
Abstract
The initial identification of long non-coding RNA myocardial infarction associated transcript (MIAT) as a genetic risk factor of myocardial infarction has made this lncRNA (designated as lncR-MIAT here) a focus of intensive studies worldwide. Emerging evidence supports that lncR-MIAT is susceptible in its expression to multiple deleterious factors like angiotensin II, isoproterenol, hypoxia, and infection and is anomaly overexpressed in serum, plasma, blood cells and myocardial tissues under a variety of cardiovascular conditions including myocardial infarction, cardiac hypertrophy, diabetic cardiomyopathy, dilated cardiomyopathy, sepsis cardiomyopathy, atrial fibrillation and microvascular dysfunction. Experimental results consistently demonstrated that upregulation of lncR-MIAT plays active roles in the pathological processes of the cardiovascular system and knockdown of this lncRNA effectively ameliorates the adverse conditions. The available data revealed that lncR-MIAT acts through multiple mechanisms such as competitive endogenous RNA, natural antisense RNA and RNA/protein interactions. Moreover, the functional domains of lncR-MIAT accounting for certain specific cellular functions of the full-length transcript have been identified and characterized. These insights will not only tremendously advance our understanding of lncRNA biology and pathophysiology, but also offer good opportunities for more innovative and precise design of agents that have the potential to be developed into new drugs for better therapy of cardiovascular diseases (CVDs) in the future. Herein, we provide an overview of lncR-MIAT, focusing on its roles in cardiovascular diseases, underline the unique cellular/molecular mechanisms for its actions, and speculate the perspectives about the translational studies on the potential diagnostic and therapeutic applications of lncR-MIAT.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Recent advances in the genome-wide analysis have revealed that only ~ 1.5% of the genome encodes proteins, and the major portion (> 66%) is actively transcribed into noncoding RNAs (ncRNAs) that have important functional roles in regulating the expression of protein-coding genes. Long non-coding RNAs (lncRNAs or lncRs) belong to one of the ncRNA superfamily. This recently discovered class of regulatory RNAs has magnetized tremendous attention from scientists worldwide and become a focus in the field of biomedical research owing to their widespread distributions in organisms and diverse functionalities in cells. Characterized by their sequence diversity (ranging from 200 bp to 10 kb), expression abundance, nuclear location, and minimal protein-coding capability, lncRNAs form a complex gene expression regulatory network orchestrating cellular phenotypic plasticity [1]. In line with their diversity in sequence and functionality, lncRNAs act by diverse mechanisms at different levels such as transcriptional regulation, chromatin organization, and post-transcriptional control [2]. While only a limited number of lncRNAs have been studied, accumulating evidence proves that lncRNAs interact with a large plethora of proteins involving a wide variety of human diseases including cardiovascular disease (CVD) [3,4,5,6,7,8,9,10].
Among these lncRNAs identified to be associated with CVD [11], myocardial infarction associated transcript (lncR-MIAT) was found to confer risk of myocardial infarction (MI) upon its upregulation by the single nucleotide polymorphism (SNP) and to predict left ventricular dysfunction in acute MI (AMI) [12]. A subsequent study indicates MIAT expression level is a significant univariate predictor of left ventricular dysfunction in patients with AMI [13]. The involvement of lncR-MIAT in the pathological angiogenesis through regulating retinal microvascular function in the setting of diabetes mellitus (DM) was demonstrated [14]. The role of lncR-MIAT in regulating cardiac hypertrophy (CH) was then documented [15]. Our research group reported that lncR-MIAT acts as a competing endogenous RNA (ceRNA) to promote cardiac fibrosis in post-infarct myocardium [16]. LncR-MIAT was also found to be involved in the pathogenesis of diabetic cardiomyopathy [17]. These initial seminal studies represent one of the milestones in the field of research on lncRNA and CVDs, and also aroused tremendous research interest in lncR-MIAT from scientists worldwide. To date, emerging data have shown that this lncRNA plays crucial roles in a wide range of CVD, including MI, CH, cardiomyopathy, atrial fibrillation (AF), and other conditions. The findings have not only advanced our understanding of the molecular function and pathophysiological roles of lncRNAs, but also provided us with novel insights and therapeutic targets for CVDs. Most notably, the published studies conducted in either animal models or patients with CVDs all point to a consistent conclusion that lncR-MIAT is a CVD-promoting lncRNA, thereby being a potential novel molecular target for the treatment of various CVDs.
In this review, we present the cellular function and pathophysiological roles of lncRNA in CVDs, its underlying cellular/molecular mechanisms, and the clinical implications and translational perspective of this lncRNA as a potential therapeutic target for the treatment of CVDs with critical analysis and summary of the most updated studies relevant to lncR-MIAT.
Identification and characterization of lncR-MIAT
Identification of lncR-MIAT
The first research associating lncR-MIAT with myocardial infarction was published as early as 2006 by Ishii et al. [12]. In this study, Ishii et al. enrolled 188 Japanese individuals with MI and 752 general Japanese populations by multiplex PCR method for detecting 52,608 gene-based SNPs, to investigate whether genetic variants were associated with MI. From this screening, they found one SNP (rs2301523) in gene FLJ25967 showed the most significant susceptibility to MI (P = 0.0006). A subsequent case–control association study from the same group by genotyping 3464 MI patients and 3819 control subjects confirmed the association of the SNP/rs2301523 with MI after excluding the other SNPs located at the same haplotype block [12]. Moreover, the authors also investigated the association between genotype information of MI patients and clinical profiles consisting of smoking, hypertension, sex, diabetes, age, and hyperlipidemia and found no positive association for each of these factors. These results prompted them to postulate that SNP/rs2301523 is directly related to the pathogenesis of MI.
To identify the full-length gene containing SNP/rs2301523 and its open reading frame, the authors went on to carry out 5′- and 3′-RACE experiments and obtained four splicing variant cDNAs: AB263414, AB263415, AB263416, and AB263417 (GenBank Accession number). Yet, none of the four transcripts was found to have a long ORF, suggesting it a non-protein coding gene. To verify this notion, they conducted an in vitro translation assay and detected no translated product from any of the four cDNA sequences, indicating this gene encodes a functional RNA. Furthermore, they also excluded it from miRNA precursor because it did not contain a possible hairpin structure. Based upon all these results, the authors believed it a novel gene and designated it MIAT for myocardial infarction associated transcript (MIAT) as it provides the genetic risk of MI.
We now know that MIAT is a member of the lncRNA family belonging to the ncRNA superfamily. Herein, we coin it lncR-MIAT for the sake of convenience and clarity. This lncRNA is also known as RNCR2 or Gomafu.
Characteristics of lncR-MIAT
LncR-MIAT is around 10-kb long polyadenylated transcripts containing seven exons, and it could produce more than 10 isoforms by alternative splicing [18]. Unlike most of the other lncRNAs, lncR-MIAT is evolutionarily conserved from mammals to amphibians [19, 20]. The largest exon of orthologous genes all contains multiple tandem UACUAAC repeats (5–8 repeats in each transcript) [19], which include the ACUAAY consensus recognition site for Quaking, a well-known RNA binding protein that regulates RNA stability [21]. In addition, lncR-MIAT is primarily and abundantly expressed in the nervous system (particularly in fetal brain) and retina [18, 19]. In neuronal cells, it is a nuclear‐located lncRNA which is tightly associated with the nuclear matrix. The transcripts are distributed throughout the nucleus and display a punctate pattern with no overlapping to the markers of known nuclear bodies.
Northern blot analysis revealed that in addition to brain, lncR-MIAT is also appreciably expressed spleen, lung, and peripheral blood leukocyte [12], but nearly null in heart, although quantitative PCR analysis demonstrated a moderate expression level of this lncRNA in blood cells and myocardium [13, 22].
LncR-MIAT as a risk factor and biomarker for cardiovascular disease
LncR-MIAT and MI
Though the original work by Ishii [12] established the association of lncR-MIAT with the pathogenesis of MI lays the groundwork for subsequent studies on this lncRNA, it does not provide a causal link between lncR-MIAT and MI but left some unanswered questions. One of these questions is whether and how the expression of lncR-MIAT is altered in the setting of MI or other CVDs. Another important question is how lncR-MIAT constitutes a risk factor for MI or how it affects cardiovascular function.
