding interactions of two active compounds 17 and 10 to active sites of E. coli topoisomerase II DNA gyrase B and C. albicans lanosterol 14α-demethylase (1.14.13.70) (CYPLI) (cytochrome P450 51) enzymes, respectively, were also examined.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Over the past two decades, bacterial and fungal multi-drug resistance has been substantially developed. Medicinal chemists are mainly attracted toward the synthetic strategies that allow assembling a large number of active compounds in short time period. Copper-catalyzed azide–alkyne cycloaddition reaction between the organic azide and terminal alkyne becomes lately a very popular synthetic tool for the efficient construction of pharmacologically active compounds (Lauria et al., 2014). This reaction leads to regioselective synthesis of 1,4-disubstituted-1,2,3-triazoles and can also be performed in aqueous medium at room temperature. 1,2,3-Triazoles-containing compounds display fascinating biological activities such as anticancer (Singh et al., 2012), antitubercular (Patpi et al., 2012, Surineni et al., 2015), antimicrobial (Genin et al., 2000, Kaushik et al., 2014, Cao et al., 2014), anti-HIV (Whitting et al., 2006), antimalarial activities (Bakunov et al., 2010).
Quantitative structure–activity relationship (QSAR) studies help in quantitative prediction of the role of substrate structure to govern biological activities. The main objective of QSAR studies is to envisage biological activity and physicochemical properties and the rational identification of new drug candidates (Puzyn et al., 2010). QSAR studies can overcome the failure of novel drug agent in clinical trials, thereby saving the cost of product development (Dudek et al., 2006).
Molecular docking studies have led to the discovery of many novel drug candidates for a large number of enzymatic targets and remain one of the most rational approaches in drug discovery. Recently, bacterial DNA gyrase has drawn much interest as a validated target for finding potent antibacterial agents. DNA gyrase is mainly inhibited by some of the antibacterial drugs including ciprofloxacin, widely used for the treatment of bacterial contagious diseases (Ostrov et al., 2007). Lanosterol 14α-demethylase (P45014DM, CYP51) is a potential antifungal drug target for various heterocyclic compounds including imidazoles and triazoles, which catalyze the removal of the 14-methyl group (C-32) of lanosterol (Na, 2011). This is required for the biosynthesis of ergosterol, and inhibition of this enzyme results in growth inhibition of fungal cell (Ji et al., 2000). We describe herein, the antimicrobial screening, QSAR and docking simulations of amide-linked 1,4-disubstituted 1,2,3-bistriazoles (Lal et al., 2014).
Materials and methods
Antimicrobial activity
All the amide-linked 1,4-disubstituted 1,2,3-bistriazoles (Lal et al., 2014) were tested in vitro for antimicrobial activity against Gram-positive bacteria viz S. aureus (MTCC 3160) and B. subtilis (MTCC 441) and Gram-negative bacteria E. coli (MTCC 7443) and C. albicans (MTCC 183) and A. niger (MTCC 282) by standard serial dilution method (Cappucino and Sherman, 1999) using a stock solution of 50 μg/mL concentration. Double-strength nutrient broth and Sabouraud dextrose broth were employed as culture media for antibacterial and antifungal activities, respectively. Dimethylsulfoxide was used as solvent control. The stock solutions of the test compounds were serially diluted to get the concentration of 25–1.56 µg/mL and then inoculated with 100 µL of suspension of respective microorganism in sterile saline. The inoculated test tubes were incubated at 37 ± 1 °C for 24 h for S. aureus, B. subtilis, E. coli and C. albicans and at 25 ± 1 °C for 120 h in case of A. niger. Ciprofloxacin and fluconazole were used as standard drug for antibacterial and antifungal activities, respectively, and were also tested under similar conditions for comparison with the test compounds.
QSAR studies
Dataset
The data given in Table 1 were used for this study. The entire dataset of 18 compounds was employed to develop QSAR models.
Structure generation
The structures of the molecules were sketched and optimized as per the procedure given in molecular modeling studies. The molecules were prepared on the same conformation of basic skeleton amide-linked 1,4-disubstituted 1,2,3-bistriazoles.
Descriptors calculation
In order to obtain a QSAR model, compounds were always represented by the molecular descriptors. The molecular descriptors (863 descriptors including one-, two-, three-dimensional descriptors) were calculated with PaDEL Descriptor 2.12 program (Yap, 2011). The different descriptors can characterize a molecule from dissimilar aspects, but a few of them may express the same meanings with the same or similar values. To minimize duplication and errors, the constant or near-constant descriptors and descriptors having value zero for even one molecule were deleted. If the pairwise correlation of two descriptors was more than 0.75, the one having higher pair correlation with other descriptors was excluded from the original matrix of variables to minimize redundant information. The remaining descriptors underwent the subsequent variable selection process.
