
Abstract
Type 1 diabetes (T1D) is characterized by an immune-mediated progressive destruction of the insulin-producing β-cells. Proinflammatory cytokines trigger endoplasmic reticulum (ER) stress and subsequent insulin secretory deficiency in cultured β-cells, mimicking the islet microenvironment in T1D. β-cells undergo physiologic ER stress due to the high rate of insulin production and secretion under stimulated conditions. Severe and uncompensated ER stress in β-cells is induced by several pathological mechanisms before onset and during T1D. We previously described that the small drug Compound A (CpdA), a selective glucocorticoid receptor (GR/NR3C1, nuclear receptor subfamily 3, group C, member 1) ligand with demonstrated inflammation-suppressive activity in vivo, is an effective modulator of effector T and dendritic cells and of macrophages, yet, in a GR-independent manner. Here, we focus on CpdA’s therapeutic potential in T1D cellular and animal models. We demonstrate that CpdA improves the unfolded protein response (UPR) by attenuating ER stress and favoring the survival and function of β-cells exposed to an environment of proinflammatory cytokines. CpdA administration to NODscid mice adoptively transferred with diabetogenic splenocytes (from diabetic NOD mice) led to a delay of disease onset and reduction of diabetes incidence. Histological analysis of the pancreas showed a reduction in islet leukocyte infiltration (insulitis) and preservation of insulin expression in CpdA-treated normoglycemic mice in comparison with control group. These new findings together with our previous reports justify further studies on the administration of this small molecule as a novel therapeutic strategy with dual targets (effector immune and β-cells) during autoimmune diabetes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The endoplasmic reticulum (ER) of β-cells plays an essential role in the production of insulin. Due to the high demand for insulin secretion during food intake, β-cells undergo physiological ER stress.
The imbalance between protein loading and folding capacities causes ER stress leading to the activation of the unfolded protein response (UPR). The UPR primarily functions to mitigate ER stress under physiological conditions and promotes insulin production and cell survival [1]. Under pathological conditions, the UPR cannot cope with the chronic hyperactivation of ER stress leading to β-cell dysfunction and eventually death.
ER stress and β-cell insulin secretory deficiency have been shown to precede the onset of autoimmune diabetes [2]. There is cumulative evidence supporting the role of proinflammatory cytokines, elevated in the islet microenvironment during autoimmune diabetes, in the activation of ER stress, oxidative stress, β-cell dysfunction and death [3,4,5]. In experimental diabetes, pharmacological restoration of UPR in β-cells has been reported as a preventive and/or therapeutic intervention [6, 7].
We have reported that Compound A (2-(4-acetoxyphenyl)-2-chloro-N-methyl-ethylammonium chloride; CpdA), described as a non-steroidal glucocorticoid receptor (GR) ligand with dissociative properties [8], is an effective modulator of effector T lymphocytes and dendritic cells [9, 10]. We provided evidence that CpdA immunomodulatory effects might be explained by a GR-independent inactivation of the NF-κB intracellular signaling pathway after TLR4 activation on dendritic cells [10].
Considering the role of the immune system in the pathogenesis of autoimmune diabetes along with our previous results on the immunomodulatory activity of CpdA, we asked whether CpdA might have beneficial effects counteracting cytokine-induced ER stress in β-cells and thus may exhibit therapeutic potential against the progression of experimental autoimmune diabetes.
Materials and methods
Reagents
Culture media, supplements and antibiotics were purchased from Gibco (Thermo Fisher Scientific, Carlsbad, CA, USA). Fetal Bovine Serum was from Natocor (Córdoba, Argentina). Compound A (2-(4-acetoxyphenyl)-2-chloro-N-methyl-ethylammonium chloride; CpdA) was synthesized as described [10, 11]. Dex and RU486 were purchased from Sigma-Aldrich. Recombinant cytokines were from R&D Systems (Minneapolis, MN, USA).
Animals
NOD, NODscid, and C57BL/6 J mice (breeders from Jackson Laboratory, Bar Harbor, ME, USA) were housed under pathogen-free controlled environment (20–22 °C, 12 h light–dark cycle) in ventilated cages, and provided with food and water ad libitum. All procedures were conducted in accordance with the Guide for the care and use of laboratory animals, Eighth edition (2011). Studies were approved by the Animal Research and Care Committee (#0001 & #0069), FCEyN, University of Buenos Aires.
INS-1E cell line
The rat β-cell line INS-1E (Prof. Wollheim, University Medical Centre, Geneva, Switzerland) was used between passages 63 and 90, and cultured at 37 °C in a humidified atmosphere containing 5% (vol./vol.) CO2 in complete RPMI 1640 medium [11 mM glucose, 10% (vol./vol.) heat-inactivated fetal bovine serum (FBS), penicillin (50 IU/ml), streptomycin (50 µg/ml), L-glutamine (2 mmol/l), 2-mercaptoethanol (50 µmol/l), HEPES (10 mmol/l) and sodium pyruvate (1 mmol/l)]. The possible presence of mycoplasma was periodically checked by PCR. INS-1E were seeded at 40 × 103 cells/cm2 in multiwell plates (Nunc, Thermo Scientific, Denmark) in complete medium with charcoal-treated FBS.
Mice islets isolation and culture
Islets (C57BL/6 J) were isolated by collagenase digestion and handpicked after density gradient centrifugation [12]. For standardization, islets with 100–125 µm in diameter were considered as an islet equivalent (IEQ). Islets were cultured on ultra-low fixation plates (Corning Costar, Kennebunk, ME, USA), at 37 °C in humidified atmosphere containing 5% (vol./vol.) CO2 in RPMI 1640 medium containing 5.5 mM glucose, 10% charcoal-treated FBS, penicillin (50 IU/ml), streptomycin (50 µg/ml), L-glutamine (2 mmol/l) and HEPES (10 mmol/l) for 16–24 h prior to performing experiments.
Human islets isolation and culture
Human pancreata were obtained from the Center for Organ Recovery and Education, Pittsburgh, PA. Islets were isolated using a semiautomated method following collagenase intraductal injection as previously described [13, 14]. Islets were purified with a COBE 2991 cell separator using discontinuous Euro-Ficoll gradients; purity was assessed by dithizone staining, as described [15]. Available characteristics of the donors as well as islet preparations are summarized in Table S1.
Islets were handpicked and cultured in ultra-low attachment plates (Corning Costar, Kennebunk, ME, USA), at 37 °C in humidified atmosphere containing 5% (vol./vol.) CO2 in RPMI 1640 medium containing 5.5 mM glucose, 10% charcoal-treated FBS, penicillin (50 IU/ml), streptomycin (50 µg/ml), L-glutamine (2 mmol/l) and HEPES (10 mmol/l). For standardization, islets with a diameter of 150–200 μm were considered as an islet equivalent (IEQ). Islets were cultivated for 24–72 h before experimentation.