Intriguingly, the initial work of Ishii et al. published in 2006 [12] did not raise close attention and major interest in the role of lncR-MIAT on CVDs, presumably because then lncRNAs were not yet a hot spot for research. Not until 8 years later in 2014, Vausort et al. [13] evaluated the expression levels of 5 lncRNAs including lncR-MIAT using the PCR technology in peripheral blood cells isolated from 414 AMI patients. The authors found that 274 patients with ST-segment-elevation MI (STEMI) had lower levels of lncR-MIAT compared to 140 patients with non-ST-segment-elevation MI (NSTEMI). Moreover, lncR-MIAT appears to be a significant univariable predictor of left ventricular (LV) dysfunction as assessed by an ejection fraction (EF) ≤ 40% at 4-month follow-up. However, this prediction capacity of lncR-MIAT gets lost in multivariate analysis. While this study suggests the potential value of lncR-MIAT as a predictor of LV dysfunction, interpretation of the results is not straightforward because of the following two issues. First, the difference in lncR-MIAT levels between STEMI and NSTEMI is mild though statistically very significant and the expression has apparent intergroup overlaps rendering the difference insufficiently discrete. Second, changes of lncR-MIAT in peripheral blood cells are likely independent of changes in cardiac cells and therefore, do not reflect ischemic myocardial injuries, as the former may have entirely different regulatory mechanism for lncR-MIAT expression from the latter.
In a separate study published in 2016, similar under-expression of lncR-MIAT in STEMI relative to NSTEMI was observed in platelets from 32 patients with AMI using the RNA-seq technology [23]. The major weakness of this study is the small sample size despite the result is in line with that reported by Vausort et al. [13].
With the aforementioned studies, it remained unclear whether lncR-MIAT can be used as a biomarker for AMI. To resolve this issue, Azat et al. [22] measured the plasma levels of lncR-MIAT using quantitative PCR analysis with the samples drawn from 58 acute STEMI patients and 50 patients with unstable angina for control. The results revealed a threefold increase of lncR-MIAT in acute STEMI patients relative to that in control patients. More importantly, the increase began as early as within 3 h of the onset of ischemic symptoms and reached a peak level at 6 h, indicating lncR-MIAT a more effective biomarker than cTnT and CK‑MB. Similarly, lncR-MIAT is remarkably upregulated in AMI rats and hypoxic cardiomyocytes. This study is the first to establish that lncR-MIAT may act as a novel biomarker for diagnosing AMI with evidence from both humans and experimental animals and from both in vivo/ex vivo and in vitro analyses. Yet, it falls short in the small sample size too.
Another similar study assessed the possibility of peripheral blood mononuclear cells (PBMC)-derived lncRNAs as biomarkers for AMI by measuring the expression levels of 10 lncRNAs known to be associated with CVDs, in PBMC samples from 132 AMI patients and 104 healthy controls using quantitative RT-PCR [24]. The report shows that the expression level of lncR-MIAT is significantly higher in patients than in controls. However, while the correlation analysis fails to link lncR-MIAT to the conventional cardiac biomarkers including troponin T, CK and CKMB, it is found negatively correlated with cardiac EF and Genisini score, an intensity index for coronary artery disease (CAD), in line with the previously published work [12, 13].
LncR-MIAT and coronary artery disease (CAD)
The study reported by Tan et al. [25] aimed to reveal the clinical significance of lncR-MIAT expression in predicting CAD. To this end, the investigators assessed the serum level of lncR-MIAT, tumor necrosis factor-α (TNF-α) as well as interleukin-6 (IL-6) in 102 CAD patients and 89 healthy controls, the results showed that the expression of lncR-MIAT is around threefold higher in CAD patients than in healthy donors. Linear correlation analysis revealed a positive correlation between lncR-MIAT, IL-6 and TNF-α in CAD. In addition, multivariate analysis indicates that except for hypertension, diabetes, and HDL-C, serum lncR-MIAT is also an independent risk factor for CAD. With these findings, the authors concluded that serum lncR-MIAT might be a biomarker for diagnosis and prognosis of CAD. Yet, the potential association of serum lncR-MIAT with the coronary risk factors such as hypertension, hyperlipidemia, diabetes, sex, and age were not analyzed in this study.
This notion was supported and extended by Toraih et al. [26] who determined the circulating level of lncR-MIAT in 110 stable CAD patients and 117 control volunteers by qPCR and correlated its expression with both clinical and laboratory data. Their results showed that the median lncR-MIAT level in patients is impressively 12-fold higher than in controls. These findings prompted the authors to propose that lncR-MIAT has a diagnostic significance in CAD patients. In this study, the investigators also excluded the diagnostic value of another lncRNA MALAT1 as the change of its level is not sufficiently reliable to have clinical applications.
LncR-MIAT and ischemic stroke (IS)
IS, also referred to as brain (cerebral) ischemia, is the most common form of stroke, accounting for more than 80% of all cases. In the study presented by Zhu et al. [27], lncR-MIAT expression level was found greatly upregulated in peripheral blood leukocytes of 189 IS patients, compared with 189 healthy controls. And this increase is tightly correlated with NIHSS scores, mRS, high-sensitivity C-reactive protein, as well as infarct volume in IS patients. Strikingly, the overall survival analysis unraveled that IS patients who have higher lncR-MIAT expression levels have poorer prognosis than those with lower levels of this lncRNA. Furthermore, multivariate analysis and ROC curves prove that lncR-MIAT could discriminate IS patients from healthy controls with high sensitivity and specificity. These results led the investigator to conclude that lncR-MIAT could be a potential diagnostic and prognostic biomarker of IS.
LncR-MIAT and diabetic cardiomyopathy
de Gonzalo-Calvo et al. [28] evaluated the potential of circulating lncRNAs as indicators of subclinical cardiac abnormalities in type 2 diabetes mellitus (T2DM). 48 patients with T2DM and 12 healthy volunteers participated in the study. The investigators detected the expression of several selected lncRNAs in serum by RT-PCR and also measured left ventricular (LV) parameters, the results showed no difference in the expression levels of these lncRNAs between patients and healthy controls. However, in patients with T2DM, lncR-MIAT level is directly related to LV mass to LV end-diastolic volume ratio, an indicator of cardiac remodeling, and was further validated in 30 well-controlled T2DM patients. Based on these findings, they concluded that lncR-MIAT is one of the independent lncRNA predictors of diastolic function and remodeling in patients with T2DM. Apparently, the same weakness of the small sample size applies to this study.
The presently available studies, though still relatively scarce and bearing some weaknesses and controversies, have provided promising and encouraging evidence in support of the value of lncR-MIAT as a biomarker and/or predictor for MI, CAD and IS (Table 1). The consistent finding is significant upregulation of lncR-MIAT in blood samples of either blood cells or serum or plasma. These pioneering works merit further investigations with a larger sample size and better-controlled criteria for inclusion of patients and controls for scrutinizing issue.
LncR-MIAT and myocardial infarction (MI)
MI is a global health epidemic that predisposes to heart failure (HF) and sudden cardiac death (SCD) due to lethal arrhythmias [29]. Despite that the advances over the past decades in the secondary treatment of MI have helped alleviation of symptoms and even improvement of survival; none can reverse the pathological process, leaving the cure a major unmet medical need. MI is a metabolic catastrophe characterized by deprivation of oxygen and nutrients to the high energy-demanding cardiomyocytes and overproduction of reactive oxygen species (ROS) and other harmful metabolites [30, 31]. MI provokes a cascade of long-term changes termed remodeling processes, beginning with adaptive cytoprotection but ending up with maladaptive deterioration. MI is accompanied by several key cellular processes such as cardiomyocyte apoptosis, cardiac fibrosis, and inflammatory response, and its progress is also regulated by a variety of factors including lncRNAs.
Alterations of lncR-MIAT level in blood documented by the studies described above, particularly with the results obtained from the case–control association study with genome-wide SNP analysis and animal models of MI, strongly insinuate the possible functional roles of lncR-MIAT in the heart. Whilst the very first study, Ishii et al. isolated the full-length sequence of lncR-MIAT and established its association with MI [12], what precise role that MIAT takes part in the pathogenesis of MI and how exactly MIAT takes its actions have not been experimentally studied. Not until 2017 with an 11 years lag, the first experimental study on the role of this lncRNA came from our research group [16]. In this work, we identified lncR-MIAT as a pro-fibrotic lncRNA governing cardiac fibrosis in a mouse model of MI. Our study was thereafter followed by rapid increases in the number of studies focusing on the functional aspect of lncR-MIAT in other CVDs. In the setting of MI, lncR-MIAT is best characterized as a key regulator of apoptosis, fibrosis and inflammation. Therefore, in this section, we focus on these findings.
