
Abstract
Activation of Takeda G-protein receptor 5 (TGR5) plays a key role in pathways associated with diabetes, metabolic syndrome, and autoimmune disease. Pharmacophore and 3D-quantitative structure–activity relationship modeling were applied to study the structure–activity relationship of TGR5 agonists. The best HypoGen pharmacophore hypothesis Hypo1 with a correlation coefficient of 0.93 consists of one hydrogen-bond acceptor, one aromatic ring and three hydrophobic features, whereas the best phase hypothesis AHHRR.1321 with favorable statistics (q 2 = 0.7613, r 2 = 0.927) has one hydrogen-bond acceptor, two hydrophobic features and two ring aromatic features. Furthermore, comparing those two models, the preferable AHHRR.1321 was employed as a novel searching tool for chemical databases to conduct virtual screening for new potential lead candidates. Consequently, refined Lipinski ‘rule of five’ and ADME properties were utilized as a filter to reduce less drug-like molecules. Among the hits, 10 non-steroidal compounds with good fitness score and physicochemical properties were identified.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The Takeda G-protein receptor 5 (TGR5) also known as G-protein bile acid receptor 1 (GPBAR1), belonging to the G-protein coupled receptor (GPCR) family, is a plasma membrane-bound bile acid receptor (Kawamata et al., 2003) found in many human and animal tissues, including liver, intestine, and brain (Kawamata et al., 2003; Vassileva et al., 2006). Some non-genomic actions of bile acid (BA) are mediated through the formation of cAMP as the activation of TGR5 (Maruyama et al., 2002). An accumulating body of evidence now demonstrates that TGR5 also acts in a number of processes important in inflammation (Wang et al., 2011). Moreover, the recent observations have revealed an unexpected role for GPBA in the nervous system, and it is a newly identified liver tumor suppressor in carcinogenesis (Duboc et al., 2014). In cell culture models, TGR5 has been linked to signaling pathways involved in metabolism, cell survival, proliferation, and apoptosis, suggesting a possible role of TGR5 in cancer development (Feng et al., 2007). Thus, its pharmacological modulation may furnish alternative therapeutic strategies to treat diabetes, obesity, other metabolic syndromes, inflammation, and so on (Gioiello et al., 2012). The recent studies also suggested that the activation of TGR5 in macrophages may be of utility in atherosclerosis (Pols, 2014).
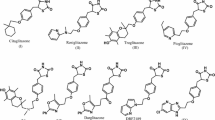
Depending on the chemical characteristics, TGR5 ligands can be classified into two types: steroidal and non-steroidal agents. The Pellicciari group reported their research for potent and selective TGR5 agonists based on natural bile acid. Among them, INT-777, one of the semisynthetic BA derivatives, exhibited good properties in vitro and in vivo and was considered as a promising anti-diabetic drug candidate in the preclinical study (Pellicciari et al., 2009). In addition, a number of pharmaceutical and biotech companies including Takeda Pharmaceuticals (2004), Kalypsys (Herbert et al., 2010a, b), GSK (SmithKline Beecham Corp., 2007), Hoffmann-La (2010), SIMM (Duan et al., 2012), Pfizer (Futatsugi et al., 2013), and Novartis have focused their efforts toward finding structurally diverse non-steroidal TGR5 agonists. Some representative structures are shown in Fig. 1. These agonists offer the potential to enable delivery of a tool compound with higher selectivity against other bile acid-mediated pathways, such as FXR, and lead to a wide range of small molecules, after all BAs derivatives vary a little (Jansen, 2010; Lavoie et al., 2010).

Reported representative TGR5 agonists
As an important method for drug discovery, computer-aided techniques have been applied in the identification of TGR5 agonists. Macchiarulo et al. (2008) developed a molecular interaction field analysis (MFA) and a 3D-quantitative structure–activity relationship study (3D-QSAR) of TGR5 agonists using a training set of 43 bile acid derivatives. Martin et al. (2013) constructed their homology model using bovine rhodopsin as template and then carried out the docking procedure. As the pharmacophore model of non-steroidal ligands has not been published, herein we focus on the uncovered area.