SDS-PAGE and Western blot
INS-1E cells were harvested on ice-cold PBS, washed and lysed in lysis buffer [50 mM Tris–HCl pH 7.4, 250 mM NaCl, 25 mM NaF, 2 mM EDTA, 0.1% Triton-X, protease inhibitors mix (Complete ULTRA, Roche)]. Protein concentration was determined using the BCA assay Kit (Pierce) and samples conserved at -20 °C. Proteins were separated by 8–12% SDS-polyacrilamyde gel electrophoresis (SDS-PAGE), blotted onto nitrocellulose or PVDF membranes (Amersham GE-Healthcare, UK) and incubated with primary antibodies: IκBα (#sc-371; Santa Cruz Biotechnology, Santa Cruz, CA, USA); iNOS (#610,332, BD Biosciences, San Jose, CA, USA); ORP150 (#ab124884), BIP (#ab21685, Abcam, Cambridge, MA, USA); phospho-IκBα (#9246), β-actina (#3700), phospho-eIF2α (#9721), eIF2α (#2103), ATF4 (#11,815), CHOP (#2895); PDI (#3501) (Cell Signaling Technology, Danvers, MA, USA). Blots were incubated with HRP-conjugated secondary antibodies (Bio-Rad, Hercules, CA, USA) and visualized using ECL (Supersignal; Thermo Fisher Scientific, Carlsbad, CA, USA).
Confocal microscopy
INS-1E were cultured 72 h onto fibronectin coated coverslips, treated as described in the figures, fixed by cold methanol and incubated with primary antibodies: NFκB p65 (RelA, #sc-8008) or GR (M-20) (#sc-1004, Santa Cruz Biotechnology). Secondary antibodies (1/200 dilution) were anti-goat or anti-rabbit Alexa Fluor 647 conjugated dye (Life Technology). The coverslips were mounted on slides with Mowiol and images were acquired on a Zeiss LSM 710 Confocal microscope (Carl Zeiss GmbH, Germany). Data acquisition was performed with ZEN Black 2011 software, and image quantification was performed using Fiji software.
Nitric oxide production
Nitrite was measured as an indicator of nitric oxide (NO) production using Griess reagent (1% sulphanilamide and 0.1% naphthyl ethylene diamine dihydrochloride in 2.5% phosphoric acid) at 570 nm [16].
Quantitative real-time PCR
Total RNA was extracted from INS-1E cells with TRIzol reagent (Thermo Fisher Scientific, Carlsbad, CA, USA) according to the manufacturer's instructions. Nucleic acid quantification and quality control were performed with a NanoDrop One (Thermo Fisher Scientific, Carlsbad, CA, USA). For cDNA synthesis, 1 μg RNA was reverse-transcribed using RevertAid Reverse Transcriptase (Thermo Fisher Scientific, Carlsbad, CA, USA) in the presence of RiboLock RNase Inhibitor (Thermo Fisher Scientific, Carlsbad, CA, USA) and oligo(dT) primers. Real-time PCR was performed on a Bio-Rad CFX96 Touch Real-Time PCR Detection System, using SYBR Green mix (Thermo Fisher Scientific, Carlsbad, CA, USA). All reactions were performed in triplicate and HPRT or RPL19 were used as normalization controls. Relative expression was calculated with the 2−ΔΔCT method [17]. Primers are listed in Table S2.
Transient transfections and luciferase reporter assays
To determine ATF6 pathway activation, we used a reporter plasmid containing the firefly luciferase gene under the control of five copies of ATF6 consensus binding site (5xATF6-LUC). To quantitatively measure XBP1 splicing, we used a splicing-specific reporter plasmid containing the coding sequence of firefly luciferase conjugated to the second ORF of XBP1u (XBP1u-LUC); luciferase is expressed only after IRE1-induced splicing removes the 26-nt intron.
Plasmids were transfected into INS-1E cells with Lipofectamine 3000 reagent (Thermo Fisher Scientific) in OptiMEM medium and cells were treated after 24 h. LUC activity in cell lysates was measured using the Luciferase measure kit (Promega) with a Junior Portable luminometer (Berthold, Bad Wild- bad, Germany). Cells were co-transfected with RSV-β-galactosidase expression vector, and β-gal activity was measured by ONPG assay as a normalization control for transfection efficiency.
Assessment of cell viability and apoptosis
For cell viability assays, INS-1E cells were seeded in 96-well plates. After treatment, medium was replaced by fresh medium containing 0.5 mg/mL MTT (Thermo Fisher Scientific, Carlsbad, CA, USA). After 3 h at 37 °C, media was replaced for 100 μL of acidified isopropanol (40 mM HCl) and incubated at room temperature 15 min. Absorbance was measured at 570 nm.
Apoptosis assessments were performed in isolated mouse islets. After treatment, islets were washed and stained with Hoechst 33,342 (10 μg/mL) and propidium iodide (PI; 5 μg/mL) for 30 min at 37 °C. Images were acquired under a Zeiss Axio Observer Z1 Inverted Phase Contrast Fluorescence Microscope (Carl Zeiss GmbH, Germany). The percentage of apoptotic cells was analyzed by two investigators blinded to the experiment using Fiji software.
Insulin quantification and Glucose-Stimulated Insulin Secretion (GSIS)
The quantification of insulin secreted by INS-1E cells and islets was performed by a sandwich ELISA [18]. For GSIS, cells/islets were incubated in Krebs–Ringer phosphate buffer (KRB: 135 mmol/L NaCl, 0.5 mmol/L NaH2PO4, 3.6 mmol/L KCl, 0.5 mmol/L MgCl2, 1.5 mmol/L CaCl2, 5 mM NaHCO3, pH 7.4), 10 mmol/L HEPES, 0.1% BSA, with 2 mmol/L glucose for a period of 2 h. Cells/islets were incubated in KRB-HEPES-BSA 2 mmol/L glucose for 1 h; the solution was collected and the cells/islets were incubated in KRB-HEPES-BSA 20 mmol/L glucose for an additional 1 h before collecting the solution. Secreted insulin was normalized to total protein content of cell/islet lysates and stimulation index (ratio between insulin released under high glucose versus low glucose conditions) was calculated. Protein concentration was determined using the BCA assay Kit (Pierce).
Adoptive transfer of diabetes in mice and CpdA treatment
Eight-week-old female NODscid mice were adoptively transferred with diabetogenic splenocytes (i.p. 5 × 106 cells/mice) isolated from diabetic NOD mice [16, 19] and injected i.p. with CpdA 100μg/200μL or vehicle (Control) three times a week from day -1 to day 50. Body weight was registered weekly and animals monitored for appearance of treatment-related adverse effects. Tail-blood glucose was measured with a glucometer (Optium Xceed®, Abbott Laboratories, North Chicago, IL, USA); diabetes was diagnosed when glycemia reached ≥ 300 mg/dl in two consecutive days. The incidence of diabetes between groups was compared by Kaplan–Meier analysis and the log-rank test.
Histological examination
The pancreata were fixed in 10% formaldehyde and embedded in paraffin. Insulin immunolabeling was performed on 7 μm tissue sections with anti-insulin (clone HB125, #MU029-UC, Biogenex, Fremont, CA, USA) and HRP-conjugated donkey anti-mouse (#715‐036–150, Jackson ImmunoResearch, Baltimore, PA, USA) and signal revealed with 3,3-diaminobenzidine (DAB) substrate; nuclei were counterstained with haematoxylin. Images were acquired under an optical microscope (Olympus CX31, Olympus, Tokyo, Japan). Two investigators blinded to the experiment scored at least 10 islets per mouse to calculate the infiltration percentage using the following criteria: 0, no insulitis; 1: < 25%; 2: 25–50%; 3: 50–75%; and 4: > 75%.