Pro-apoptotic property of lncR-MIAT
It is known that apoptosis, a form of programmed cell death, is one of the central events in response to the metabolic disturbances in MI, such as insufficient energy supply and oxidative stress, which damages cardiac function and structural integrity of myocardium in MI, and cytoprotection against apoptosis has long been considered a strategy for the management of MI [32, 33]. The cell death initiates the adverse structural remodeling process of the heart because the adult mammalian cardiomyocytes have a rather limited ability to regenerate themselves after injury. Better understanding of cardiac cell death is therefore of paramount importance for tackling the cardiac problems associated with cell death.
In the study aiming to evaluate lncR-MIAT as a biomarker of AMI as already described above [22], the group also examined the cellular function of this lncRNA in cultured neonatal rat cardiomyocytes. Their results from TUNEL assay exhibited that knockdown of lncR-MIAT by RNAi technology significantly inhibits cardiomyocyte apoptosis. They subsequently demonstrated that silencing lncR-MIAT significantly decreases the expression of Bax and increases the expression of Bcl2; however, how it affects the expression of these proteins was not assessed in this study.
Chen et al. described similar results in their study conducted in a mouse model of myocardial ischemia and reperfusion injury (myocardial I/R injury) and a H9C2 cell model of hypoxia/reoxygenation (H/R) [34]. Specifically, knockdown of lncR-MIAT by virus-mediated short-hairpin RNA technology was found to decrease LDH release and myocardial infarct size, and also improve the cardiac function with partial restoration of reduced EF and fractional shortening (FS). These beneficial effects of lncR-MIAT knockdown are accompanied by enhanced cardiomyocyte viability and diminished caspase 3/7 activities at the cellular level, indicating a cytoprotective effect against apoptosis. The investigators then went further to delineate the signaling mechanisms and observed that targeting lncR-MIAT blocks p65 nuclear translocation, mitigates the protein levels of NF-κB/p65, p53 upregulated modulator of apoptosis (PUMA), cleaved caspase-3, and pro-apoptotic protein Bax, while elevates the anti-apoptotic protein Bcl-2. These findings suggest that silencing lncR-MIAT alleviates H9c2 cardiomyoblast H/R in vitro and myocardial I/R injuries in vivo by inhibiting apoptosis through suppressing the NF-κB and PUMA signal pathway. Whilst this is a quite comprehensive research, a missing link is how lncR-MIAT could possibly elicit all the detrimental effects on cardiac injuries and regulate the expression of multiple signaling molecules and how exactly it does all these jobs.
In an effort to decipher the mechanism by which catechin, a bioactive polyphenol extracted from green tea, produces cytoprotection against apoptosis in a H9C2 cell model of H/R injury and a rat model of myocardial I/R injury, Cong et al. found that catechin improves cardiac function, inhibit apoptosis and meanwhile downregulates lncR-MIAT transcripts in myocardium [35]. The beneficial action of catechin at the cellular level can be abrogated by transfection of lncR-MIAT-overexpressing plasmid. These authors further identified transcription factor CREB as a negative regulator of lncR-MIAT expression. Moreover, lncR-MIAT inhibits Akt/Gsk-3β activation and promotes apoptosis in H9C2 cells. Similar to the study by Chen et al. [34] introduced above, the mechanistic link is missing on how lncR-MIAT regulates Akt/Gsk-3β activation.
These three reports confirm the pro-apoptotic property of lncR-MIAT with the first two using loss-of-function approaches and the latter with a gain-of-function strategy. The results advanced our understanding of the cellular function of lncR-MIAT that underlies at least partially the increased risk of MI associated with the presence of lncR-MIAT. Unfortunately, none of these studies provides mechanistic insight into the link between lncR-MIAT and the downstream effectors modulated by this lncRNA, nor do they look into the possible expression deregulation of lncR-MIAT in ischemic myocardium. To solve these issues, we conducted a research in MI mice created by occluded the left descending coronary artery and in neonatal mouse ventricular cardiomyocytes (NMVCs) cultured under hypoxic conditions to simulate MI.
One of the problems of infarcted myocardium is reduced supply of and relatively increased demand for energy. Mitochondria are cellular organelles serving as the powerhouse of cells, which through oxidative phosphorylation produce more than 95% of the energy for cardiac cells under normal physiological conditions. Mitochondrial dysfunction is an important hallmark of multiple cardiovascular disorders, particularly ischemic heart disease [36,37,38]. Mitochondrial function is critically and delicately determined by its membrane integrity for maintaining the appropriate membrane potential (ΔΨm). Normal ΔΨm is required for many proteins that are anchored in the mitochondrial membrane to function normally, of which the translocator protein (TSPO) that spans the inner and outer mitochondrial membranes is highly expressed in the cardiac cells, has been found to play a key role in regulating mitochondrial function through modulating the mitochondrial membrane potential [39,40,41,42]. In addition to the well-known mitochondrial death pathway regulated by Bcl-2 and Bax proteins [43], TSPO has also been found to participate in determining cell survival and death pathways. Particularly, deregulation of TSPO can cause mitochondrial ΔΨm disruption leading to apoptosis [44, 45].
Our experimental results from PCR quantification demonstrated that lncR-MIAT was enormously upregulated (fivefold increase) in myocardium MI mice and in hypoxic NMVCs (threefold increase) as well [46]. Experimental knockdown of lncR-MIAT by lentivirus-mediated RNAi tremendously improved cardiac function and inhibited cardiomyocyte apoptosis in MI. At the molecular levels, lncR-MIAT positively regulated TPSO expression and interacted directly with TSPO to induce opening of mitochondrial permeability transition pore (mPTP), resulting in a decrease of mitochondrial membrane potential with declined ΔΨm, and eventually leading to apoptosis. Knockdown of lncR-MIAT mitigated all these deleterious alterations.
Furthermore, we identified a 226-nt region that is homologous among the four variants of Homo sapiens lncR-MIAT (Accession: NR_003491.3, NR_033319.2, NR_033320.2 and NR_033321.2) and conserved between human sequences and one Mus musculus lncR-MIAT sequence (Accession: NR_033657.1), we named this fragment the functional domain of lncR-MIAT (MIAT-FD for abbreviation), and further experimentally validated it reproduced the deleterious effects of full-length lncR-MIAT to bind TSPO and induce cardiomyocyte apoptosis.
The present study revealed a new cellular function of lncR-MIAT as a pro-apoptotic lncRNA in the setting of MI. One of the important findings of this work is the short stretch of lncR-MIAT encompassing the TSPO binding motif, that is, MIAT-FD, is sufficient to replace full-length lncR-MIAT to perform corresponding functions, another potential implication of this study is that knockdown of lncR-MIAT offered remarkable amelioration of myocardial injuries, suggesting the possibility of lncR-MIAT as a molecular target for the development of new strategies for MI treatment.
Pro-fibrotic property of lncR-MIAT
Cell death in AMI initiates the structural remodeling if the heart with enhanced cardiac fibrogenesis to fill the intramyocardial gaps left. Cardiac fibrosis is a pathological process characterized by abnormal accumulation and excessive deposition of extracellular matrix (ECM) [47,48,49]. Generation of ECM is a repair response after cell death, which initially brings about benefit for protecting the relative integrity of the heart, but eventually leads to cardiac pathological remodeling.
In one of our published studies [16], we found that lncR-MIAT is remarkably upregulated in MI heart and isolated cardiac fibroblasts treated with serum or angiotensin II (AngII), meanwhile, there is an accumulation of cardiac interstitial fibrosis in a mouse model of MI, which was later reproduced in our recent study [46]. Masson trichrome staining revealed that knocking down lncR-MIAT markedly reduces the infarct size in MI mice compared with the control group, along with pronounced decreases in the protein levels of collagens I and III in peri-infarct area of MI mice. The upregulation of lncR-MIAT is accompanied by abnormal upregulation of several fibrosis-related regulators including Furin and TGF-β1, whereas knockdown of lncR-MIAT by its siRNA corrects this deregulation.