At first, we planned to generate a common pharmacophore model on the basis of TGR5 agonists containing BAs, terpenes, and non-steroidal molecules. However, the obtained model turned out to be too simple to differentiate TGR5 agonists from negative ones, it only has four common features: two hydrophobic spheres, one aromatic ring features, and one hydrogen-bond acceptor (HBA) (Fig. 2). The decoy set validation came to a bad result with EF value less than 1. Although the cost and correlation value of this number of false pharmacophore model were reasonable enough, the model obviously lacked specific information and might lead to an increasing uncredible virtual screening results. Furthermore, SAR studies have shown that BAs, terpenes, and non-steroidal are characterized with different pharmacophoric elements for TGR5 activation. Steroidal and non-steroidal ligands may bind to a common orthosteric site involving different interacting residues, or they may interact with different regions of the TGR5 binding pockets. Therefore, it was not proper for the further study. Due to those deficiencies, we aimed at the non-steroidal ligands for constructing the pharmacophore, which has been attracting more medicinal chemists.

Hypo1 geometric constraint generated by steroidal and non-steroidal compounds consists of one aromatic rings (R), two hydrophobic (H) features, and one hydrogen-bond acceptor (HBA). Pharmacophore features are color represented as blue for hydrophobic, green for hydrogen-bond acceptor, and yellow for ring aromatic feature (Color figure online)
In this work, we combined two 3D-quantitative SAR (QSAR) modeling tools to investigate key pharmacophoric features. We carry out both HypoGen and Phase in the generation of pharmacophore models to obtain the most significant features by comparison of both models (Zhang et al., 2009). These two kinds of models were further validated by various approaches to justify its qualification. Furthermore, we employed the preferable Phase pharmacophore model as a novel searching tool for chemical databases to conduct virtual screening for new potential lead candidates, with due cognizance of the refined Lipinski ‘rule of five’ and absorption, distribution, metabolism, excretion (ADME) properties. This information is relevant to extend, on a quantitative basis, the current structure–activity relationships of non-steroidal compounds as TGR5 modulators and will be beneficial to design new potent and selective agonists of the receptor.
Results and discussion
HypoGen pharmacophore study
A data-set of 29 TGR5 agonists belonging to several structural classes (Fig. 3) was collected and randomly divided in 17 training set and 10 test set compounds (Gioiello et al., 2012; Herbert et al., 2010a, b; Duan et al., 2012; Zhu et al., 2013a, b). Top 10 pharmacophore hypotheses were exported based on the activity values of the training set molecules. Hypogen produces three cost values: fixed cost, total cost, and null cost. The cost difference between null cost and fixed cost was found to be 106.81, and it was more than 70 bits. Configuration cost should be smaller than 17 for a good pharmacophore hypotheses since it represents the complexity of the hypotheses (Bharatham et al., 2006). Various cost values, correlation coefficient, RMS deviation, and pharmacophore features of 10 hypotheses are summarized in Table 1. Hypo1 consists of one HBA, three hydrophobic features (H), and one aromatic ring (R), which establishes the highest cost difference (88.05), best correlation coefficient (0.93), maximum fit value (13.54), and lowest root mean square (RMS) of 1.37. The fixed and the null cost values for the 10 hypotheses were 67.1629 and 173.97, respectively. Higher cost difference and correlation value with low RMS and error values have been observed for Hypo1 when compared with other hypotheses. Hence, Hypo1 was selected as a best hypothesis and employed for further analyses. Figure 4 shows the Hypo1 chemical features. Figure 5a and b represents the best pharmacophore model aligned with the most active and inactive molecules 6 and 23 with EC50 of 0.3 and 5,100 nm, respectively. The pharmacophore features are mapped well to the active molecule in the Fig. 5a. On the other hand, the feature of HBA in Fig. 5b could not fit well since it is a low active molecule. Our results indicated that the HBA moiety seemed to be essential for TGR5 agonists.