Statistical analysis
Results are presented as mean ± SD. Comparison between groups was carried out using paired or unpaired Student´s t-test or ANOVA followed by Bonferroni´s multiple comparison test, as appropriate. A p < 0.05 was considered to indicate a statistically significant difference. All statistical analyses were performed using GraphPad Prism version 6.0 Software.
Results
CpdA inhibits cytokine-triggered NF-κB pathway activation and reduces nitric oxide production in INS-1E cells
To assess the impact of CpdA on cytokine-induced β-cell dysfunction we first evaluated the effect of CpdA on the NF-κB pathway (Fig. 1a–e). CpdA pretreatment inhibited CYT-triggered IκBα phosphorylation (Fig. 1a,b) protecting its degradation (Fig. 1a,c) and hampering, NF-κB nuclear translocation in INS-1E cells (p < 0.05; Fig. 1d,e); a pathway related to CYT-induced NO production and β-cell apoptosis. Dex showed weaker inhibition on IκBα phosphorylation and degradation compared to CpdA, and did not prevent CYT-triggered NF-κB nuclear translocation in INS-1E cells (Fig. 1a,c). NO is an inducer of IL-1β-mediated ER stress and apoptosis of β-cells. CpdA reduced cytokine-triggered NO secretion by INS-1E cells (p < 0.01; Fig. 1f). This effect was observed as early as 6 h after challenge (Fig. S1a) and could be explained by the reduction in inducible Nitric Oxide Synthase (iNOS) mRNA (p < 0.01; Fig. 1g) and protein (p < 0.01; Fig. 1h,i) expression. 5-methylisothiourea sulfate (SMT), an inhibitor of iNOS activity, abrogated CYT-induced NO production in INS-1E (Fig. S1b).

CpdA inhibits cytokine-triggered NF-κB pathway activation and reduces nitric oxide (NO) production in INS-1E cells. a–c INS-1E cells were pretreated with vehicle, CpdA 10 μM or dexamethasone (Dex) 0.1 μM for 1 h and then challenged or not with IL-1β 100 pg/mL and IFN-γ 5 ng/mL (CYT). After indicated time, levels of phospho-IkBα and total IkBα were analyzed by Western blot. Representative blots (a) and quantitative analysis of phospho-IkBα (b) and IkBα (c) protein expression expressed as mean ± SEM of n = 4 independent experiments; β-actin was used as loading control. (*) p < 0.05 vs. vehicle. d INS-1 cells were treated as described in (a) with 30 min CYT stimulation. NF-κB (RelA) cellular localization was analyzed by immunofluorescence staining. A representative confocal microscopy pictures of INS-1E cells immunostained for NF-κB (red) in different experimental conditions as indicated; nuclei were stained with DAPI (blue); scale bars 10 μm. e Quantification of nuclear:cytoplasmic ratio of NF-kB staining from analysis of 5 separate high-power field images for each experimental condition. Data are shown as mean ± SD of n = 3 independent experiments. f INS-1E were treated as described in (a) with 16 h CYT stimulation. NO secretion was assessed by Griess reaction. Data are shown as mean ± SD of n = 5 independent experiments. g–i INS-1E were treated as described in (a) with 6 h CYT stimulation, iNOS mRNA and protein expression were analyzed by RT-qPCR and Western blot, respectively. Relative iNOS mRNA levels (g) normalized to HPRT expressed as mean ± SD of n = 3 independent experiments. Representative blots (h) and quantitative analysis of iNOS (i) protein expression expressed as mean ± SD of n = 3 independent experiments; β-actin was used as loading control. d–i (†) p < 0.05 vs. vehicle; (*) p < 0.05, (**) p < 0.01, (***) p < 0.001 vs. vehicle + CYT
CpdA hampers the cytokine-induced activation of ER stress and favors UPR pathways in INS-1E cells
The PERK/eIF2α/ATF4/CHOP pathway is one of the ER stress-induced signaling branches related to apoptosis in β-cells. CpdA treatment impaired the phosphorylation of eIF2α (p < 0.01; Fig. 2a,b) and decreased ATF4 (p < 0.01; Fig. 2a,c) and CHOP (p < 0.05; Fig. 2a,d) expression in CYT-challenged INS-1E cells. A similar effect was observed with the addition of Dex (p < 0.05, Fig. 2).

CpdA hampers the cytokine-induced activation of ER stress related pathways and favors unfolded protein response (UPR) pathways in INS-1E cells. a–d INS-1E cells were pretreated with vehicle, CpdA 10 μM or dexamethasone (Dex) 0.1 μM for 1 h and then challenged or not with IL-1β 100 pg/mL and IFN-γ 5 ng/mL (CYT). After 16 h, levels of phospho- and total eIF2α, ATF4 and CHOP were analyzed by Western blot. Representative blots (a) and quantitative analysis of phospho- and total eIF2α (b), ATF4 (c) and CHOP (d) protein expression expressed as mean ± SD of n = 3/4 independent experiments; β-actin was used as loading control. e–f INS-1 cells were transiently transfected with XBP1u-LUC (e) or 5xATF6-LUC (f) and RSV-βGal reporter plasmids. At 24 h post-transfection, cells were treated as described in (a). After 16 h, cells were collected and firefly luciferase (LUC) activity was measured and normalized against β-galactosidase (β-Gal) activity for transfection efficiency. Relative LUC activity is expressed as mean ± SD of n = 3 independent experiments; (†) p < 0.05 vs. vehicle; (*) p < 0.05, (**) p < 0.01, (***) p < 0.001 vs. vehicle + CYT
IRE1α-XBP1 and ATF6 pathways are an integral part of the UPR, involved in the regulation of ER chaperones expression and aimed at restoring homeostasis. We observed that CpdA favors the UPR stimulating IRE1-mediated XBP1 splicing (2.8-fold increase vs. Veh, p < 0.05 and 3.6-fold increase vs. CYT, p < 0.001; Fig. 2e) and counteracts a decreased (0.5 ± 0.1 vs. vehicle, p < 0.05) transcriptional activity of ATF6 in CYT-challenged INS-1E cells (1.5-fold increase vs. CYT, p < 0.01; Fig. 2f). Dex slightly upregulated IRE1-mediated Xbp1 splicing; an effect that was only significant in the absence of cytokines (1.7-fold increase, p < 0.05; Fig. 2e). On the other hand, a 1.4- and 2.4-fold increase of ATF6 transcriptional activity was observed in the presence of Dex in comparison with vehicle and CYT-challenged conditions, respectively (p < 0.05; Fig. 2f).
In the presence of CpdA the levels of BIP, a key UPR modulator, were restored in CYT-treated INS-1E cells (p < 0.05; Fig. 3a,b). Additionally, an enhancement in the expression of PDI and ORP150, chaperones involved in insulin folding and processing, was observed both in presence and absence of pro-inflammatory cytokines (between 1.3- and 2.26-fold increase, p < 0.05; Fig. 3a,c,d). No significant effect was observed on the level of these proteins under Dex treatment.