QSAR modeling and validation
The models were developed by multiple linear regression (MLR), and the stepwise multiple linear regression variable subset selection was applied for variable selection using SPSS software package (SPSS, 1996). Different parameters were utilized to validate the models. The correlation coefficient R was used as a measure of the goodness of fit. Other fitting criteria used were R 2adj, R 2 − R 2adj, RMSE (root-mean-squared error), MAE (mean average error), s (standard error of estimate) and F (Fisher’s value). For confirmation of model robustness and predictive power, the cross-validation coefficient \(Q_{\text{loo}}^{2}\) (leave one out) was used, where a model is built with n-1 compounds and the nth compound is predicted. Each compound iteratively held out from the set used for model development and predicted in turn. A sign of the model performance is achieved from the cross-validation coefficient, which may be defined as \(Q_{\text{loo}}^{2}\) = (1 − PRESS/TSS).
Where TSS is the total sum of squares. PRESS (predictive error sum of squares) is the sum of the squared difference between the observed and the predicted values when the compound is left from the process of fitting. The model with high \(Q_{\text{loo}}^{2}\) value is assumed to have high predictive power. This is the sole technique that uses all the information available and is very relevant mainly in small datasets as is our case (Gramatica, 2007).
Docking simulations
To elucidate the mechanism by which the triazole derivatives induce antibacterial activity, the inhibitory activities of selected compounds were examined against DNA gyrase. Reported procedure (Kumar et al., 2013) was used for performing docking studies. 2D structures of the compounds were made with MavinSketch 5.10 (Mavin Sketch, 2012). Afterward, these structures were optimized, changed into 3D and cleaned with gradient optimization. The X-ray crystallographic structure of E. coli topoisomerase II DNA gyrase B along with co-crystallized ligand CBN (PDB ID: 1KZN) was taken from Brookhaven Protein Data bank (http://www.rcsb.org/pdb). Molecular modeling tool UCSF Chimera1.9 [(Pettersen et al., 2004) was utilized for preparation of protein for docking studies. All solvent molecules and co-crystallized ligands were deleted, and imperfect side chains were corrected with Dunbrack rotamer library (Dunbrack, 2002). After that, Gasteiger charges were calculated with Antechamber (Wang et al., 2006). The prepared molecules were saved in pdb format for further work out. These structures of ligands and proteins were transformed into pdbqt format with AutoDock Tools (AutoDock Tools, 2010). AutoDock Vina program was used for docking simulations. The Vina search space chosen was center_x = 19.1474427293, center_y = 28.3592144362, center_z = 36.8274474404, size_x = 25.0, size_y = 25.0, size_z = 25.0 and center_x = 25.6113317536, center_y = 9.04563751488, center_z = 14.3998135289, size_x = 33.3016475075, size_y = 25.0, size_z = 25.0 for E. coli topoisomerase II DNA gyrase and C. albicans lanosterol 14α-demethylase, respectively. The exhaustiveness was set to be 8.
Docking protocols were validated using reported crystal structures of protein–ligand complex. The root-mean-square deviation (RMSD) between the conformations of the CBN from the X-ray crystal structure and those from AutoDock Vina was <2 Å, signifying that the protocols chosen for the AutoDock Vina docking studies were practical to imitate the X-ray structure. These protocols were utilized for docking of the compounds under study into the active site of DNA gyrase B. Results were visualized with the help of Discovery Studio version 3.5 (Discovery Studio, 2012) and PyMOL (2006).
Homology modeling of CaCYP51
The sequences of C. albicans lanosterol 14α-demethylase (1.14.13.70) (CYPLI) (cytochrome P450 51) was retrieved from UniProt database (P10613) and submitted to SWISS-MODEL workspace. The SWISS-MODEL (Arnold et al., 2006, Benkert et al., 2011, Biasini et al., 2014) template library (SMTL version 2014-09-10, PDB release 2014-09-05) was searched with Blast and HHBlits for evolutionary-related structures matching the target sequence. For each identified template, the template’s quality was predicted from features of the target–template alignment. The template 4k0f (lanosterol 14α-demethylase, seq identity = 65.067) with the highest quality was chosen for building of model. Promod-II was used for model building based on the target–template alignment. Conserved coordinates between the template and the target were copied from the template to the model. Remodeling of insertions and deletions was accomplished using a fragment library, and side chains were reconstructed. At last, the geometry of the resulting model was cleaned. The QMEAN scoring function (Benkert et al., 2011) was used for evaluation of global and per-residue model quality.