2D chemical structure of reported TGR5 agonists in the training set and test set together with their biological activity values (EC50)

Hypo1 geometric constraint generated by non-steroidal compounds consists of one hydrogen-bond acceptor (HBA), one ring aromatic (R), and three hydrophobic (H) features. Pharmacophore features are color represented as blue for hydrophobic, green for hydrogen-bond acceptor, and yellow for ring aromatic feature (Color figure online)

Best pharmacophore model Hypo1 aligned with training set compounds. a With most active compound 6 (EC50 = 0.3 nM). b With least active compound 23 (EC50 = 5 100 nM). Pharmacophore features are color represented as blue for hydrophobic, green for hydrogen-bond acceptor, and yellow for ring aromatic feature (Color figure online)
Random hypotheses built by Fischer validation (confidence of level of 95 %) are illustrated in Fig. 6. None of the exported pharmacophores had lower cost than the original hypotheses. It clearly showed that the Hypo1 hypothesis was not generated by chance, because its statistics were superior to all random hypotheses. Moreover, decoy set was generated to ensure whether Hypo1 could pick out active molecules from inactive compounds. EF and GH were calculated to evaluate the hypotheses. Decoy set contains active and inactive compounds of TGR5 agonists. Parameters such as total number of compounds in the hit list (H t), number of active percent of yields (%Y), percent ratio of actives in the hit list (%A), EF, false negatives, false positives, and GF were calculated (Nagarajan et al., 2011). The false positives and false negatives are 357 and 2, respectively. The EF and GF are calculated to be 3.784 and 0.25, respectively, which are very good indications of the high efficiency of the screening (Table 2). According to all the validations, we drew the conclusion that Hypo1 can be taken as further analyses such as virtual screening.

Fisher’s randomization test results
Phase pharmacophore study
As a first step, common features of pharmacophore hypotheses were generated, scored, and ranked by Phase. Four highly active compounds of the set (6, 9, 3, and 10 in Fig. 3) were selected, aiming at the definition of a reliable and not subjective alignment rule for the subsequent 3D-QSAR development. The top-ranked hypothesis (AHHRR. 1321) was formed by five features: one hydrogen-bond acceptors (A), two aromatic rings (R), and two hydrophobic features (H) (Fig. 7). Pharmacophore AHHRR.1321 (shown in Fig. 8 superimposed to the compound 6 and the compound 23) was allowed to confirm the perfect and worse match, related to the corresponding TGR5 activity. Taking into account this and comparation of the above SAR studies, it could be assumed that pharmacophore AHHRR.1321 actually accounts for relevant interactions between agonists and TGR5. Consequently, it is not arbitrary to state that matching the pharmacophore may indicate binding to this receptor.

AHHRR. 1321 consists of one hydrogen-bond acceptor (HBA), two ring aromatic (R), and two hydrophobic (H) features. Pharmacophore features are color represented as blue for hydrophobic, red for hydrogen-bond acceptor, and yellow for ring aromatic feature (Color figure online)

Pharmacophoric features aligned to the (a) highest active ligand (b) least active. Pharmacophore features are color represented as blue for hydrophobic, red for hydrogen acceptor donor, and yellow for ring aromatic feature (Color figure online)
Pharmacophore AHHRR.1321 was then used to align the molecules for the development of an atom-based 3D-QSAR analysis. Models containing one to three PLS factors were generated, whose statistical parameters are reported in Table 3. The model with three PLS factors was preferred and selected, since it performed better on the whole than those with fewer factors. The high correlation and cross-validated correlation coefficients (R 2 = 0.927 and Q 2 = 0.7613, respectively) together with the high Pearson R value (R-Pearson = 0.8704) suggested a close correspondence between predicted and actual EC50 activity values, indicative of a model with strong predictive power and significance. A scatter plot of experimental against predicted activities was created to assess the results (see Fig. 9), which showed that EC50 values were effectively predicted for both training and test set molecules. These features, along with the small number of PLS factors, the large F value supported the reliability of the approach. It is significant to mention that, all the test set compounds, the differences between experimental and calculated EC50 values were within one order of magnitude for all the compounds, demonstrating that the 3D-QSAR model was reasonably efficient in the estimation of TGR5 activity.