CpdA enhances the expression of ER chaperones in INS-1E cells. a–d INS-1E cells were pretreated with vehicle, CpdA 10 μM or dexamethasone (Dex) 0.1 μM for 1 h and then challenged or not with IL-1β 100 pg/mL and IFN-γ 5 ng/mL (CYT). After 16 h, levels of BIP, PDI and ORP150 were analyzed by Western blot. Representative blots (a) and quantitative analysis of BIP (b), PDI (c) and ORP150 (d) protein expression expressed as mean ± SD of n = 4 independent experiments; β-actin was used as loading control. (†) p < 0.05 vs. vehicle; (*) p < 0.05, (**) p < 0.01 vs. vehicle + CYT
CpdA acts independently of the GR in INS-1E cells
CpdA was originally described as a GR-ligand with dissociative properties. Interestingly, using indirect immunofluorescence localization, we demonstrated that treatment of INS-1E cells with CpdA did not support nuclear translocation of GR (Fig. 4a,b). Dex treatment induced GR translocation to the nucleus in INS-1E cells, both in the absence or presence of CYT (p < 0.05; Fig. 4a,b). In addition, while the effect of Dex on NO secretion and CHOP expression was abolished in the presence of the potent GR antagonist RU486 (p < 0.01; Fig. 4c,d), the activity of CpdA was not affected under this experimental condition (Fig. 4c,d).

CpdA acts independently of the GR-complex. a–b INS-1E cells were pretreated with vehicle, CpdA 10 μM or dexamethasone (Dex) 0.1 μM for 1 h and then challenged or not with IL-1β 100 pg/mL and IFN-γ 5 ng/mL (CYT). After 30 min, glucocorticoid receptor (GR) expression was analyzed by immunofluorescence staining. a Representative confocal microscopy pictures of INS-1E cells immunostained for GR (red) in the different experimental conditions as indicated; nuclei were stained with DAPI (blue); scale bars 10 μm. b Quantification of nuclear:cytoplasmic ratio of GR staining was performed from analysis of 5 separate high-power field images for each experimental condition. Data are shown as mean ± SD of n = 3 independent experiments. (†) p < 0.05 vs. vehicle; (***) p < 0.001 vs. vehicle + CYT. c–d INS-1E were treated as described in (a) with or without the GCs antagonist RU486 (1 μM) and 16 h of CYT stimulation. c NO secretion was assessed by Griess reaction. Data are shown as mean ± SD of n = 3 independent experiments. d Levels of CHOP protein expression were analyzed by Western blot; quantitative analysis of blots is expressed as mean ± SD of n = 3 independent experiments; β-actin was used as loading control. c–d (**) p < 0.01, (***) p < 0.001 between indicated experimental conditions; NS not significant differences
CpdA attenuates CYT-induced apoptosis and preserves glucose-stimulated insulin secretion in β-cells
Proinflammatory cytokine-induced ER stress can lead to β-cell dysfunction and death, thus we explored the effects of CpdA on β-cell function and survival. CpdA partially reduced the decline in β-cell viability observed under CYT challenge (p < 0.05; Fig. 5a) and attenuated the activation of apoptotic pathways exerted by CYT in INS-1E cells. Bax and Bcl-2 genes are involved in the control of apoptosis; the ratio between both genes constitutes a rheostat that can predict the response of a cell toward life or death under an apoptotic stimulus. We showed that CpdA reduced by 50% the 6.4 -fold increase in Bax/Bcl-2 mRNA expression ratio triggered by CYT in INS-1E cells (p < 0.01; Fig. 5b). Analogously, CpdA treatment diminished by 38,5% the 16.8-fold enhancement of death protein 5 (DP5) mRNA expression (p < 0.05; Fig. 5c), one of the key pro-apoptotic BH3-only proteins involved in CYT-induced β-cell death [20]. A similar effect on Bax/Bcl-2 mRNA expression ratio (p < 0.01; Fig. 5b) and DP5 mRNA expression (p < 0.01; Fig. 5c) was observed in CYT-challenged INS-1E cells under Dex pre-treatment. However, Dex did not show protective effects on INS-1E cell viability under CYT challenge (Fig. 5a).

CpdA attenuates CYT-induced apoptosis and preserves glucose stimulated insulin secretion in β-cells. a–f INS-1E cells were pretreated with vehicle, CpdA 10 μM or dexamethasone (Dex) 0.1 μM for 1 h and then challenged or not with IL-1β 100 pg/mL and IFN-γ 5 ng/mL (CYT) for 16 h. a Cell viability was assessed by MTT assay. Viability in control group (Veh) has been considered as 100%. Data are shown as mean ± SD of n = 3 independent experiments. b Bax mRNA to Bcl-2 mRNA ratio, c DP5 mRNA and d TNF-α mRNA expression were analyzed by RT-qPCR. Relative mRNA levels normalized to HPRT are expressed as mean ± SD of n = 3/4 independent experiments. e Cumulative insulin secretion was determined in the conditioned media (11 mM glucose) by specific ELISA. Values were normalized to total protein content measured by BCA. Relative insulin secretion is shown as mean ± SD of n = 3 independent experiments. f Glucose-Stimulated Insulin Secretion (GSIS) was assessed by ELISA in the conditioned media of cells cultured in the presence of low (2 mM) or high (20 mM) glucose. Insulin secretion index (20 mM/2 mM) is shown as mean ± SD of n = 4 independent experiments. g–h Murine islets (5 IEQ/well) were pretreated with vehicle, CpdA 10 μM or dexamethasone (Dex) 0.1 μM for 1 h and then challenged or not with IL1-β 100 pg/mL + IFN-γ 5 ng/mL + TNF-α 8 ng/mL for 16 h. (g) Apoptosis was assessed by Hoechst and PI double fluorescence staining. Percentages of apoptotic cells in islets are expressed as mean ± SD of n = 3 independent experiments. h GSIS was assessed in isolated mice islets as described in (f). Insulin secretion index (20 mM/2 mM) is shown as mean ± SD of n = 4 independent experiments. (†) p < 0.05 vs. vehicle; (*) p < 0.05, (**) p < 0.01, (***) p < 0.001 vs. vehicle + CYT
TNF-α gene expression is upregulated in β-cells in response to pro-inflammatory cytokine exposure contributing to islet inflammation [21]. CpdA prevented the increase in TNF-α mRNA expression (45% reduction vs CYT, p < 0.01; Fig. 5d) triggered by CYT in INS-1E cells; a more pronounced effect was observed with Dex (p < 0.001; Fig. 5d).
In addition to the protective effect observed on cell viability, CpdA displayed a beneficial effect on β-cell function under CYT-induced ER stress. CYT exposure of INS-1E cells induced a reduction in the cumulative secreted insulin over a period of 16 h (75% reduction, p < 0.05; Fig. 5e); CpdA counteracted the CYT effect (p < 0.01; Fig. 5e). The GSIS that was severely affected by CYT exposure (0.75-fold CYT vs. 3.57-fold veh, p < 0.05; Fig. 5f) was partially recovered by CpdA (p < 0.05; Fig. 5f). Dex showed a similar beneficial effect on cumulative insulin secretion as well as under glucose-stimulated conditions (Fig. 5e,f).