Results and discussion
Antimicrobial activity
In vitro antimicrobial activity of amide-linked 1,4-disubstituted 1,2,3-bistriazoles (1–18) was tested against two Gram-positive bacteria Staphylococcus aureus (MTCC 3160) and Bacillus subtilis (MTCC 121), Gram-negative bacteria Escherichia coli (MTCC 40) and two fungal strains viz Aspergillus niger (MTCC 281) and Candida albicans (MTCC 183) by standard serial dilution method (Cappucino and Sherman, 1999) using a stock solution of 50 μg/mL concentration. Ciprofloxacin and fluconazole were used as standard drugs for antibacterial and antifungal activity, respectively. Minimum inhibitory concentrations (MIC) were determined in µM/mL and are presented in Table 2.

The antibacterial activity data indicated that most of the compounds exhibited good to high activity. In case of S. aureus, compounds 3 (MIC, 0.0029 µM/mL), 4 (MIC, 0.0026 µM/mL), 16 (MIC, 0.0026 µM/mL) and 17 (MIC, 0.0026 µM/mL) displayed almost twofold activity to that of standard ciprofloxacin (MIC, 0.0047 µM/mL), while compounds 1b (MIC, 0.0058 µM/mL), 5 (MIC, 0.0052 µM/mL), 7 (MIC, 0.0058 µM/mL), 9(MIC, 0.0058 µM/mL), 11 (MIC, 0.0052 µM/mL), 12 (MIC, 0.0052 µM/mL), 15 (MIC, 0.0058 µM/mL) and 18 (MIC, 0.0052 µM/mL) have been found to exhibit comparable activity to the standard. Further, most of the nitro-substituted bistriazoles were found to be more effective than the methyl-substituted one.
In case of B. subtilis, compound 1 (MIC, 0.0029 µM/mL) was found to be most active and exhibited almost twofold activity to that of standard (MIC, 0.0047 µM/mL). Compounds 4(MIC, 0.0052 µM/mL), 6 (MIC, 0.0052 µM/mL), 8 (MIC, 0.0058 µM/mL), 10 (MIC, 0.0052 µM/mL), 12 (MIC, 0.0052 µM/mL), 15 (MIC, 0.0058 µM/mL), 16 (MIC, 0.0052 µM/mL) and 17 (MIC, 0.0052 µM/mL) displayed almost equal potency to that of reference. Many nitro-substituted bistriazoles were found to be more potent than the methyl-substituted one.
Likewise, compounds 7 (MIC, 0.0029 µM/mL), 14 (MIC, 0.0029 µM/mL) and 17 (MIC, 0.0026 µM/mL) showed almost twofold activity compared to the standard (MIC, 0.0047 µM/mL) against E. coli. Compounds 4 (MIC, 0.0052 µM/mL), 8 (MIC, 0.0058 µM/mL), 9 (MIC, 0.0058 µM/mL), 10 (MIC, 0.0052 µM/mL), 11 (MIC, 0.0052 µM/mL), 15 (MIC, 0.0058 µM/mL), 16 (MIC, 0.0052 µM/mL) and 18 (MIC, 0.0052 µM/mL) have comparable activity to that of reference. Notably, all the analogues derived from pyridyl bisalkyne were very potent against the Gram-negative bacteria E. coli except 13. In most of the cases, the activity of these bistriazoles was found to be superior to analogues derived from isophthaloyl, terephthaloyl bisalkynes. The presence of nitro group in all bistriazoles leads to an increase in activity except in case of compounds derived from terephthaloyl bisalkyne. Furthermore, compounds 4, 15, 16 and 18 exhibited very good and broad spectrum activity against all the three bacterial strains under study.
The perusal of antifungal activity data shows that most of the compounds exhibited good activity. In case of C. albicans, compounds 2 (MIC, 0.0058 µM/mL), 3 (MIC, 0.0058 µM/mL), 4 (MIC, 0.0052 µM/mL), 7 (MIC, 0.0058 µM/mL), 8 (MIC, 0.0058 µM/mL), 10 (MIC, 0.0052 µM/mL), 11 (MIC, 0.0052 µM/mL), 13 (MIC, 0.0058 µM/mL), 8 (MIC, 0.0058 µM/mL) and 16 (MIC, 0.0052 µM/mL) showed almost equal potency to reference drug fluconazole (MIC, 0.0051 µM/mL). Likewise, compounds 10 (MIC, 0.0052 µM/mL) and 12 (MIC, 0.0052 µM/mL) with nitro substituent at phenyl rings were found to be most active against A. niger than other compounds and showed comparable activity to that of standard. As evident from the activity data, all the tested compounds with 3-methyl and 2-nitro substituent showed good inhibitory activity against C. albicans. The activity data reflect that most of the bistriazoles under study were found to be more potent against C. albicans than A. niger.