Correlation graph between experimental and predicted TGR5 activity using pharmacophore-based QSAR model. a Training set. b Test set
The 3D-QSAR model represents 3D characteristics as cubes that signify the model and color according to the positive or negative coefficients. The 3D-QSAR results were visualized using 3D plots of the crucial volume elements occupied by ligands. The 3D plot representation of the model as a whole, superimposed to derivatives 6 and 23, is shown in Fig. 8. In this representation, blue and red cubes indicate positive and negative coefficients, respectively, that is volumes in which the occupying atoms of the ligands cause an increase or a decrease of activity. Cubes having small positive and negative coefficients, which therefore did not greatly affect activity, were filtered out by setting a 1.7e−02 coefficient threshold. Notably, compound 6, showing the most potent inhibition against TGR5, mainly occupies blue regions (Fig. 10a), while the less active compound 23 occupies mainly the red regions (Fig. 10b). Figure 11a shows that electron-withdrawing favorable effects (blue cubes) are present close to carbonyl. A few electron-withdrawing unfavorable effects (pink cubes) were found to be distributed in a discrete fashion, and these are not quite reliable for any prediction. Figure 11b depicts that hydrophobic favorable effects (blue cubes) are located adjacent to three aromatic rings.

Atom-based 3D-QSAR model visualized in the context of the most active ligand (a) and least active ligand (b) in the training set

3D pictorial representation of the cubes generated using the QSAR model. Blue cubes indicate favorable regions and red cubes indicate unfavorable region for the activity. Atom-based 3D-QSAR model visualized in context of the ligand 6 (a) cubes for electron-withdrawing groups (b) cubes for hydrophobic regions (Color figure online)
After the generation of the 3D-QSAR model and in order to perform its validation, a decoy set which was the same as the Hypo1 validation was selected from the literature. Forty-five active TGR5 agonists were also included in the decoy set to calculate the statistical parameters such as goodness of hit score (GH) and enrichment factor (EF). GH and EF are the two main parameters which play an important role in predicting the capability of the pharmacophore hypothesis. The EF and GF are calculated to be 33.48 and 0.95, respectively, which are very good indications of the high efficiency of the screening (Table 4).This result provided further evidence that the correlation shown by the model was not accidental.
Comparison of the pharmacophore models
Both HypoGen and Phase would contain a workflow of selecting a training set, generating conformers, finding hypotheses from actives, and scoring hypotheses. Nevertheless, there are also some differences between these two methodologies, for instance rejecting hypotheses using inactives and building QSAR models would be the specific steps in HypoGen and Phase, respectively (Zask et al., 2009). Fewer features would be generated by HypoGen, attributing to the rejection of inactives. Meanwhile, maximum features and minimum interfeature distance would make the same point. Contrarily, less essential character may be summarized by Phase, so that the selection of candidates and validation of the chosen hypotheses are extremely significant. However, different generations of conformers and scoring algorithm would lead to different hypotheses. Therefore, it seems more accurate by utilizing models and screening queries when analogical conclusion restricted by various approaches (Evans et al., 2008). The difference between those two models is obvious. Comparing the superposition of Hypo1, AHHRR.1321, and most active compound 1, only H5 and R11 of AHHRR.1321 are located in the same site as the features in Hypo1. One hydrophobic feature is substituted by R12. As we all know, the ring aromatic feature is a particular case of hydrophobic feature. In short, the R12 in AHHRR.1321 was more specific than Hypo1 with regard to TGR5. With regard to the decoy set validation, the EF value of AHHRR.1321 is 33.48, which is almost tenfold than that of Hypo1. We can conclude that AHHRR.1321 is more effective to identify active compounds from inactive ones. Thus, AHHRR1321 was used as the protocol for virtual screening.
Database screening
Pharmacophore screening
Virtual screening is valuable for discovering lead compounds in a more cost-efficient, less resource-intensive manner compared with experimental methods (Marcu et al., 2000). By employing this pharmacophore model as a search template, we have performed a database search for potential TGR5 agonists from Specs database of over 200,000 compounds. Totally, compounds satisfied all the critical features in AHHRR.1321 and 932 compounds were considered for further analyses based on the cut-off fitness value of 1.00.