The protective effect of CpdA on INS-1E cells was confirmed in isolated murine islets; a pronounced reduction in the percentage of apoptotic cells (12.5 ± 4.8% vs. 30.3 ± 6.9%, p < 0.01; Fig. 5g) and enhancement of GSIS (p < 0.01; Fig. 5h) were observed in CYT-stressed islets.
The efficacy of CpdA in improving β-cell functionality under the challenge of proinflammatory cytokines was confirmed with human isolated islets from three organ donors. CpdA treatment improved GSIS in CYT-challenged human islets (n = 2, Fig. 6a,b). Previous reports indicated that ER stressors like proinflammatory cytokines or palmitate [21, 22] induce the expression of cytokines in human islets. CpdA pre-treatment hampered the increment observed in IL-6 (p < 0.01; Fig. 6c), IL-1β (p < 0.01; p < 0.01; Fig. 6d) and TNF-α (p < 0.001; Fig. 6e) mRNA in CYT-exposed isolated human islets.

CpdA improves glucose response in cytokine-challenged isolated human islets. a–h Isolated human islets were pretreated with vehicle, CpdA 10 μM or dexamethasone (Dex) 0.1 μM for 1 h and then challenged or not with IL1-β 250 pg/mL + IFN-γ 50 ng/mL (CYT) for 16 h. a–b Glucose-Stimulated Insulin Secretion (GSIS) was assessed by ELISA in the conditioned media of islets (5 IEQ/well) cultured in the presence of low (2 mM) or high (20 mM) glucose. Values were normalized to total protein content measured by BCA. Islets from two non-diabetic donors were tested in independent experiments with similar results. Data are shown as insulin secretion index (20 mM/2 mM). c IL-6 mRNA d IL-1β mRNA and e TNF-α mRNA expression were analyzed by RT-qPCR. Islets (50 IEQ/well) from three non-diabetic donors were tested in independent experiments (n = 3); relative mRNA levels normalized to RPL19 are expressed as mean ± SD. (†) p < 0.05 vs. vehicle; (**) p < 0.01, (***) p < 0.001 vs. vehicle + CYT
CpdA delays the onset of hyperglycemia and reduces the number of diabetic mice after adoptive transfer of disease
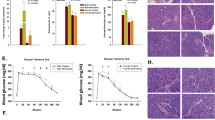
Vehicle-treated mice developed hyperglycemia from day 21 and reached an incidence of 100% (10/10) on day 39 with a median of 24 days. CpdA delayed the onset of diabetes with a median of 44 days and led to 38% (8/21) of diabetes-free mice at day 100 (p < 0.0001 vs. control) (Fig. 7b). CpdA neither affected mice´s body weight (Fig. S2a) nor displayed any other adverse side effects.

Beneficial effects of CpdA administration in the adoptive transfer of autoimmune diabetes in mice. a Experimental scheme. Non-obese diabetic (NODscid) mice were adoptively transferred with diabetogenic splenocytes (at day 0 i.p. 5 × 106 cells/mice) and treated with CpdA (i.p. 100μg, n = 21) or vehicle (Control, n = 10) three times a week from day – 1 to day 50. b Kaplan–Meier plot of cumulative diabetes incidence. p < 0.0001 vs. vehicle, by log-rank (Mantel–Cox) test. c Graph representing the classification of pancreatic islets according to the severity of leukocyte infiltration (insulitis) in each experimental group. Grade: 0, no insulitis; 1: < 25% infiltrate; 2: 25–50% infiltrate; 3: 50–75% infiltrate; and 4: > 75% infiltrate. Bars show mean ± SEM of independent individuals. Control d28 after adoptive transfer (n = 2), CpdA-treated d28 (n = 3), CpdA-treated d100 (n = 3), NODscid sham control (n = 2). d Islet immunostaining for insulin expression from each experimental group. Representative islets are shown. Scale bar 50 μm. Arrows (➤) indicate the infiltrating leukocytes. Insulitis: NODscid sham, grade 0; Control d28, grade 4; CpdA-treated d28, grade 2; CpdA-treated d100, grade 1
To determine whether the delay in the onset of hyperglycemia in CpdA-treated mice was due to changes in insulin sensitivity, an intraperitoneal insulin tolerance test (IITT) was performed in adoptively transferred NODscid mice (non-diabetic) after 40 days of CpdA-treatment, hereby revealing no significant differences in glucose clearance when compared to two different control groups: non-transferred age-matched NODscid mice and young 4-week-old NOD mice (Fig. S2b).
At the end of the study, an intraperitoneal glucose tolerance test (IGTT) was performed to assess the physiological capacity of β-cells in maintaining glucose homeostasis in CpdA-treated NODscid mice (n = 3, Fig. S2c). For comparison, we utilized non-manipulated female young 4-week-old NOD mice (n = 2). IGTT showed that normoglycemic CpdA-treated NODscid mice were able to reach basal glycemia levels after 120 min of glucose bolus, similarly to young NOD mice (Fig. S2c, AUC: 24,553 ± 7607 vs. 13,302 ± 653).
Taken together, these results indicate that CpdA delayed the onset of hyperglycemia and improved glycemic control in this autoimmune diabetes model. Furthermore, IITT results suggest that CpdA treatment did not affect glucose uptake in peripheral tissues, as has been previously shown [45].
CpdA treatment reduces leukocyte infiltration and preserves insulin expression in islets
Islet infiltration by leukocytes (insulitis) initiates the β-cell destruction and autoimmune diabetes. To investigate the mechanism exerted by CpdA on the beneficial effects in the diabetes adoptive transfer model, we harvested pancreas for histological analysis at 28 days after splenocyte transfer and at the end of the experiment (100 days) (Fig. 7c,d). At day 28, diabetic mice from the control group (vehicle-treated) presented 75% of islets with infiltration grade 4, 16.5% with grade 3 and 8.5% with grade 1 (Fig. 7c). Most of the islets in diabetic mice did not express immune-reactive insulin (Fig. 7d). In animals treated with CpdA, we observed at day 28, 13% of islets infiltrated with grade 4, 32% with grade 3, 19,5% grade 2 and 15,5% grade 0. CpdA-treated mice that reached 100 days normoglycemic showed 79% of islets without infiltration (grade 0) and 21% with grade 1. In the latter group, insulin staining was normal and islets conserved an intact architecture with normal size (Fig. 7d). Aged-matched NODscid mice (sham treated) presented strong insulin staining without leukocyte infiltration (Fig. 7c,d). These results showed that CpdA reduced the severity of insulitis compared with diabetic non-treated mice, preserving insulin stores in β-cells.
Discussion
Here, we describe the ability of CpdA to effectively attenuate ER stress induced by proinflammatory cytokines in β-cells, to improve metabolic fitness of inflamed β-cells and to display clear therapeutic benefit in an aggressive murine model of autoimmune diabetes (Fig. 8).