Furthermore, MI downregulates miR-24 in the heart and cardiac fibroblasts, whereas knockdown of lncR-MIAT upregulates it. Luciferase reporter gene assay supports the direct interaction between lncR-MIAT and miR-24. We experimentally established Furin, an activating factor of TGF-β1, as a target gene for miR-24. Furin suppresses fibroblast proliferation and collagen production. All these data revealed that there is a new pathway regulating cardiac fibrosis: MI → lncR-MIAT↑ → miR-24↓ → Furin/TGF-β 1↑ → cardiac fibrosis↑.
Our study therefore identifies lncR-MIAT as a pro-fibrotic lncRNA by acting as a ceRNA of miR-24 to derepress Furin that in turn activates TGF-β1, leading to fibrogenesis. Together with its pro-apoptotic effect as described above, it is possible that aberrant upregulation of lncR-MIAT in MI induces cardiomyocyte apoptosis and then stimulates fibrosis to compensate for the loss of cardiac cells; nevertheless it does so overly to cause excessive fibrosis exaggerating MI injuries. These findings advocate the potential of lncR-MIAT as a biomarker for cardiac fibrosis. Nonetheless, a question that remains unanswered is how lncR-MIAT produces the opposite actions on different cell types: promoting death of cardiac cells but growth of cardiac fibroblasts. This notion is worthy of future studies for clarification.
A recent study generated the data in support of the fibrosis-promoting property of lncR-MIAT [50]. This study aimed to delineate the mechanism for the increased cardiac fibrosis in the offspring heart with perinatal exposure to nicotine in a well-established rat model and cultured primary neonatal rat cardiac fibroblasts. These authors first demonstrated that perinatal nicotine exposure causes significant deposition of collagens I and III and concomitant increase in lncR-MIAT along with the decrease in miR-29 family in offspring heart. This fibrogenesis is reversed by miR-29 overexpression. Experimental knockdown of lncR-MIAT upregulates miR-29 family and decreases the levels of collagens I and III, suggesting nicotine-induced excessive collagen deposition in offspring heart is mediated by lncR-MIAT which acts as a sponge or ceRNA on miR-29.
Pro-inflammatory property of lncR-MIAT
A recent study reported by Xing et al. [51] demonstrated that lncR-MIAT plays an important role in oxidative stress and inflammation during the septic cardiomyopathy. Despite that this work was not performed in a MI model, the results have profound implications in MI. This is because inflammation is one of the driving forces for the pathogenesis of atherosclerosis, affecting the MI size and clearing cellular debris thereby recovery from MI injuries [52]. Dysregulation of or persistent pro-inflammatory response may induce adverse post-MI cardiac remodeling and major adverse clinical events [53].
The investigators looked into the roles of lncR-MIAT and miR-330-5p in regulating oxidative stress and inflammatory responses in a mouse model of lipopolysaccharide (LPS, also named endotoxin)-induced septic cardiomyopathy and LPS-stimulated HL-1 cells (a mouse atrial cell line) as well. They found that in septic mice, lncR-MIAT is upregulated along with increased levels of pro-inflammation cytokines; in contrast, miR-330-5p is downregulated. Knockdown of lncR-MIAT or overexpression of miR-330-5p suppresses inflammatory reaction induced by LPS in vitro. Further experiments revealed that lncR-MIAT could directly interact with miR-330-5p by the ceRNA mechanism to activate the TRAF6/NF-κB signaling axis, thereby promoting inflammation response in LPS-induced septic cardiomyopathy. The results clearly indicate lncR-MIAT is a pro-inflammatory lncRNA.
LncR-MIAT and cardiac hypertrophy
The involvement of lncR-MIAT in the development of cardiac hypertrophy (CH) has been demonstrated by several studies using different animal and cellular models. The results from these studies all point to a same conclusion that lncR-MIAT is a pro-hypertrophic growth stimulator in heart.
The first of these studies was published in 2016 by Zhu et al. [15], who employed a mouse model of CH stimulated by AngII as well as in vitro model with rat heart-derived H9c2 cells. They found lncR-MIAT is significantly increased by 5–6 folds in CH of both animal and cell models. Silencing of lncR-MIAT by siRNA substantially alleviates the upregulation of hypertrophic biomarkers ANP, BNP and β-MHC in H9c2 cells and markedly attenuates the increase of the cell surface area and the protein synthesis induced by AngII. Overexpression of lncR-MIAT in H9c2 cells downregulates miR-150 expression, whereas knockdown of lncR-MIAT upregulates it. Moreover, forced expression of miR-150 inhibits the expression level of hypertrophic marker genes and suppresses hypertrophic phenotypes, which is in line with other studies shown the anti-hypertrophic action of this miRNA [54]. However, the downstream mediators to miR-150 were not characterized in this study, and the relationship between lncR-MIAT and miR-150 was not experimentally established either.
The second investigation on the same topic was documented by Li et al. [55] who followed up the study by Zhu et al. [15] with specific research goal to decipher the underlying mechanism of lncR-MIAT regulates the development of CH. In this report, lncR-MIAT and miR-93 showed opposite expression patterns in AngII-treated cardiomyocytes, with high expression of lncR-MIAT and low expression of miR-93. The reciprocal alterations of expression urged the workers to establish the targeting relationship between lncR-MIAT and miR-93 using the combination of qPCR, luciferase assay and RNA immunoprecipitation (RIP) techniques. Their results verified lncR-MIAT as a ceRNA of miR-93 and further regulated the TLR4 expression. Silencing of lncR-MIAT inhibits the expression levels of hypertrophic marker proteins, accompany with the reduced cell surface area in AngII-treated cardiomyocytes. Their data therefore indicate that lncR-MIAT knockdown inhibits cardiac hypertrophy through the miR-93/TLR4/PI3K/Akt/mTOR pathway.
In another study, the researchers confirmed the pro-hypertrophic property of lncR-MIAT in a mouse model of CH stimulated by intraperitoneal isoproterenol (ISO) and a cellular model of cardiomyocyte hypertrophy with neonatal rat ventricular myocytes (NRVMs) [56]. Their results showed an around 4.5-fold increase in lncR-MIAT transcripts with ISO stimulation both in vivo and in vitro. Knockdown of lncR-MIAT decreases the expression levels of hypertrophic marker genes and cell surface area. They also provided the evidence supporting the ability of lncR-MIAT to inhibit miR-150 and went further to identify P300, an acetyltransferase and a transcriptional coactivator that is known to promote cardiac growth thereby driving CH [57], as a target gene for miR-150. Next, they established that lncR-MIAT decreases P300 expression via absorbing miR-150.
Still, another group described their work on evaluating the effects of berberine, an effective natural supplement compound extracted from several plants, on CH in a rat model with constriction of abdominal aorta and H9C2 cells stimulated with AngII to induce cardiomyocyte hypertrophy [58]. They depicted that berberine significantly relives CH and cardiomyocyte enlargement and expression of hypertrophy marker proteins. Most notably, berberine suppresses the expression of lncR-MIAT in CH. In addition, berberine downregulates phosphorylated mTOR, phosphorylated AMPK and LC3 and upregulates the autophagy-related genes. While the authors concluded that berberine exerts beneficial effects on CH via decreasing lncR-MIAT expression and enhancing autophagy, they did not provide any link between lncR-MIAT and autophagy; neither did they elucidate how berberine suppresses the expression of lncR-MIAT.
LncR-MIAT and cardiomyopathy
Cardiomyopathy represents a group of heterogeneous diseases that negatively impact cardiac function leading to HF and sudden death. Cardiomyopathies can be separated into primary (genetic, acquired, or mixed) and secondary categories. Primary cardiomyopathies specifically target the myocardium and may arise from genetic etiology, such as arrhythmogenic right ventricular cardiomyopathy/dysplasia, hypertrophic cardiomyopathy and mitochondrial cardiomyopathy or from genetic and acquired mixed etiology like diabetic cardiomyopathy, dilated cardiomyopathy (DCM), and restrictive cardiomyopathy or from acquired etiology like septic cardiomyopathy (SCM) [59, 60]. Recent studies have linked lncR-MIAT to a range of cardiomyopathies including diabetic cardiomyopathy, dilated cardiomyopathy (DCM), and septic cardiomyopathy (SCM).
Diabetic cardiomyopathy
Diabetic cardiomyopathy is a special type of heart disease that occurs independently of other heart risk factors, such as CAD, hypertension, it is caused by insulin resistance, hyperinsulinemia, and hyperinsulinemia in the cardiac metabolic tissues. The pathophysiological factors contributing to the development of diabetic cardiomyopathy cover oxidative stress, autophagy, inflammation, apoptosis, etc. These metabolic disorders lead to heart remodeling, myocardial fibrosis, diastolic dysfunction, and eventually heart failure [61, 62].