Drug-like filter
Drug-likeness properties are an important indicator for selecting the compounds for in vitro studies, which includes molecular or physicochemical properties that contribute to favorable Lipinski’s rule of five. Hence, we further sorted these 932 compounds using the refined Lipinski’s rule of five and finally 301 compounds were further considered for ADME studies. The percentage of the human oral absorption of published compounds was found to be 68–100 %. Through calculating the QPlog Po/w, QPlog S, and human oral absorption, 10 hits were retrieved in the end (Fig. 12). For selected lead compounds, the partition coefficient (QPlog Po/w) and water solubility (QPlog S) was within the permissible range of 4.47–5.95 and −5.06 to −8.04, respectively, and the human oral absorption was 100 %.

2D chemical structure of 10 retrieved compounds
Conclusion
In summary, we built two different pharmacophore models by Phase and HypoGen separately using the same training set and test set of TGR5 non-steroidal agonists. It was the first time that a typical pharmacophore hypothesis of TGR5 agonists belonging to the class of bile acid derivatives was reported. Hypo1 indicated that one HBA, three HY, and one R feature would be the common features of potential TGR5 agonists. Meanwhile, features required by Phase hypothesis AHHRR.1321 is different, one HBA, two HY, and two ring aromatic features. Moreover, a number of approaches could be applied for validating each hypothesis. Fischer validation and decoy set validation suggest that both pharmacophore hypotheses were reliable for the discovery of novel TGR5 agonists. Comparing the two results, the AHHRR.1321 was applied for further screening and a different filter strategy was endeavored. Finally, 10 non-steroidal compounds were identified which deserved further study. It is our hope that the pharmacophore generated will be valuable for researchers seeking to develop novel TGR5 agonists. Interestingly, three compounds 34, 35, and 39 bear the same skeleton benzopyrimidine. Given that the reported TGR5 agonists do not hold this moiety, it may be promising to lead to a new series of TGR5 modulators.
Materials and methods
Collection of data-set
TGR5 agonists were gained from reported paper with EC50 ranging from 0.3 nM to 5.1 μM. Prior to the establishment of the models, all molecules were minimized and modified by ChemBio 3D elementarily. The data-set of 29 compounds was then divided randomly into training and test set, respectively in such a way that both sets consisted of highly active, medium active, and least active compounds. Training and test sets consisted of 17 and 10 compounds, respectively. The in vitro inhibitory activity data are reported as EC50.
HypoGen pharmacophore model
Pharmacophore modeling is one of the most potent and rapid method to discover a novel scaffold. The automatic generation procedure using the HypoGen module was adopted for generation of the hypotheses. HypoGen uses the activity values of the small compounds in the training set to generate the hypothesis. The hypothesis may reveal the critical features for binding. Considering the chemical features of the compounds included in the training set, four features were selected: HBA, hydrogen-bond donor (HBD), hydrophobic (HY), and ring aromatic (RA).
The training set of 17 compounds was used to construct HypoGen pharmacophores. The best mode of conformation generation algorithm was used for generating conformations. The related parameters which were chosen for generating conformations were as follows: energy threshold: 20 kcal/mol and maximum conformations: 255. The minimum and maximum features in the hypothesis run were sets 1 and 5, respectively. The default Uncertainty value 3 had been changed to 2 for effectively correlating the training set with their activity.
In terms of validation, firstly Fischer’s randomization method was used to measure the statistical significance of our model. In this model, 19 random spreadsheets were generated to obtain the 95 % of confidence level. Secondly, decoy set was used. In the decoy set method, a database of 1,539 decoys was obtained from a collection offered by DrugBank (subset of random FDA-approved small molecule drug structures without TGR5 reported). Forty-five active TGR5 agonists were also included in the decoy set to calculate the statistical parameters such as goodness of hit score (GH) and enrichment factor (EF). GH and EF are the two main parameters which play an important role in predicting the capability of the pharmacophore hypothesis. All queries were performed using the Ligand Pharmacophore Mapping protocol. Fast and flexible approach was used.