QSAR studies
To shed light on the structure–activity relationships, QSAR studies were carried out for antibacterial and antifungal activities. Biological activity data determined as MIC values were first changed into pMIC values and used as dependent variables in QSAR studies and are shown in Table 1. Different 2D and 3D descriptors were calculated and used as independent variables in QSAR studies. The values of descriptors used in successful model development are shown in Table 1. In the present QSAR study, a dataset containing eighteen compounds was used and underwent multiple linear regression analysis for generation of QSAR models. Correlation matrix to indicate correlation between activity and descriptors as well as intercorrelation between descriptors is shown in Table 3. No monoparametric model was found to be suitable for description of these activities. Therefore, multi-parametric models were developed and best QSAR equation for S. aureus was as follows:
QSAR model for antibacterial activity against S. aureus
(n = 18; R = 0.852; R 2adj = 0.666; R 2−R 2adj = 0.059; s = 0.155; F = 12.318; \(Q_{\text{loo}}^{2}\) = 0.597; RMSE = 0.137; MAE = 0.111; RMSEcv = 0.166; MAEcv = 0.138)
Here and afterward, n = number of compounds, R = correlation coefficient, R 2adj = adjusted coefficient of determination, s = standard error, F = Fisher’s value, \(Q_{\text{loo}}^{2}\) = cross-validated coefficient (leave one out), RMSE = root-mean-squared error, and MAE = mean average error.
From the statistical data of the model, it can be said that it is a good QSAR model and has good predictive ability with value of \(Q_{\text{loo}}^{2}\) = 0.597. The value of \(Q_{\text{loo}}^{2}\) more than 0.5 is the necessary condition for a valid QSAR model. Low values of RMSE and MAE prove that the model is free from errors. The predicted activities by this equation are shown in Table 4 along with observed activities, and both are compared in the plot displayed in Fig. 1. The plot between observed activities and residuals (Fig. 2) shows that the values of residuals are spread on both positive and negative sides of the zero line, which proves that the developed model is free from systematic error. The positive sign of the coefficient of ALOGP2 shows that the activity is directly correlated with this parameter, while negative signs of coefficients of WLambda2.mass and Weta3.eneg indicate that the activity is correlated with these descriptors in inverse manner. The standardized coefficients of these parameters are 0.5699 (ALOGP2), −0.6774 (WLambda2.mass) and −0.3951 (Weta3.eneg). It can be clearly seen that the WLambda2.mass is most contributing descriptor followed by ALOGP2 and Weta3.eneg. WLambda2.mass is a directional WHIM parameter, weighted by atomic masses. Weta3.eneg is a directional WHIM, weighted by Mulliken atomic electronegativites (Todeschini and Gramatica, 1998). ALOGP2 is square of ALogP (Ghose-Crippen LogKow) and represents the lipophilicity of the molecules (Ghose and Crippen, 1986, 1987).

Plot of observed pMICsa versus calculated pMICsa for QSAR model described by Eq. 1

Plot of observed pMICsa versus residual pMICsa for QSAR model described by Eq. 1
The antibacterial activity against B. subtilis was explained by the following QSAR equation.
QSAR model for antibacterial activity against B. subtilis
(n = 14; R = 0.905; R 2adj = 0.786; R 2 − R2adj = 0.033; s = 0.079; F = 24.926; \(Q_{\text{loo}}^{2}\) = 0.670; RMSE = 0.070; MAE = 0.061; RMSEcv = 0.094; MAEcv = 0.081)
Four compounds (1, 3, 11 and 12) were found to be outlier; hence, these were excluded in model development procedure. It is a biparametric model in which both descriptors Wnu1.mass and WPSA-1 are correlated with the observed activity in a positive manner as the signs of coefficients of both parameters are positive. The standardized coefficients for Wnu1.mass and WPSA-1 are 0.858 and 0.512, respectively, showing higher importance of Wnu1.mass as compared to WPSA-1 in determination of activity against B. subtilis. Wnu1.mass is a directional WHIM, weighted by atomic masses (Todeschini and Gramatica, 1998). WPSA-1 is a charged partial surface area (CPSA) parameter calculated by following formula: PPSA-1 * total molecular surface area/1000, where PPSA-1 is partial positive surface area, i.e., sum of surface area on positive parts of molecule (Stanton and Jurs, 1990).