Compound A impacts several cell targets with potential therapeutic effects on autoimmune diabetes. Schematic outline of results. We previously reported that CpdA is an effective modulator of effector T and dendritic cells, and macrophages in vitro and in vivo. In this study, we found that CpdA improves UPR and attenuates ER stress-related apoptotic pathways, favoring the survival and function of β-cells exposed to an environment of proinflammatory cytokines. CpdA administration to NODscid mice adoptively transferred with diabetogenic splenocytes attenuated the progress of the autoimmune attack leading to a delay of disease onset and reduction of diabetes incidence. These findings together with our previous reports justify further studies on the administration of this small molecule as a novel therapeutic strategy with dual targets (effector immune and β-cells) during autoimmune diabetes
The ER is key in the production and secretion of insulin in accordance with the physiological demand and poses a continuous great challenge for the β-cell to maintain its homeostasis. Islet inflammation contributes to the pathogenesis of both type 1 and type 2 diabetes [23, 24]. Infiltrating effector immune cells during type 1 diabetes cause cellular dysfunction that may ultimately culminate in β-cell demise. IL-1β, TNF-α and IFN-γ were shown to induce ER stress [3, 25,26,27]. IL-1β mediated ER stress and apoptosis in β-cells are partially triggered by an increase in NO production through NF-κB pathway activation, although its magnitude is species-specific [27, 28]. We found that one of the mechanisms by which CpdA reduces cytokine-induced ER stress and apoptosis in INS-1E is by impairing NF-κB signaling, iNOS transcription and translation, and ultimately NO generation. In this same direction, insulin promoter-driven overexpression of the iNOS transgene induced diabetes, while genetic ablation of iNOS abrogates streptozotocin-induced diabetes in mice [29, 30].
Moreover, inhibition of the NF-κB-iNOS-NO axis protects β-cells from cytokine-induced apoptosis in vitro and in experimental diabetes [31,32,33,34]. The latter agrees with the fact that the action of CpdA by decreasing NO production protects INS-1E cells from inflammatory cytokines. It remains to be determined whether the same protective mechanisms exerted by CpdA occur in our model of adoptively transferred autoimmune diabetes.
ER stress links inflammation to initiation of β-cell dysfunction and activates UPR. In pathological conditions, UPR can lead to β-cell dysfunction and death [1]. In addition, NO contributes to the change in UPR signaling toward survival or death [26]. CpdA modulates UPR by counteracting ER stress induced by proinflammatory cytokines, as demonstrated by the significant reduction of the ER stress arm (ATF4/CHOP) and increases in XBP1 and ATF6 that promote the adaptive/restorative UPR phase. The importance of restorative UPR in β-cells is manifested in experimental [35, 36] and type 1 diabetes [37]. ER stress markers have been detected in islets of naturally occurring diabetic mice [2] and type 1 diabetic patients [38]. In type 1 diabetes models, the stress-relieving UPR improves the function and extends β-cell survival [6]. As a first step for stress adaptation, the UPR transiently restores ER homeostasis by decreasing protein translation through p-eIF2α. We found that CpdA reduces p-eIF2α suggesting the return of protein synthesis in cytokine-perturbed β-cells.
The canonical ER stress transducer IRE1 facilitates XBP1s expression, expands ER and induces the expression of foldases and chaperones all as useful adaptations to restore ER homeostasis [39]. Marked observations distinguished CpdA from Dex actions. We found that CpdA but not Dex attenuates the cytokine-mediated reduction of BiP in INS-1E cells. Also, the ER-resident chaperones ORP150 (also called Grp170) and PDI were specifically increased in CpdA-treated β-cells under basal as well as cytokine-stimulated conditions. Taking these results into account, CpdA helps maintain or even increase ER chaperone levels under the challenge of proinflammatory cytokines in INS-1E cells.
All these CpdA actions on the activation of the adaptation phase of cytokine-challenged β-cells were positively reflected in their viability, lowered apoptosis levels and improvement of GSIS. Βeta-cells are highly sensitive to apoptotic stimuli when faced with additional cellular stress, in part due to the constitutive low expression of anti-apoptotic proteins and free radical scavenging enzymes [15, 40].
The relative expression of Bax and Bcl-2 proteins regulates apoptosis [41]. Also, DP5 is induced by cytokines leading to caspase-3 activation and β-cell death [20]. The reduction of both Bax/Bcl-2 ratio and DP5 mRNA expression contribute to CpdA improvement of INS-1E cells viability, when impaired by cytokine stimulation.
The synthesis of TNF-α by IL-1β stimulation amplifies the inflammatory response in β-cells [21]. We found that CpdA reduced TNF-α mRNA transcription, which could disrupt the positive loop of Bax/Bcl-2 ratio stimulation and may reduce apoptosis through NF-κB signaling [42]. The latter observation reflected the recovery of cumulative insulin secretion, as well as that stimulated by glucose in INS-1E-CpdA cells challenged with cytokines. Also, CpdA improved GSIS and reduced inflammatory cytokine mRNA expression in cytokine-stimulated isolated murine and human islets.
GCs are widely used in the clinic. However, chronic administration presents deleterious side effects and resistance to GCs. CpdA was shown to interfere with the activity of NF-κB by a GR-dependent transrepression mechanism explaining its anti-inflammatory activity [8, 43, 44]. Unlike Dex, CpdA does not induce GR-mediated transactivation. Accordingly, the administration of CpdA did not induce diabetogenic or HPA axis-suppressive side effects in vivo [45]. Here, we show GR-independent effects of CpdA in β-cells, similarly to what was previously reported for other cell types [10, 46, 47].
Recently, autophagy induction by CpdA was described as a contributory mechanism to its anti-inflammatory phenotype in stimulated macrophages [48]. Opposite regulations between CpdA and Dex at the level of the autophagy receptor SQSTM1 (p62) could attribute to CpdA’s anti-inflammatory effects, regardless of the binding to GR [48]. Therefore, it would be important to determine whether the induction of autophagy by CpdA, in addition to reducing ER stress, may additionally contribute to an efficient protection of β-cells against inflammatory stimuli.
The beneficial effect of CpdA on diabetes by streptozotocin (STZ) administration has been previously reported [49]. However, STZ induces the death of β-cells by chemical toxicity with the appearance of necrotic β-cells and insulitis as early as 2-4 h and 3–4 days after its administration, respectively [50]. We used the transfer of diabetogenic splenocytes as a model to resemble the state of immune activation of an individual at the time of diagnosis of type 1 diabetes. We found that CpdA administration delayed and, in some cases, completely halted the progression of hyperglycemia by diminishing islets infiltration of inflammatory cells characteristic of insulitis. Likewise, CpdA has been successful in other animal and cellular disease models showing potent anti-inflammatory properties [44, 45, 51, 52].
We reported that CpdA affects dendritic cells that subsequently generate weak contact hypersensitivity response and T lymphocytes, favoring the Th2-type response over Th1 [9, 10]. These findings suggest that CpdA could also act in vivo by affecting both antigen-presenting cells and effector T lymphocytes responsible for β-cell damage, as well as explain the reduction of insulitis in normoglycemic mice that received diabetogenic splenocytes.