Phase pharmacophore model
A pharmacophore-based 3D-QSAR study was carried out using PHASE implemented in the Maestro 9.4 modeling package (Schrodinger, Inc., LLC, New York, USA). Like HypoGen, Phase could be utilized for pharmacophore hypothesis generation, activity estimate, and virtual screening. As for the development of Phase pharmacophore and 3D-QSAR models, a total of 29 ligands were adopted. Several sets of pharmacophore sites for all ligands were then created using a set of available pharmacophore features (hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobe (H), negative ionizable (N), positive ionizable (P), and aromatic ring (R)). Retain specified chiralities of stereoisomers and neutralize of ionization were set in beginning preparation. Conformations search was carried out by ConfGen and a thorough sampling method applied MMFFs force field. The number of 100 conformers per rotatable bond and maximum number of 1,000 conformers per structure were set. Preprocess and postprocess minimization steps were 1,000 and 500, respectively. For each molecule, a maximum of 1,000 conformers was generated with a relative energy difference of 20 kcal/mol, the redundant conformers being removed after setting the root-mean-square deviation (RMSD) value at 1 Å. Four highly active compounds were used to build the pharmacophores. In this study, compounds with pEC50 above 8.50 were defined as ‘active’, while below 6.5 as ‘inactive’, which sorted 4 actives and 5 inactives. While finding the common pharmacophore, Nsites was set as the available maximum to be five, and all the four active compounds were required to find and score hypotheses (Zhang et al., 2009). Scoring of pharmacophore with respect to activity of ligand was conducted using default parameters for site, vector, and volume terms. The selected of hypotheses focused on both survival score and the alignment of indispensable sites (Almerico et al., 2010). Common pharmacophore hypotheses were identified, scored, and ranked using conformational analysis and a tree-based partitioning technique.
Atom-based QSAR models were generated for TGR5 hypothesis using the 17-member training set and a grid spacing of 1.0 Å. QSAR models containing one to three PLS factors were generated. Best 3D-QSAR model was selected based on the correlation coefficient values in training set molecules, which was further validated by predicting activities of 10 test set molecules. Three external test set predictors, namely Q 2, Pearson R, and RMSE, were used to validate the developed model. The same training set and test set were used for the construction and validation of the Phase pharmacophore study. The generated hypotheses were assessed by statistical parameters and correlated with the observed and estimated activity for the training set of 17 compounds and test set of 10 compounds. The best hypothesis was chosen for the alignment of compounds for further 3D-QSAR study. To validate the hypothesis, decoy set method was used as the same as Hypogen, a database of 1,539 decoys was obtained from a collection offered by DrugBank (subset of random FDA-approved small molecule drug structures without TGR5 agonists reported). Forty-five active TGR5 agonists were also included in the decoy set to calculate the statistical parameters such as goodness of hit score (GH) and enrichment factor (EF). All queries were performed using the Advanced Pharmacophore Screening protocol in Maestro 9.4.
Virtual screening protocol
The validated QSAR model was used as a 3D structural query for retrieving potential inhibitors from Specs database of 207,018 molecules. The Specs databases were downloaded from their official website. Multiple conformations of the databases were generated using Schrödinger. The Specs database was then screened with the pharmacophore models in Phase module using the following running conditions: (a) generated conformers during search by a rapid sampling method. (b) The maximum number of conformers was set as 50 and retained up to 5 conformers per rotatable bond. (c) returned at most 1 hit per molecule, 10,000 hits total. (d) must match on all five site points. The rest of options and parameter were set as default.
Drug-like analysis
At a glance, the inspection of some physicochemical properties (lipophilicity (clog P), molecular weight (MW), HBAs, and hydrogen-bond donors (HBD)) of these three structural classes of TGR5 ligands reveals that all of them are characterized by relatively high MW and lipophilicity. Indeed, 90 % of non-steroidal ligands fall in a shorter HBA between 1 and 12, with an average value of 6.02 ± 2.80. The combination of the above properties results in different compliance of each structural class of TGR5 ligands to Lipinski’s ‘rule of five’. Therefore, refined Lipinski’s ‘rule of five’ filter [(1) hydrogen-bond donors should be less than 2, (2) HBAs should be more than 1 and less than 6, (3) molecular weight should be less than 550 Da, and (4) log P should be more than 3 and less than 6] was utilized to exclude false-positive drug-like compounds. As oral absorbability is a significant factor, the ADME properties were calculated by QikProp which predicts required principle and physiochemical descriptors of possible drug compounds. The program was processed in normal mode-predicted principle descriptors and physiochemical properties for all known and screened compounds with detailed analysis of the log P (octanol/water), QPlog S (predicted aqueous solubility), QPlog BB (predicted brain/blood barrier partition co efficient), and percentage human oral absorption.