The parameters Wlambda2.unity along with MDEC-23 described the antibacterial activity of the compounds against E. coli, and the best QSAR model obtained is depicted in Eq. 3.
QSAR model for antibacterial activity against E. coli
(n = 16; R = 0.920; R 2adj = 0.823; R 2 − R 2adj = 0.023; s = 0.095; F = 35.893; \(Q_{\text{loo}}^{2}\) = 0.758; RMSE = 0.086; MAE = 0.067; RMSEcv = 0.108; MAEcv = 0.084)
Two compounds (13 and 16) were found to be outliers in this QSAR model. Negative sign of both Wlambda2.unity and MDEC-23 proved that the activity is negatively correlated with these descriptors. The standardized coefficients for Wlambda2.unity and MDEC-23 are −0.610 and −0.812, respectively, which shows that MDEC-23 is more contributing toward activity than Wlambda2.unity. MDEC-23 is molecular distance edge (MDE) parameter describing molecular distance edge between all secondary and tertiary carbons (Liu et al., 1998). Wlambda2.unity is a directional WHIM descriptor, weighted by unit weights (Todeschini and Gramatica, 1998).
Best QSAR model developed for describing antifungal activity against C. albicans was biparametric model derived for 15 compounds. Three compounds (1, 3 and 11) were found to be outlier and so not used in development of QSAR model. The equation is shown below:
QSAR model for antifungal activity against C. albicans
(n = 15; R = 0.975; R 2adj = 0.942; R 2 − R 2adj = 0.008; s = 0.034; F = 114.586; \(Q_{\text{loo}}^{2}\) = 0.923; RMSE = 0.033; MAE = 0.029; RMSEcv = 0.041; MAEcv = 0.036)
The coefficient of VPC-5 is positive, showing that the activity is correlated with this parameter in positive manner, while VP-7 is correlated in negative way to the activity as the coefficient of this descriptor is negative in the equation. The standardized coefficients for VPC-5 and VP-7 are 0.904 and −1.790, respectively, proving more contribution of VPC-5 toward activity determination as compared to VP-7. VPC-5 is a chi path cluster descriptor describing valence path cluster of order 5, while VP-7 is a chi path descriptor discussing valence path of order 7 (Kier and Hall, 1976).
The predicted activities, residuals along with observed activities for all above discussed QSAR models, are displayed in Table 1. The plots between observed and calculated activities as well as between observed activity and residuals are shown in Figs. 3, 4, 5, 6, 7, 8 for QSAR models 2, 3 and 4, respectively. All these plots show that the developed models have good predictive ability and are free from errors. No statistically significant model was found for activity against A. niger.

Plot of observed pMICbs versus calculated pMICbs for QSAR model described by Eq. 2

Plot of observed pMICbs versus residual pMICbs for QSAR model described by Eq. 2

Plot of observed pMICec versus calculated pMICec for QSAR model described by Eq. 3

Plot of observed pMICec versus residual pMICec for QSAR model described by Eq. 3

Plot of observed pMICca versus calculated pMICca for QSAR model described by Eq. 4

Plot of observed pMICca versus residual pMICca for QSAR model described by Eq. 4
All the models discussed above are statistically significant models as all the statistical parameters are in valid range. The value of correlation coefficient is more than 0.9 for models 2, 3 and 4, while it is more than 0.85 for model 1. Also, the values of R 2adj are very high and the difference R 2 − R 2adj is very less for all models, showing that R 2adj is very near to variance explained by the models. The values of standard error are very low for all models. All these facts prove that the developed models have very good fitting ability. The predictive ability of all these models is confirmed by high value of leave-one-out cross-validation coefficient (\(Q_{\text{loo}}^{2}\)), which is more than 0.5 (Golbraikh and Tropsha, 2002), and low values of RMSE and MAE. The difference between RMSE and RMSEcv is very less establishing the fact that described models have sufficient generalizability (Gramatica, 2007). Different compounds were found to be outliers for different QSAR models, and these were response outliers for which the reference value of response is invalid as QSAR models developed including these molecules displayed high residual values for these compounds (Furusjo et al., 2006).