We have introduced the work by de Gonzalo-Calvo et al. [28] in an earlier section that in patients with T2DM, lncR-MIAT level is directly associated with LV mass to LV end-diastolic volume ratio indicating this lncRNA as an independent predictor of diabetic cardiomyopathy. One year after this study, Zhou et al. [17] presented a functional study with a rat model of diabetic cardiomyopathy stimulated by streptozotocin (type 1 diabetes mellitus) and demonstrated that lncR-MIAT is upregulated by ~ 4-times in diabetic heart. Noticeably, TUNEL assay and Annexin V-FITC/PI staining showed that lncR-MIAT knockdown by shRNA-carrying lentivirus reduces cardiomyocyte apoptosis in diabetic rats and neonatal rat cardiomyocytes incubated with high glucose. Echocardiographic examination generated the results that silencing lncR-MIAT attenuates hypertrophic responses with remarkable improvement of LV function. Luciferase assay and RNA immunoprecipitation assay established lncR-MIAT as a target of miR-22-3p and DAPK2 as another direct target gene for miR-22-3p. Thus, the increase in miR-22-3p represses the expression of both lncR-MIAT and DAPK2. Overexpression of lncR-MIAT mediated by pcDNA plasmid counteracts the repressive effect of miR-22-3p on DAPK2. These data suggest lncR-MIAT is a pro-apoptotic lncRNA in the setting of diabetic cardiomyopathy as in MI which was already described earlier in this review. One apparent contradiction of this study is that if miR-22-3p can indeed repress lncR-MIAT at the post-transcriptional level, then decrease in this lncRNA should weaken its ceRNA action on miR-22-3p ultimately leading to a loss of lncR-MIAT function. This point needs to be clarified to ensure the credibility of the observed results about the relationship between lncR-MIAT and miR-22-3p.
Dilated cardiomyopathy
Chagas disease is a parasitic infection caused by the protozoan Trypanosoma cruzi [63, 64], it is clinically divided into acute phase and chronic phase, dilated chronic cardiomyopathy (CCC) is the most severe manifestation of chronic Chagas disease that is characterized by HF, ventricular arrhythmias, heart blocks, thromboembolic phenomena, and sudden death. The research report by Frade et al. [65] describes that lncR-MIAT was specifically upregulated on CCC myocardial samples than in idiopathic dilated cardiomyopathy (DCM) and control subjects, using a whole-transcriptome analysis. lncR-MIAT level is 3–49 folds greater than that in control subjects and 2–20-folds higher than in subjects with DCM. The differential expressions of lncR-MIAT were validated by quantitative PCR analysis on a larger set of heart tissue samples. Furthermore, the overexpression of lncR-MIAT was also confirmed in myocardial specimens of mice infected with T. cruzi. The authors believe that lncR-MIAT is a specific biomarker of CCC and they also hypothesize that enhanced lncR-MIAT expression in Chagas disease is related to endothelial dysfunction based upon the recognized role of this lncRNA in regulating microvascular function and the pathogenic role of endothelial dysfunction in Chagas disease. However, the functional analysis for lncR-MIAT on CCC has not been experimented in this report.
Septic cardiomyopathy
Sepsis, a disorder of the immune and inflammatory response to infection, constitutes a life-threatening multi-organ failure [66, 67]. Cardiac dysfunction, also referred to as septic cardiomyopathy (SCM), is a frequent complication of severe sepsis, usually manifested as systolic and diastolic dysfunction associated with worse clinical outcomes. As already introduced in a previous section, lncR-MIAT promotes inflammation and oxidative stress in endotoxin-induced septic cardiomyopathy through targeting the miR-330-5p/TRAF6/NF-κB axis [51], indicating that in addition to diabetic cardiomyopathy and DCM, lncR-MIAT is also an important regulator of SCM.
LncR-MIAT and atrial fibrillation
Atrial fibrillation (AF) is the most common sustained arrhythmia with high morbidity and mortality. Atrial remodeling is the basis for the occurrence and maintenance of atrial fibrillation, which is manifested as atrial electrical remodeling (AER) in the early stage and structural remodeling in the late stage [68]. AER is caused by changes in the expression and/or function of cardiac ion channels, especially the decrease of L-type Ca2+ current (ICaL) or the increase of inward rectifying K+ current (IK1), or the combination of the two, is the key electrophysiological changes in AER [69,70,71,72,73,74,75]. On the one hand, ICaL is the main determinant of length of the plateau phase of cardiac atrial action potential duration (APD), and reduction of ICaL density can cause profound APD shortening, particularly the plateau phase. Reduction of ICaL density in AER may be consequent to either functional decrease in activities of L-type Ca2+ channels (Cav1.2) or expression downregulation of CACNA1C, the gene encoding the α1c subunit of Cav1.2 that carries ICaL [70, 72, 74, 75]. On the other hand, IK1 is the principal K+ current, with the function of setting and maintaining the resting membrane potential, as well as governing the late phase of repolarization. This current is carried by Kir2 subfamily (Kir2.1, Kir2.2, Kir2.3 and Kir2.4), of which Kir2.1 is the main subunit expressed in myocardium. Significant increase in IK1 current density has been always detected in AF [69, 71, 73, 75]. Similar to ICaL, enhancement of IK1 in AF can be explained either by functional impairment and expression abnormality; increased expression of KCNJ2 mRNA and its encoded Kir2.1 protein (which carries cardiac IK1) have been reported in AF patients [71, 73, 76, 77]. Taken together all these facts, it is clear that decrease in ICaL and/or increase in IK1 are causative factors for AER thereby AF. However, the molecular mechanisms underlying abnormal downregulation of CACNA1C/Cav1.2 and upregulation of KCNJ2/Kir2.1 remain incompletely understood, which constitutes an obstacle for effective therapy and clinical management of AF.
Additionally, the development of atrial fibrosis is the hallmark of structural remodeling in AF and is considered the substrate for AF perpetuation [78,79,80]. It is known pharmacological therapies of AF suffer from poor efficacy and increased risk of serious complications, especially pro-arrhythmia in the ventricle. Therefore, a new strategy targeting the processes involved in developing the substrate that supports AF has been recently evolving as a mainstream research topic and clinical consideration. AF in many clinical settings is characterized by significant atrial fibrosis, and thus targeting at the fibrotic substrate itself may be more effective in the management of AF.
An interesting report was published very recently by Yao et al. [81] demonstrated that expression of lncR-MIAT was found dramatically increased (around fivefold increase 1 week after AF induction) in atrial tissues of a rat model of AF induced by rapid electrical pacing on right atrium, along with a marked decrease of miR-133a-3p. Similarly, lncR-MIAT is also significantly upregulated whereas the miR-133a-3p is downregulated in peripheral blood leukocytes from 35 patients with AF relative to that from 30 healthy control volunteers. Knockdown of lncR-MIAT by lentivirus vector carrying a lncR-MIAT shRNA prolongs atrial effective refractory period (AERP) and shortens the duration of AF. Besides, silencing lncR-MIAT effectively mitigates AF-induced atrial apoptosis and atrial fibrosis as indicated by reduced the number of atrial myocytes with TUNEL-positive staining and decreased collagen deposition in the right atrium uncovered by Mason staining. Furthermore, knockdown of lncR-MIAT decreases expression of collagens I and III, TGF-β1 and CTGF in the right atria of AF rats. These results were contrary to those of rats with inhibition of miR-133a-3p. The authors then observed that the beneficial effects of lncR-MIAT downregulation on AF are efficiently reversed by miR-133a-3p antisense inhibitor. Luciferase reporter gene assay verified miR-133a-3p as a direct target of lncR-MIAT for eliciting its ceRNA action. Their findings established a new signaling pathway leading to AF: lncR-MIAT↑ → miR-133a-3p↓ → CTGF↑ → TGF-β1↑ → collagens↑ → AF↑, and lncR-MIAT is therefore a pro-AF lncRNA that promotes AF via restricting the functional availability of miR-133a-3p by the ceRNA mechanism.