References
Almerico AM, Tutone M, Lauria A (2010) 3D-QSAR pharmacophore modeling and in silico screening of new Bcl-xl inhibitors. Eur J Med Chem 45:4774
Bharatham N, Bharatham K, Lee KW (2006) Pharmacophore identification and virtual screening for methionyl-tRNA synthetase inhibitors. J Mol Graph Model 25:813–823
Duan H, Ning M, Chen X, Zou Q, Zhang L, Feng Y, Zhang L, Leng Y, Shen J (2012) Design, synthesis, and antidiabetic activity of 4-phenoxynicotinamide and 4-phenoxypyrimidine-5-carboxamide derivatives as potent and orally efficacious TGR5 agonists. J Med Chem 55:10475–10489
Duboc H, Taché Y, Hofmann AF (2014) The bile acid TGR5 membrane receptor: from basic research to clinical application. Dig Liver Dis 4:302–312
Evans DA, Doman TN, Thorner DA, Bodkin MJ (2008) 3D QSAR methods: phase and catalyst compared. J Chem Inf Model 47:1248–1257
Feng J, Zhang Z, Wallace MB, Stafford JA, Kaldor SW, Kassel DB, Navre M, Shi L, Skene RJ, Asakawa T, Takeuchi K, Xu R, Webb DR, Gwaltney SL (2007) Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J Med Chem 50:2297–2300
Futatsugi K, Bahnck KB, Brenner MB, Buxton J, Chin JE, Coffey SB, Dubins J, Flynn D, Gautreau D, Guzman-Perez A, Hadcock JR, Hepworth D, Herr M, Hinchey T, Janssen AM, Jennings SM, Jiao W, Lavergne SY, Li B, Li M, Munchhof MJ, Orr STM, Piotrowski DW, Roush NS, Sammons M, Stevens BD, Storer G, Wang J, Warmus JS, Wei L, Wolford AC (2013) Optimization of triazole-based TGR5 agonists towards orally available agents. Med Chem Comm 4:205–210
Gioiello A, Rosatelli E, Nuti R, Macchiarulo A, Pellicciari R (2012) Patented TGR5 modulators: a review (2006–present). Expert Opin Ther Pat 22:1399–1414
Herbert MR, Pinkerton AB, Siegel DL (2010a) Heterocyclic modulators of tgr5 for treatment of disease. WO 2010016846 A1
Herbert MR, Siegel DL, Staszewski L, Cayanan C, Banerjee U, Dhamija S, Anderson J, Fan A, Wang L, Rix P, Shiau AK, Rao TS, Noble SA, Heyman RA, Bischoff E, Guha M, Kabakibi A, Pinkerton AB (2010b) Synthesis and SAR of 2-aryl-3-aminomethylquinolines as agonists of the bile acid receptor TGR5. Bioorg Med Chem Lett 20:5718–5721
Hoffmann-La R (2010) Novel phenyl amide or pyridyl amide derivatives and their use as GPBAR1 agonists. WO 2010049302 A1
Jansen PLM (2010) A new life for bile acids. J Hepatol 52:937–938
Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M (2003) AG protein-coupled receptor responsive to bile acids. J Biol Chem 278:9435–9440
Lavoie B, Balemba OB, Godfrey C, Watson CA, Vassileva G, Corvera CU, Nelson MT, Mawe GM (2010) Hydrophobic bile salts inhibit gallbladder smooth muscle function via stimulation of GPBAR1 receptors and activation of KATP channels. J Physiol 588:3295–3305
Macchiarulo A, Gioiello A, Thomas C, Massarotti G, Nuti R, Rosatelli E, Sabbatini P, Schoonjans K, Auwerx J, Pellicciari R (2008) Molecular field analysis and 3D-quantitative structure–activity relationship study (MFA 3D-QSAR) unveil novel features of bile acid recognition at TGR5. Journal of chemical information and modeling. J Chem Inform Model 48:1792–1801
Marcu MG, Chadli A, Bouhouche I, Catelli N, Neckers LM (2000) The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J Biol Chem 275:37181–37186