Type 1 diabetes remains a disease without a cure and multidrug therapy has been suggested as an option. The use of small-molecules with a dual anti-inflammatory targeting potential are of high relevance in strategies to combat autoimmune diabetes. CpdA meets the criteria of what is called combined therapy for autoimmune diabetes by modulating effector immune cells to dampen islet inflammation and also by protecting β-cells.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CpdA:
-
Compound A
- eIF2α:
-
Eukaryotic translation initiation factor 2α
- ER:
-
Endoplasmic reticulum
- GR:
-
Glucocorticoid receptor
- GSIS:
-
Glucose-stimulated insulin secretion
- SEGRAM:
-
Selective glucocorticoid receptor agonists and modulators
- STZ:
-
Streptozotocin
- T1D:
-
Type 1 diabetes
- UPR:
-
Unfolded protein response
References
Clark AL, Urano F (2016) Endoplasmic reticulum stress in β-cells and autoimmune diabetes. Curr Opin Immunol 43:60–66. https://doi.org/10.1016/j.coi.2016.09.006
Tersey SA, Nishiki Y, Templin AT et al (2012) Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 61(4):818–827. https://doi.org/10.2337/db11-1293
Cardozo AK, Ortis F, Storling J et al (2005) Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic b-cells. Diabetes 54:452–461. https://doi.org/10.2337/diabetes.54.2.452
Miani M, Colli ML, Ladrière L, Cnop M, Eizirik DL (2012) Mild endoplasmic reticulum stress augments the proinflammatory effect of IL-1β in pancreatic rat β-cells via the IRE1α/XBP1s pathway. Endocrinology 153(7):3017–3028. https://doi.org/10.1210/en.2011-2090
Mandrup-Poulsen T, Pickersgill L, Donath MY (2010) Blockade of interleukin 1 in type 1 diabetes mellitus. Nat Rev Endocrinol 6(3):158–166. https://doi.org/10.1038/nrendo.2009.271 (PMID: 20173777)
Engin F, Yermalovich A, Nguyen T et al (2013) Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci Transl Med 5(211):211ra156. https://doi.org/10.1126/scitranslmed.3006534
Fu S, Yalcin A, Lee GY et al. (2015) Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci Transl Med 7(292):292ra98. https://doi.org/10.1126/scitranslmed.aaa9134
De Bosscher K, Vanden Berghe W, Beck IM et al (2005) A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A 102(44):15827–15832. https://doi.org/10.1073/pnas.0505554102
Liberman AC, Antunica-Noguerol M, Ferraz-de-Paula V et al (2012) Compound A, a dissociated glucocorticoid receptor modulator, inhibits T-bet (Th1) and induces GATA-3 (Th2) activity in immune cells. PLoS ONE 7(4):e35155. https://doi.org/10.1371/journal.pone.0035155
Barcala Tabarrozzi AE, Andreone L, Deckers J et al (2016) GR-independent down-modulation on GM-CSF bone marrow-derived dendritic cells by the selective glucocorticoid receptor modulator Compound A. Sci Rep 6:36646. https://doi.org/10.1038/srep36646
Louw A, Swart P, de Kock SS, van der Merwe KJ (1997) Mechanism for the stabilization in vivo of the aziridine precursor 2-(4-acetoxyphenyl)-2-chloro-N-methyl-ethylammonium chloride by serum proteins. Biochem Pharmacol 53(2):189–197. https://doi.org/10.1016/s0006-2952(96)00661-2
Ricordi and Rastellini C (2000) Methods in pancreatic islet separation. In: Ricordi C, (Ed.) Methods in cell transplantation. Austin, TX: RG Landes 2000; 433–438
Ricordi C, Lacy PE, Scharp DW (1989) Automated islet isolation from human pancreas. Diabetes 38(1):140–142. https://doi.org/10.2337/diab.38.1.s140
Lakey JR, Warnock GL, Shapiro AM, Korbutt GS, Ao Z, Kneteman NM, Rajotte RV (1999) Intraductal collagenase delivery into the human pancreas using syringe loading or controlled perfusion. Cell Transplant 8(3):285–292. https://doi.org/10.1177/096368979900800309
Bottino R, Balamurugan AN, Bertera S, Pietropaolo M, Trucco M, Piganelli JD (2002) Preservation of human islet cell functional mass by anti-oxidative action of a novel SOD mimic compound. Diabetes 51(8):2561–2567. https://doi.org/10.2337/diabetes.51.8.2561
Castro CN, Barcala Tabarrozzi AE, Winnewisser J et al (2014) Curcumin ameliorates autoimmune diabetes. Evidences in accelerated murine models of type 1 diabetes. Clin Exp Immunol 177(1):149–160. https://doi.org/10.1111/cei.12322
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4):402–408. https://doi.org/10.1006/meth.2001.1262
Atorrasagasti C, Onorato A, Gimeno ML et al (2019) SPARC is required for the maintenance of glucose homeostasis and insulin secretion in mice. Clin Sci (Lond) 133(2):351–365. https://doi.org/10.1042/CS20180714
Perone MJ, Larregina AT, Shufesky WJ et al (2006) Transgenic galectin-1 induces maturation of dendritic cells that elicit contrasting responses on naïve and activated T cells. J Immunol 176(12):7207–7220. https://doi.org/10.4049/jimmunol.176.12.7207
Gurzov EN, Ortis F, Cunha DA et al (2009) Signaling by IL-1beta+IFN-gamma and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic beta-cell apoptosis. Cell Death Differ 16(11):1539–1550. https://doi.org/10.1038/cdd.2009.99
Burke SJ, Lu D, Sparer TE, Karlstad MD, Collier JJ (2014) Transcription of the gene encoding TNF-α is increased by IL-1β in rat and human islets and β-cell lines. Mol Immunol 62(1):54–62. https://doi.org/10.1016/j.molimm.2014.05.019
Igoillo-Esteve M, Marselli L, Cunha DA et al (2010) Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 53(7):1395–1405. https://doi.org/10.1007/s00125-010-1707-y
Eguchi K, Nagai R (2017) Islet inflammation in type 2 diabetes and physiology. J Clin Invest 127(1):14–23. https://doi.org/10.1172/JCI88877
Donath MY, Størling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T (2008) Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev 29(3):334–350. https://doi.org/10.1210/er.2007-0033
Størling J, Binzer J, Andersson AK et al Nitric oxide contributes to cytokine-induced apoptosis in pancreatic beta cells via potentiation of JNK activity and inhibition of Akt. Diabetologia 48(10):2039–50. https://doi.org/10.1007/s00125-005-1912-2.