While the above-described study touches the structural remodeling associated with AF, our study focuses on atrial electrical remodeling (AER) [82]. We found that lncR-MIAT was significantly downregulated in a mouse model of AF, Knockdown (KD) of MIAT with lentivirus-mediated siRNA-transfer increased AF vulnerability in healthy mice. MIAT-KD decreased the expression of CACNA1c/Cav1.2 and ICaL, increased the expression of KCNJ2/Kir2.1 and IK1, leading to a shortened APD of cardiomyocytes. Searching for the underlying mechanism, we found that MIAT could be acted as a ceRNA to absorb miR-135a and then affect the expression of target gene CACNA1c/Cav1.2 and ICaL. Meanwhile, MIAT also served as an antisense RNA (asRNA) [83] to decrease KCNJ2 expression [82]. Further two distinct functional domains were identified as one is responsible for absorbing miR-135a and the other for binding to KCNJ2. Experimentally proved that the two functional domains could not only well mimic the regulatory effect of full-length lncR-MIAT, but also reproduce electrical remodeling and associated AF susceptibility in mice. Therefore we speculated that lncR-MIAT might be a vital player in controlling AF and MIAT replacement might be considered a potential therapeutic option for AF.
Interestingly, MIAT may be a tissue-specific lncRNA, as it is significantly upregulated in the ventricle but robustly downregulated in the atrium. This suggests that there are probably distinct regulatory mechanisms for lncR-MIAT expression in the ventricle and atrium to maintain relative homeostasis between different tissues. Indeed, the work by García-Padilla et al. unraveled the expression of several lncRNAs including lncR-MIAT being in different temporal and tissue-restricted patterns during cardiogenesis and in adult hearts [84].
It is noted that our findings are in apparent contradiction to those documented by Yao et al. [81]. We found lncR-MIAT is downregulated in AF and it elicits anti-AF effect, whereas Yao et al. identified lncR-MIAT as a pro-AF factor with its expression upregulated in AF. There could be several possible explanations for the controversies. First, the animal models used are different between the two studies with Yao’s work with rats and ours with mice. Second, for the measurements of lncR-MIAT levels in humans, Yao et al. used peripheral blood leukocytes but we used plasma samples, and there might be cell type-specific regulations of lncR-MIAT expression. Third, Yao et al. focused on atrial fibrosis but we aimed to investigate the changes of ion channels. Forth, Yao et al. determined AF at least 1 week after AF induction while we did it immediately after AF induction with pretreatment of lncR-MIAT-related constructs.
LncR-MIAT and other cardiovascular conditions
In addition to the results described in previous sections, lncR-MIAT has also been shown to elicit other effects, e.g. regulating diabetes mellitus-induced microvascular dysfunction [14] and being involved in cardiac development [84] and endurance exercise [85].
A study by Yan et al. [14] investigated the role of lncR-MIAT in regulating microvascular dysfunction in retinas in the setting of DM induced by intraperitoneal injection of streptozotocin. Quantitative PCR analysis showed a significantly higher retinal lncR-MIAT level in DM rats and endothelial cells incubated in high glucose than in non-DM control animals and endothelial cells cultured under normal conditions. Knockdown of lncR-MIAT could not only alleviate visual function and retinal vascular leakage in vivo, but also affect retinal microvascular stability in vitro. Besides, they also found vascular endothelial growth factor (VEGF) was greatly upregulated in the diabetic retinas, which could be partially reduced by silencing of lncR-MIAT. Further they proved that lncR-MIAT could promote VEGF expression by binding with miR-150-5p, and the lncR-MIAT-miR-150-5p-VEGF regulatory loop is essential for endothelial cell function. This study highlights the importance of lncR-MIAT in pathological angiogenesis, which bears tremendous therapeutic implications because deregulated angiogenesis is a critical cause of diseases, such as atherosclerosis, ocular disorders and cancers [86].
The involvement of lncR-MIAT in cardiac development was described by García-Padilla et al. [84] in mice. The authors found that lncR-MIAT is preferentially expressed in a chamber-specific fashion at embryonic stages and in adult hearts; specifically, the expression of lncR-MIAT is more prominent in the ventricular chambers of embryonic hearts and nearly exclusively in adult hearts. However, this study stays at the level of expression alterations without going insight into the functional assessment to elucidate the actual roles of lncRNAs in cardiac development.
An interesting study conducted in MI male Wistar rats demonstrated that 4 weeks of moderate intensity endurance exercise causes considerable alterations of cardiac function indices and hemodynamic parameters, myocardial fibrotic areas and apoptosis, and expression of genes encoding lncRNAs [85]. Endurance exercise significantly reduces the expression of lncR-MIAT in myocardial tissues of both MI and sham-operated control rats. However, the implications of such changes were not evaluated.
Mechanisms of action by lncR-MIAT
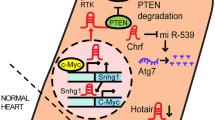
The molecular mechanisms by which lncRNAs elicit their effects are very diverse and include regulation of gene expression at the epigenetic, transcriptional, post-transcriptional and post-translational levels, and of protein function by direct interactions [1,2,3, 5]. Important to note is that an individual lncRNA may well take its actions by multiple mechanisms, even on a same target gene. Different lncRNAs may act via an identical mechanism to regulate a same gene. As far as lncR-MIAT in CVDs per se, the mechanisms of actions can be summarized as the following categories: ceRNA, naturally occurring antisense RNA (asRNA), and RNA/protein interaction (Fig. 1).

Cartoon diagram depicting the mechanisms of action of lncR-MIAT. ceRNA competitive endogenous RNA, asRNA antisense RNA
ceRNA via the lncRNA‐miRNA‐mRNA axis
The ceRNA hypothesis proposes that transcripts with shared miRNA binding sites in a gene/mRNA compete for post-transcriptional regulation of this gene [87]. This hypothesis has been believed a universal mechanism for lncRNAs and unifying function of lncRNAs existing commonly in organisms and for other regulatory RNAs as well, which is supported by empirical evidence from enormous studies [8].
From the published data to date, lncR-MIAT has been associated with 8 miRNAs, including miR-133a-3p, miR-135a, miR-150-5p, miR-22-3p, miR-24, miR-29, miR-330-5p, and miR-93, which function as the intermediators of the cellular function of lncR-MIAT (Table 2). The ceRNA action of lncR-MIAT causes derepression or upregulation of the target protein-coding genes of the relevant miRNAs via limiting the functional availability of its targeted miRNAs, leading to subsequent changes of cellular function accordingly. This information has been introduced in the relevant contexts of this review.
Nonetheless, it needs to be noted that the ceRNA hypothesis is not without major skepticism and criticisms. Theoretically, lncRNAs carrying miRNA binding sites can all act as ceRNAs; but in fact this is not always the case. Several renowned research groups performed well control and rigorously designed in-depth investigations and provided strong evidence against the validity of the ceRNA hypothesis [88,89,90,91,92]. In our laboratories, we have also examined no less than 50 lncRNAs for their possibility to act like ceRNAs; however, the results showed that most of these lncRNAs could not act as molecular sponges to their target miRNAs. There was no reciprocal relationship between them although they have one or more binding sites. Therefore, the prediction of sequence complementarity alone did not represent the existence of a real regulatory relationship, and further experimental verification was crucial. Especially, lncR-MIAT is a low-abundance lncRNA in the myocardium; in fact, it could hardly be detected with less sensitive techniques like Northern blot [12]. Despite that its level is upregulated in many CADs, whether the copy number of this lncRNA in a cardiac cell is sufficient to elicit ceRNA action remains yet to be verified and validated.
Direct interaction with proteins
In our study linking lncR-MIAT to MI injury, we showed that this lncRNA could bind directly to TSPO protein to stabilize it and in this way, lncR-MIAT induces opening of mPTP and further leading to apoptosis. On top of this, we identified a 226-nt region that encompasses the sequence motif of lncR-MIAT that binds TSPO and is homologous among various species and experimentally verified this fragment as the FD of lncR-MIAT, which can essentially replicate the pathophysiological function of the full-length lncR-MIAT on TSPO, mitochondria, apoptosis and cardiac function [46].
One advantage associated with the mechanism of direct lncRNA: protein interaction is that it offers an opportunity for identifying and characterizing the functional domain of the targeted protein, which enables the gain-of-function approach for basic researches and clinical applications of lncRNAs that are otherwise too long in their sequences to penetrate the cells to take their actions. Another advantage of the mode of action is that it allows one to get into deeper insight into the mechanisms of cellular functions and pathological roles of lncRNAs [8, 93, 94].
asRNA to affect the stability of target mRNAs
Antisense RNA (asRNA) is a class of RNA molecules that specifically binds to mRNA through the principle of complementary base pairing, and inhibits the expression of target genes at the transcriptional and translational levels. It was first discovered in the prokaryotic Escherichia coli, and subsequent studies confirmed the presence of antisense RNA in eukaryotic cells [83, 95,96,97]. Jayprokas et al.’s latest study showed that lncRNA can be used as antisense RNA to regulate gene expression level through binding with mRNA, and play an important role in the development of cancer [83].