Martin RE, Bissantz C, Gavelle O, Kuratli C, Dehmlow H, Richter HGF, Sander UO, Erickson SD, Kim K, Pietranico-Cole SL, Alvarez-Sánchez R, Ullmer C (2013) 2-Phenoxy-nicotinamides are potent agonists at the bile acid receptor GPBAR1 (TGR5). ChemMedChem 8:569–576
Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K (2002) Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 298:714–719
Nagarajan S, Ahmed A, Choo H, Cho YS, Oh KS, Lee BH, Shin KJ, Pae AN (2011) 3D QSAR pharmacophore model based on diverse IKKbeta inhibitors. J Mol Model 17:209–218
Pellicciari R, Gioiello A, Macchiarulo A, Thomas C, Rosatelli E, Natalini B, Sardella R, Pruzanski M, Roda A, Pastorini E, Schoonjans K, Auwerx J (2009) Discovery of 6α-ethyl-23 (S)-methylcholic acid (S-EMCA, INT-777) as a potent and selective agonist for the TGR5 receptor, a novel target for diabesity. J Med Chem 52:7958–7961
Pols TW (2014) TGR5 in inflammation and cardiovascular disease. Biochem Soc Trans 42:244–249
SmithKline Beecham Corp. Chemical compounds (2007) Use of sulfonamide derivatives in the treatment of disorders of the metabolism and the nervous system. WO 2007127505 A3
Takeda Pharmaceuticals (2004) Screening method. WO 2004043468 A1
Vassileva G, Golovko A, Markowitz L, Abbondanzo SJ, Zeng M, Yang S, Hoos L, Tetzloff G, Levitan D, Murgolo DJ, Keane K, Davis HR Jr, Hedrick J, Gustafson EL (2006) Targeted deletion of Gpbar1 protects mice from cholesterol gallstone formation. Biochem J 398:423–430
Wang YD, Chen WD, Yu D, Forman BM, Huang W (2011) The G-Protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-κB) in mice. Hepatology 54:1421–1432
Zask A, Kaplan J, Verheijen JC, Richard DJ, Curran K, Brooijmans N, Bennett EM, Toral-Barza L, Hollander I, AyralKaloustian S, Yu K (2009) Morpholine derivatives greatly enhance the selectivity of mammalian target of rapamycin (mTOR) inhibitors. J Med Chem 52:7942–7945
Zhang J, Liu G, Tang Y (2009) Chemical function-based pharmacophore generation of selective κ-opioid receptor agonists by catalyst and phase. J Mol Model 15:1027–1041
Zhu J, Ning M, Guo C, Zhang L, Pan G, Leng Y, Shen J (2013a) Design, synthesis and biological evaluation of a novel class of potent TGR5 agonists based on a 4-phenyl pyridine scaffold. Eur J Med Chem 69:55–68
Zhu J, Ye Y, Ning M, Mandi A, Feng Y, Zou Q, Kurtan T, Leng Y, Shen J (2013b) Design, synthesis, and structure–activity relationships of 3,4,5-trisubstituted 4,5-dihydro-1,2,4-oxadiazoles as TGR5 agonists. ChemMedChem 8:24
Acknowledgments
This work was supported by the the National Natural Science Foundation of China (Nos. 30801428, 81273378), Specialized Research Fund for the Doctoral Program of Higher Education (No. 200803161011), the Natural Science Foundation of Jiangsu Province (No. BK2011622), and the Fundamental Research Funds for the Central Universities (No. ZJ13051).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Zhou, C., Zou, F., Xu, Y. et al. Identification of new non-steroidal TGR5 agonists using virtual screening with combined pharmacophore models. Med Chem Res 24, 2561–2572 (2015). https://doi.org/10.1007/s00044-014-1310-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00044-014-1310-8