From the above discussed QSAR models, it can be clearly observed that antibacterial activity of the compounds is generally explained by WHIM (weighted holistic invariant molecular) descriptors which are invariant to molecule rotation and translation in space and no alignment of compounds is needed. They include information regarding the complete 3D molecular structure in terms of their size, shape, symmetry and atom distribution. Calculation of these indices is accomplished from (x, y, z) coordinates of a 3D structure of the compound, generally from a spatial minimum energy conformation, within dissimilar weighting schemes, in a simple way and signifies a very general approach to portray molecules in a unitary theoretical framework (Todeschini and Gramatica, 1994, 1997, 1998).
These parameters offer the invariant information by making use of the principal component analysis (PCA) on the centered coordinates of the atoms forming the molecule. This changes the molecule into the space that confines nearly all the variance. In this space, several statistics along with variance, proportions, symmetry and kurtosis are calculated and these act as directional descriptors. By merging the directional descriptors, non-directional descriptors are also described. The input of each atom can be weighted by a certain chemical property, giving different principal components capturing variation within the specified property. The atoms can be weighted by van der Waals volume, mass, atomic polarizability, atomic electronegativity, molecular electrostatic potential and Kier and Hall’s electrotopological index (Dudek et al., 2006).
The antifungal activity is mainly explained by chi connectivity indices. Interestingly, the significance of WHIM, MDE as well as CPSA descriptors in antibacterial and chi parameters in antifungal activity points to the fact that dissimilar structural necessities are required for a molecule to be active against bacterial and fungal targets.
Docking studies
The foregoing studies point out that some compounds seem to be promising antimicrobial agents. Therefore, in order to understand the enzyme–inhibitor key interactions that contribute to the most stable complex conformation, a series of docking simulations was conducted to identify the binding mode of two potent compounds (17 and 10) to E. coli DNA gyrase II and fungal lanosterol 14α-demethylase, respectively (Trott and Olson, 2010). The X-ray crystallographic structures of E. coli DNA gyrase B protein (PDB code: 1KZN) were retrieved from protein data bank, while theoretical structure of fungal lanosterol 14α-demethylase (CYP51) was prepared by homology modeling. Lanosterol 14α-demethylase (1.14.13.70) (CYPLI) (cytochrome P450 51) was submitted to SWISS-MODEL workspace, and it was aligned with the template 4k0fA having 65.067 % sequence identity with the target. The global and per-residue model quality was checked by the QMEAN scoring function whose value was found to be −2.091.
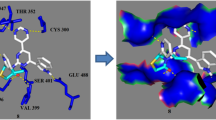
Three-dimensional structures of the most active compounds under study were docked into the active site of these proteins. The best ranked solution of compound 17 in DNA gyrase exhibited hydrogen bonding, electrostatic and hydrophobic interactions with the active site residues (Figs. 9, 10). 3-Nitro groups created hydrogen bond interactions web with Val 120, Ser 121 and Thr 165, while both hydrogens of both amido groups formed strong hydrogen bonds with carbonyl oxygen of Asp 49. One of the 3-nitrophenyl rings was involved in pi–donor-type hydrogen bonding with Asn 46, whereas one triazole ring was sandwiched between Asp 49 and Arg 76 by electrostatic-type interactions (pi–anion and pi–cation, respectively). Asp 49 also showed a pi–sigma interaction with this pyrazole ring. Distances of all these interactions are depicted in Table 4. The theoretical binding affinity of this conformation was −9.0 kcal/mol. All these residues are also involved in stable complex formation of clorobiocin with the enzyme. Therefore, it can be assumed that the compounds under study inhibit the DNA gyrase B successfully, which may be the reason behind their antibacterial action.

Compound 17 docked into active site of 1KZN showing various types of interactions, i.e., hydrogen bond (green lines), electrostatic (orange lines), pi–sigma (purple lines) and pi–alkyl (light pink) (Color figure online)

Surface diagram of 1KZN along with docked molecule 17
The lowest energy complex between 10 and lanosterol 14α-demethylase showed hydrophobic interactions primarily (Figs. 11, 12). Interestingly, one of the triazole rings was stacked against phenyl ring of Phe 233 showing pi–pi stacking interactions, while one 2-nitrophenyl ring was involved in T-shaped pi–pi stacking interactions with heme molecule present in the active site, showing phenyl–heme interaction. Leu 376 exhibits two pi–sigma interactions, with triazole ring and 2-nitrophenyl ring. One electrostatic (pi–cation) interaction was observed between Lys 90 and 2-nitrophenyl ring. Pro 230, Met 508, Leu 87 and Lys 90 created pi–alkyl interactions with the compound under study. Distances of all these interactions are shown in Table 5. The theoretical binding affinity of this molecule was −11.6 kcal/mol. All these interactions prove the efficacy of compound under study as a good antifungal agent.