Our study revealed that lncR-MIAT bears a short sequence domain antisense to KCNJ2. In order to investigate the possibility of a direct interaction between MIAT and KCNJ2 transcripts, we artificially synthesized this sequence and transfected it into Neonatal Mouse Atrial Myocytes (NMAMs) to see if it could cause any changes of KCNJ2 transcript levels. The results showed that after transfection, KCNJ2 was significantly downregulated, as well as the low expression of Kir2.1 protein, which bears implications in AF.
Clinical implications and translational perspective
Given the facts that the expression of lncR-MIAT is nearly exclusively upregulated in various CVDs and it actively participates in regulating CAD-related genes to fulfill its pathophysiological roles, it does not seem to have any doubt that this lncRNA bears implications in clinical applications as a biomarker for MI and predictor for LV function in MI and as a molecular target for new drug development for the treatment of CVDs. The relatively high conservation of lncR-MIAT sequence across species offers a unique advantage for translational studies for the potential future clinical applications.
Actually, there are two ways for a lncRNA to be a molecular target for the treatment of cardiovascular disease: Loss-of-function and gain-of-function. In consideration of the properties of lncR-MIAT, we tend to formulate the following protocols for the translational studies on this lncRNA.
First, we can use RNAi technology to silence the expression of lncR-MIAT or knock down lncR-MIAT. This approach is rational and feasible given the fact that aberrant upregulation of lncR-MIAT is common to diverse CVDs. The siRNA approach for specifically inhibiting protein-coding genes by artificially synthesized siRNAs has been implemented for basic and pre-clinical studies. However, the low efficacy, potency, and stability of siRNAs and their chemical derivatives, as well as the risk of off-target effects, limit their therapeutic application.
Second, except for RNAi approach, we could also use antisense oligodeoxynucleotides (ODNs) to block the function of lncRNAs. For example, we have obtained the data that an antisense fragment to the functional domain (FD) of lncR-MIAT (MIAT-FD) effectively disrupts the lncR-MIAT:TSPO binding and abolishes the cellular function and pathological effects of MIAT-FD and full-length lncR-MIAT. These results suggest that antisense is an effective and potent strategy for suppressing the detrimental action of lncR-MIAT in heart.
Third, in some cases, the abnormal low expression of lncRNA may be an important reason for the occurrence and development of the disease. Therefore, exogenous supplementation of the lncRNA to increase its level can correct this abnormality and could become one of the treatment approaches. However, if the length of lncRNA is too long to be carried by the plasmid or viral vectors, it will limit the application of lncRNA replacement therapy. One solution is to search for the functional domain (FD) of lncRNAs, which are small in length but can retain the cellular function of full-length lncRNAs for a particular disease entity. For instance, if lncR-MIAT is indeed downregulated in atrial tissue of AF patients as revealed by our study described above, then lncR-MIAT replacement is highly desirable. Unfortunately, the full-length lncR-MIAT is too long in its sequence (almost 10 kb) to be carried by any vectors currently available, which forms a great obstacle to delivering this lncRNA into cells. Fortunately, this barrier can be surmounted.
Our group has successfully identified the FDs of three different lncRNAs, and experimentally confirmed that these FDs could replace full-length lncRNAs to perform corresponding biological functions [93, 94, 98]. This indicates that this strategy is effective and feasible for the lncRNAs which are directly interact with proteins. One obvious advantage of this approach over ceRNA is that lncRNA FD:protein interaction is more specific, for lncRNA FD specifically embedded in the three-dimensional spatial structure of the target protein, thus having a lower off-target effect.
Knowledge gap and future directions
Despite the important role that lncR-MIAT plays in a variety of CVDs, it should be noted that our present understanding of the cellular functions, pathological roles, and molecular mechanisms is still far from being complete. Many questions remain unanswered.
First, as a nuclear-retained lncRNA, lncR-MIAT is proposed to regulate alternative splicing of transcripts involved in neuronal activation and pathology of schizophrenia. For example, Barry et al. demonstrate that lncR-MIAT could function as a splicing factor sponge to absorb QKI and SRSF1, resulting in a remarkable decrease in expression of schizophrenia-related genes DISCI and ERBB4 and their alternatively spliced variants. Chung et al. reconfirmed that abnormal elevation of lncR-MIAT transcript level alters ErbB4 splicing, causing lower activity of parvalbumin interneurons and cognitive dysfunction in patients with schizophrenia. However, it has not been reported that whether lncR-MIAT contributes to the development of various CVDs by altered alternative splicing, until Yang et al. first revealed that knockout of MIAT significantly reduces RNA splicing events in hypertrophic hearts and affects the alternative splicing of genes associated with cardiac muscle contraction and morphogenesis. Unfortunately, they didn’t look into the exact mechanisms of how MIAT takes part in cardiac RNA splicing. Besides, Whether MIAT can be involved in the development of other CVDs through alternative splicing regulation remains to be further explored.
Second, lncR-MIAT is known to be a nuclear-retained lncRNA, but then how it could interact with miRNAs and proteins located in the cytoplasm is yet to be clarified.
Third, although we have identified a sequence motif, MIAT-FD, which is well conserved between mouse and human, could replace full-length lncR-MIAT to induce cardiomyocyte apoptosis by interacting with TSPO, the conclusion drawn from our animal study may not be directly applied to man, further validated experiments and rigorous studies are still required to make it applicable to clinical practice.
Conclusions
Overall, the expression of lncR-MIAT is aberrantly upregulated in various pathological conditions of the heart, including myocardial infarction, coronary artery disease, cardiac hypertrophy, diabetic cardiomyopathy, and dilated cardiomyopathy, sepsis cardiomyopathy, atrial fibrillation, and ischemic stroke. Upregulation of lncR-MIAT can be caused by multiple deleterious factors such as angiotensin II, isoproterenol, hypoxia, ROS, nicotine, and infection. It is thus quite possible that lncR-MIAT is an effective biomarker for MI and other cardiovascular conditions and a valuable predictor of LV function in MI.
LncR-MIAT primarily produces deleterious impacts on heart and plays critical roles in the pathological processes of multiple CVDs (Fig. 2). An important note to take here is that in all published studies involving the functional assessment of lncR-MIAT, knockdown of lncR-MIAT by siRNA or virus-mediated silencing techniques effectively abrogates the detrimental effects induced by this lncRNA. This point strongly suggests the validity of lncR-MIAT as a molecular target for the treatment of various CVDs.

Summary of the roles of lncR-MIAT in cardiovascular diseases
LncR-MIAT acts through multiple mechanisms including ceRNA, asRNA and RNA:protein binding. These insights will not only tremendously advance our understanding of lncRNA biology and pathophysiology, but also offer good opportunities for more innovative and precise design of agents that have the potential to be developed into new drugs for better therapy of CVDs in the future. Because of the high profile of lncR-MIAT in CVDs, it definitely merits future studies and the potential of this lncRNA for clinical applications will no doubt remain it a focus of research attention.