Chan JY, Cooney GJ, Biden TJ, Laybutt DR (2011) Differential regulation of adaptive and apoptotic unfolded protein response signalling by cytokine-induced nitric oxide production in mouse pancreatic beta cells. Diabetologia 54(7):1766–1776. https://doi.org/10.1007/s00125-011-2139-z
Brozzi F, Nardelli TR, Lopes M et al (2015) Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 58:2307–2316. https://doi.org/10.1007/s00125-015-3669-6
Mandrup-Poulsen T (2001) beta-cell apoptosis: stimuli and signaling. Diabetes 50(Suppl 1):S58-63. https://doi.org/10.2337/diabetes.50.2007.s58
Takamura T, Kato I, Kimura N, Nakazawa T, Yonekura H, Takasawa S, Okamoto H (1998) Transgenic mice overexpressing type 2 nitric-oxide synthase in pancreatic beta cells develop insulin-dependent diabetes without insulitis. J Biol Chem 273(5):2493–2496. https://doi.org/10.1074/jbc.273.5.2493
Flodström M, Tyrberg B, Eizirik DL, Sandler S (1999) Reduced sensitivity of inducible nitric oxide synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes 48(4):706–713. https://doi.org/10.2337/diabetes.48.4.706
Jiang X, Zhou Y, Wu KK, Chen Z, Xu A, Cheng KK (2017) APPL1 prevents pancreatic beta cell death and inflammation by dampening NFκB activation in a mouse model of type 1 diabetes. Diabetologia 60(3):464–474. https://doi.org/10.1007/s00125-016-4185-z
Patel S, Santani D (2009) Role of NF-kappa B in the pathogenesis of diabetes and its associated complications. Pharmacol Rep 61:595–603
Grey ST, Arvelo MB, Hasenkamp W, Bach FH, Ferran C (1999) A20 inhibits cytokine-induced apoptosis and nuclear factor kappaB-dependent gene activation in islets. J Exp Med 190(8):1135–1146. https://doi.org/10.1084/jem.190.8.1135
Cardozo AK, Heimberg H, Heremans Y et al (2001) A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J Biol Chem 276(52):48879–48886. https://doi.org/10.1074/jbc.M108658200
Harding HP, Zeng H, Zhang Y et al (2001) Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell 7(6):1153–1163. https://doi.org/10.1016/s1097-2765(01)00264-7
Back SH, Kaufman RJ (2012) Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem 81:767–793. https://doi.org/10.1146/annurev-biochem-072909-095555
Brozzi F, Eizirik DL (2016) ER stress and the decline and fall of pancreatic beta cells in type 1 diabetes. Ups J Med Sci 121(2):133–139. https://doi.org/10.3109/03009734.2015.1135217
Marhfour I, Lopez XM, Lefkaditis D et al (2012) Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 55(9):2417–2420. https://doi.org/10.1007/s00125-012-2604-3
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8(7):519–529. https://doi.org/10.1038/nrm2199
Tiedge M, Lortz S, Drinkgern J, Lenzen S (1997) Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 46(11):1733–1742. https://doi.org/10.2337/diab.46.11.1733
Dillon CP, Green DR (2016) Molecular cell biology of apoptosis and necroptosis in cancer. Adv Exp Med Biol 930:1–23. https://doi.org/10.1007/978-3-319-39406-0_1
Baker RG, Hayden MS, Ghosh S (2011) NF-κB, inflammation, and metabolic disease. Cell Metab 13(1):11–22. https://doi.org/10.1016/j.cmet.2010.12.008
Rauner M, Thiele S, Sinningen K et al (2013) Effects of the selective glucocorticoid receptor modulator compound A on bone metabolism and inflammation in male mice with collagen-induced arthritis. Endocrinology 154(10):3719–3728. https://doi.org/10.1210/en.2012-2221
Dewint P, Gossye V, De Bosscher K et al (2008) A plant-derived ligand favoring monomeric glucocorticoid receptor conformation with impaired transactivation potential attenuates collagen-induced arthritis. J Immunol 180(4):2608–2615. https://doi.org/10.4049/jimmunol.180.4.2608
van Loo G, Sze M, Bougarne N (2010) Antiinflammatory properties of a plant-derived nonsteroidal, dissociated glucocorticoid receptor modulator in experimental autoimmune encephalomyelitis. Mol Endocrinol 24(2):310–322. https://doi.org/10.1210/me.2009-0236
Gavrila A, Chachi L, Tliba O, Brightling C, Amrani Y (2015) Effect of the plant derivative Compound A on the production of corticosteroid-resistant chemokines in airway smooth muscle cells. Am J Respir Cell Mol Biol 53(5):728–737. https://doi.org/10.1165/rcmb.2014-0477OC
Gossye V, Elewaut D, Bougarne N et al (2009) Differential mechanism of NF-kappaB inhibition by two glucocorticoid receptor modulators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 60(11):3241–3250. https://doi.org/10.1002/art.24963
Mylka V, Deckers J, Ratman D et al (2018) The autophagy receptor SQSTM1/p62 mediates anti-inflammatory actions of the selective NR3C1/glucocorticoid receptor modulator compound A (CpdA) in macrophages. Autophagy 14(12):2049–2064. https://doi.org/10.1080/15548627.2018.1495681
Saksida T, Vujicic M, Nikolic I, Stojanovic I, Haegeman G, Stosic-Grujicic S (2014) Compound A, a selective glucocorticoid receptor agonist, inhibits immunoinflammatory diabetes, induced by multiple low doses of streptozotocin in mice. Br J Pharmacol 171(24):5898–5909. https://doi.org/10.1111/bph.12892
Like AA, Rossini AA (1976) Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science 193(4251):415–417. https://doi.org/10.1126/science.180605
Zhang Z, Zhang ZY, Schluesener HJ (2009) Compound A, a plant origin ligand of glucocorticoid receptors, increases regulatory T cells and M2 macrophages to attenuate experimental autoimmune neuritis with reduced side effects. J Immunol 183(5):3081–3091. https://doi.org/10.4049/jimmunol.0901088
Lesovaya E, Yemelyanov A, Swart AC, Swart P, Haegeman G, Budunova I (2015) Discovery of Compound A-a selective activator of the glucocorticoid receptor with anti-inflammatory and anti-cancer activity. Oncotarget 6(31):30730–44. https://doi.org/10.18632/oncotarget.5078.
Acknowledgements
The 5xATF6-LUC and XBP1u-LUC plasmids were a kind gift from Prof. Dr. Sarah Gerlo (Univ. Ghent, Belgium).
Funding
This work was supported by grants from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT #2018–1577 to MJP and #2018–719 to LA); from Universidad Austral (#2020 to MJP and LA); and from Sociedad Argentina de Diabetes (#2018 to MJP and #2019 to LA). Also, we thank the support of Fundación Marjorie para la Investigación en Diabetes (www.fumdiab.org.ar).
Author information
Authors and Affiliations
Contributions
LA and MJP conceived and designed the work. LA, FF and CS performed the in vitro experiments and acquired data. LA, MJP and AEBT performed the animal experiments and acquired data. RB provided de human islets. MSO contributed to sample preparation and data acquisition. LA and MJP interpreted the data and wrote the manuscript. RAD made important contributions in the interpretation of the results. KDB provided CpdA and research tools supported by FWO-Vlaanderen (G044217N) and contributed to the interpretation of data. All authors gave final approval of the version to be published. MJP is the guarantor of this work, has full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
All experimental procedures were conducted in accordance with the Guide for the care and use of laboratory animals, Eighth edition (2011) and were approved by the Animal Research and Care Committee (#0001 & #0069), FCEyN, University of Buenos Aires.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Andreone, L., Fuertes, F., Sétula, C. et al. Compound A attenuates proinflammatory cytokine-induced endoplasmic reticulum stress in beta cells and displays beneficial therapeutic effects in a mouse model of autoimmune diabetes. Cell. Mol. Life Sci. 79, 587 (2022). https://doi.org/10.1007/s00018-022-04615-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04615-5