Compound 10 docked into active site of lanosterol 14α-demethylase showing various types of interactions, i.e., hydrogen bond (green lines), pi–pi (pink lines), electrostatic (orange lines), pi–sigma (purple lines) and pi–alkyl (light pink) (Color figure online)

Ribbon diagram of lanosterol 14α-demethylase along with docked molecule 10 (blue). Heme molecule is shown in yellow green color (Color figure online)
Conclusion
In conclusion, amide-linked 1,4-disubstituted 1,2,3-bistriazoles were screened for antimicrobial activity and were found to posses potent activities. Some of the compounds displayed better or comparable activities to that of standards used. QSAR model developed had good fitting ability and predictive power. QSAR studies revealed that the WHIM, MDE and CPSA parameters are important for antibacterial activity, while chi descriptor explained the antifungal activity trends. Further, various interactions of 17 and 10 to active sites of E. coli topoisomerase II DNA gyrase B and C. albicans lanosterol 14α-demethylase (1.14.13.70) (CYPLI) (cytochrome P450 51) enzymes, respectively, clearly indicate the high potency of these compounds. These results can be used for the development of novel triazole-based drug agents.
References
Arnold K, Bordoli L, Kopp J, Schwede T (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22:195–201
AutoDock Tools (version 1.5.6 rc2), Stefano Forte. Molecular Graphics Laboratory, Department of Molecular Biology, The Scripps Research Institute, (1999–2010) http://mgltools.scripps.edu
Bakunov SA, Bakunova SM, Wenzler T, Ghebru M, Werbovetz KA, Brun R, Tidwell RR (2010) Synthesis and antiprotozoal activity of cationic 1,4-diphenyl-1H-1,2,3-triazoles. J Med Chem 53(1):254–272
Benkert P, Biasini M, Schwede T (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27(3):343–350
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:252–258
Cao X, Sun Z, Cao Y, Wang R, Cai T, Chu W, Hu W, Yang Y (2014) Design, synthesis, and structure-activity relationship studies of novel fused heterocycles-linked triazoles with good activity and water solubility. J Med Chem 57(9):3687–3706
Cappucino JG, Sherman N (1999) Microbiology—a laboratory manual, 4th edn. Addison Wesley Longman Inc., Harlow, p 263
Discovery Studio v4.0 client, Accelrys Software Inc. (2005–2014)
Dudek AZ, Arodzb T, Gálvez J (2006) Computational methods in developing quantitative structure-activity relationships (QSAR): a review. Comb Chem High Throughput Screen 9(3):213–228
Dunbrack RL (2002) Rotamer libraries in 21st century. Curr Opin Struct Biol 12(4):431–440
Furusjo E, Svenson A, Rahmberg M, Andersson M (2006) Chemosphere 63(1):99–108
Genin MJ, Allwine DA, Anderson DJ, Barbachyn MR, Emmert DE, Garmon SA, Graber DR et al (2000) Substituent effects on the antibacterial activity of nitrogen-carbon-linked (azolylphenyl)oxazolidinones with expanded activity against the fastidious gram-negative organisms Haemophilus influenzae and Moraxella catarrhalis. J Med Chem 43(5):953–970
Ghose AK, Crippen GM (1986) Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships. I. Partition coefficients as a measure of hydrophobicity. J Chem Inf Comput Sci 7:565–577
Ghose AK, Crippen GM (1987) Atomic physicochemical parameters for three-dimensional-structure-directed quantitative structure-activity relationships. Modeling dispersive and hydrophobic interactions. J Chem Inf Comput Sci 27(1):21–35
Golbraikh A, Tropsha A (2002) Beware of q2! J Mol Graph Model 20(4):269–276
Gramatica P (2007) Principles of QSAR models validation: internal and external. QSAR Comb Sci 26(5):694–701
Ji H, Zhang W, Zhou Y, Zhang M, Zhu J, Song Y, Lu¨ J, Zhu J (2000) A three-dimensional model of Lanosterol 14α-Demethylase of Candida albicans and its interaction with azole antifungals. J Med Chem 43(13):2493–2505
Kaushik CP, Lal K, Kumar A, Kumar S (2014) Synthesis and biological evaluation of amino acid-linked 1,2,3-bistriazole conjugates as potential antimicrobial agents. Med Chem Res 23(6):2995–3004
Kier LB, Hall LH (1976) Molecular connectivity in chemistry and drug research. Academic Press, New York
Kumar A, Kumar S, Jain S, Kumar P, Goyal R (2013) Study of binding of pyridoacridine alkaloids on Topoisomerase II using in silico tools. Med Chem Res 22(11):5431–5441
Lal K, Kaushik CP, Kumar K, Kumar A, Qazi AK, Hamid A, Jaglan S (2014) One-pot synthesis and cytotoxic evaluation of amide-linked 1,4-disubstituted 1,2,3-bistriazoles. Med Chem Res 23(8):4761–4770
Lauria A, Delisi R, Mingoia F, Terenzi A, Martorana A, Barone G, Almerico AM (2014) 1,2,3-Triazole in heterocyclic compounds, endowed with biological activity, through 1,3-dipolar cycloadditions. Eur J Org Chem 16:3289–3306
Liu S, Cao C, Li Z (1998) Approach to estimation and prediction for normal boiling point (NBP) of alkanes based on a novel molecular distance edge (MDE) Vector, lambda. J Chem Inf Comput Sci 38:387–394
Na YM (2011) Synthesis and activity of novel 1-halogenobenzylindole linked triazole derivatives as antifungal agents. Bull Korean Chem Soc 32(1):307–310
Mavin Sketch 5.10.1 ChemAxon Ltd. (1998–2012) http://www.chemaxon.com
Ostrov DA, Prada JAH, Corsino PE, Finton KA, Le N, Rowe TC (2007) Discovery of novel DNA gyrase inhibitors by high-throughput virtual screening. Antimicrob Agents Chemother 51(10):3688–3698
Patpi SR, Pulipati L, Yogeeswari P, Sriram D, Jain N, Sridhar B, Murthy R, Devi AT et al (2012) Design, synthesis, and structure-activity correlations of novel dibenzo[b, d]furan, dibenzo[b, d]thiophene, and N-methylcarbazole clubbed 1,2,3-triazoles as potent inhibitors of Mycobacterium tuberculosis. J Med Chem 55(8):3911–3922
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera a visualization system for exploratory research and analysis. J Comput Chem 25(13):1605–1612
Puzyn T, Leszczynski J, Cronin MTD (2010) Recent advances in QSAR studies: methods and applications. Cronin MTD quantitative structure–activity relationships (QSAR)—applications and methodology, 8th edn. Springer, New York, pp 3–11
PyMOL(TM) Molecular Graphics System, Version 0.99rc6, Copyright © 2006 DeLano Scientific LLC
Singh P, Raj R, Kumar V, Mahajan MP, Bedi PM, Kaur T, Saxena AK (2012) 1,2,3-Triazole tethered β-lactam-chalcone bifunctional hybrids: synthesis and anticancer evaluation. Eur J Med Chem 47(1):594–600
Stanton DT, Jurs PC (1990) Development and use of charged partial surface area structural descriptors in computer assisted quantitative structure property relationship studies. Anal Chem 62(21):2323–2329
Surineni G, Yogeeswari P, Sriram D, Kantevari S (2015) Rational design, synthesis and evaluation of novel-substituted 1,2,3-triazolylmethyl carbazoles as potent inhibitors of Mycobacterium tuberculosis. Med Chem Res 24(3):1298–1309
Todeschini R, Gramatica P (1997) The Whim theory: new 3D molecular descriptors for QSAR in environmental modeling. SAR and QSAR Environ Res 7:89–115
Todeschini R, Gramatica P (1998) New 3D molecular descriptors: the WHIM theory and QAR applications. Persepect Drug Discov 9:355–380
Todeschini R, Lasagni M, Marengo E (1994) New molecular descriptors for 2D and 3D structures. J Chemom 8:263–272
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31(2):455–461
Wang J, Wang W, Kollman PA, Case DA (2006) Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25(2):247–260
Whiting M, Tripp JC, Lin YC, Lindstorm W, Olson AJ, Elder JH, Sharpless KB, Fokin VV (2006) Rapid discovery and structure activity profiling of novel inhibitors of human immunodeficiency virus type 1 protease enabled by the copper(I)-catalyzed synthesis of 1,2,3-triazoles and their further functionalization. J Med Chem 49(26):7697–7710
Yap CW (2011) PaDEL-Descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 32(7): 1466–1474
Acknowledgments
Authors are highly thankful for financial assistance from University Grants Commission, New Delhi.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lal, K., Kaushik, C.P. & Kumar, A. Antimicrobial evaluation, QSAR and docking studies of amide-linked 1,4-disubstituted 1,2,3-bistriazoles. Med Chem Res 24, 3258–3271 (2015). https://doi.org/10.1007/s00044-015-1378-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-015-1378-9