Availability of data and materials
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Batista PJ, Chang HY (2013) Long noncoding RNAs: cellular address codes in development and disease. Cell 152:1298–1307. https://doi.org/10.1016/j.cell.2013.02.012
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: insights into functions. Nat Rev Genet 10:155–159. https://doi.org/10.1038/nrg2521
Hobuss L, Bar C, Thum T (2019) Long Non-coding RNAs: at the heart of cardiac dysfunction? Front Physiol 10:30. https://doi.org/10.3389/fphys.2019.00030
Lozano-Vidal N, Bink DI, Boon RA (2019) Long noncoding RNA in cardiac aging and disease. J Mol Cell Biol 11:860–867. https://doi.org/10.1093/jmcb/mjz046
Maass PG, Luft FC, Bahring S (2014) Long non-coding RNA in health and disease. J Mol Med (Berl) 92:337–346. https://doi.org/10.1007/s00109-014-1131-8
Schonrock N, Harvey RP, Mattick JS (2012) Long noncoding RNAs in cardiac development and pathophysiology. Circ Res 111:1349–1362. https://doi.org/10.1161/CIRCRESAHA.112.268953
Uchida S, Dimmeler S (2015) Long noncoding RNAs in cardiovascular diseases. Circ Res 116:737–750. https://doi.org/10.1161/CIRCRESAHA.116.302521
Zhang Y, Du W, Yang B (2019) Long non-coding RNAs as new regulators of cardiac electrophysiology and arrhythmias: molecular mechanisms, therapeutic implications and challenges. Pharmacol Ther 203:107389. https://doi.org/10.1016/j.pharmthera.2019.06.011
Gast M, Rauch BH, Haghikia A, Nakagawa S, Haas J, Stroux A, Schmidt D, Schumann P, Weiss S, Jensen L, Kratzer A, Kraenkel N, Muller C, Bornigen D, Hirose T, Blankenberg S, Escher F, Kuhl AA, Kuss AW, Meder B, Landmesser U, Zeller T, Poller W (2019) Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc Res 115:1886–1906. https://doi.org/10.1093/cvr/cvz085
Bar C, Chatterjee S, Falcao Pires I, Rodrigues P, Sluijter JPG, Boon RA, Nevado RM, Andres V, Sansonetti M, de Windt L, Ciccarelli M, Hamdani N, Heymans S, Figuinha Videira R, Tocchetti CG, Giacca M, Zacchigna S, Engelhardt S, Dimmeler S, Madonna R, Thum T (2020) Non-coding RNAs: update on mechanisms and therapeutic targets from the ESC Working Groups of Myocardial Function and Cellular Biology of the Heart. Cardiovasc Res 116:1805–1819. https://doi.org/10.1093/cvr/cvaa195
Bar C, Chatterjee S, Thum T (2016) Long noncoding RNAs in cardiovascular pathology, diagnosis, and therapy. Circulation 134:1484–1499. https://doi.org/10.1161/CIRCULATIONAHA.116.023686
Ishii N, Ozaki K, Sato H, Mizuno H, Susumu S, Takahashi A, Miyamoto Y, Ikegawa S, Kamatani N, Hori M, Satoshi S, Nakamura Y, Tanaka T (2006) Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet 51:1087–1099. https://doi.org/10.1007/s10038-006-0070-9
Vausort M, Wagner DR, Devaux Y (2014) Long noncoding RNAs in patients with acute myocardial infarction. Circ Res 115:668–677. https://doi.org/10.1161/CIRCRESAHA.115.303836
Yan B, Yao J, Liu JY, Li XM, Wang XQ, Li YJ, Tao ZF, Song YC, Chen Q, Jiang Q (2015) lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res 116:1143–1156. https://doi.org/10.1161/CIRCRESAHA.116.305510
Zhu XH, Yuan YX, Rao SL, Wang P (2016) LncRNA MIAT enhances cardiac hypertrophy partly through sponging miR-150. Eur Rev Med Pharmacol Sci 20:3653–3660
Qu X, Du Y, Shu Y, Gao M, Sun F, Luo S, Yang T, Zhan L, Yuan Y, Chu W, Pan Z, Wang Z, Yang B, Lu Y (2017) MIAT is a pro-fibrotic long non-coding RNA governing cardiac fibrosis in post-infarct myocardium. Sci Rep 7:42657. https://doi.org/10.1038/srep42657
Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X (2017) lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell Death Dis 8:e2929. https://doi.org/10.1038/cddis.2017.321
Sone M, Hayashi T, Tarui H, Agata K, Takeichi M, Nakagawa S (2007) The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J Cell Sci 120:2498–2506. https://doi.org/10.1242/jcs.009357
Rapicavoli NA, Poth EM, Blackshaw S (2010) The long noncoding RNA RNCR2 directs mouse retinal cell specification. BMC Dev Biol 10:49. https://doi.org/10.1186/1471-213X-10-49
Tsuiji H, Yoshimoto R, Hasegawa Y, Furuno M, Yoshida M, Nakagawa S (2011) Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells 16:479–490. https://doi.org/10.1111/j.1365-2443.2011.01502.x
Galarneau A, Richard S (2005) Target RNA motif and target mRNAs of the Quaking STAR protein. Nat Struct Mol Biol 12:691–698. https://doi.org/10.1038/nsmb963
Azat M, Huojiahemaiti X, Gao R, Peng P (2019) Long noncoding RNA MIAT: a potential role in the diagnosis and mediation of acute myocardial infarction. Mol Med Rep 20:5216–5222. https://doi.org/10.3892/mmr.2019.10768
Eicher JD, Wakabayashi Y, Vitseva O, Esa N, Yang Y, Zhu J, Freedman JE, McManus DD, Johnson AD (2016) Characterization of the platelet transcriptome by RNA sequencing in patients with acute myocardial infarction. Platelets 27:230–239. https://doi.org/10.3109/09537104.2015.1083543
Wang XM, Li XM, Song N, Zhai H, Gao XM, Yang YN (2019) Long non-coding RNAs H19, MALAT1 and MIAT as potential novel biomarkers for diagnosis of acute myocardial infarction. Biomed Pharmacother 118:109208. https://doi.org/10.1016/j.biopha.2019.109208
Tan J, Liu S, Jiang Q, Yu T, Huang K (2019) LncRNA-MIAT increased in patients with coronary atherosclerotic heart disease. Cardiol Res Pract 2019:6280194. https://doi.org/10.1155/2019/6280194
Toraih EA, El-Wazir A, Alghamdi SA, Alhazmi AS, El-Wazir M, Abdel-Daim MM, Fawzy MS (2019) Association of long non-coding RNA MIAT and MALAT1 expression profiles in peripheral blood of coronary artery disease patients with previous cardiac events. Genet Mol Biol 42:509–518. https://doi.org/10.1590/1678-4685-GMB-2018-0185
Zhu M, Li N, Luo P, Jing W, Wen X, Liang C, Tu J (2018) Peripheral blood leukocyte expression of lncrna miat and its diagnostic and prognostic value in ischemic stroke. J Stroke Cerebrovasc Dis 27:326–337. https://doi.org/10.1016/j.jstrokecerebrovasdis.2017.09.009
de Gonzalo-Calvo D, Kenneweg F, Bang C, Toro R, van der Meer RW, Rijzewijk LJ, Smit JW, Lamb HJ, Llorente-Cortes V, Thum T (2016) Circulating long-non coding RNAs as biomarkers of left ventricular diastolic function and remodelling in patients with well-controlled type 2 diabetes. Sci Rep 6:37354. https://doi.org/10.1038/srep37354
Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ (2006) Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet 367:1747–1757. https://doi.org/10.1016/S0140-6736(06)68770-9
Reed GW, Rossi JE, Cannon CP (2017) Acute myocardial infarction. Lancet 389:197–210. https://doi.org/10.1016/S0140-6736(16)30677-8
Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, Joint ESCAAHAWHFTFfUDoMI, Authors/Task Force Members C, Thygesen K, Alpert JS, White HD, Biomarker S, Jaffe AS, Katus HA, Apple FS, Lindahl B, Morrow DA, Subcommittee ECG, Chaitman BR, Clemmensen PM, Johanson P, Hod H, Imaging S, Underwood R, Bax JJ, Bonow JJ, Pinto F, Gibbons RJ, Classification S, Fox KA, Atar D, Newby LK, Galvani M, Hamm CW, Intervention S, Uretsky BF, Steg PG, Wijns W, Bassand JP, Menasche P, Ravkilde J, Trials, Registries S, Ohman EM, Antman EM, Wallentin LC, Armstrong PW, Simoons ML, Trials, Registries S, Januzzi JL, Nieminen MS, Gheorghiade M, Filippatos G, Trials, Registries S, Luepker RV, Fortmann SP, Rosamond WD, Levy D, Wood D, Trials, Registries S, Smith SC, Hu D, Lopez-Sendon JL, Robertson RM, Weaver D, Tendera M, Bove AA, Parkhomenko AN, Vasilieva EJ, Mendis S, Guidelines ESCCfP, Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck-Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, Document R, Morais J, Aguiar C, Almahmeed W et al (2012) Third universal definition of myocardial infarction. J Am Coll Cardiol 60:1581–1598. https://doi.org/10.1016/j.jacc.2012.08.001
Jose Corbalan J, Vatner DE, Vatner SF (2016) Myocardial apoptosis in heart disease: does the emperor have clothes? Basic Res Cardiol 111:31. https://doi.org/10.1007/s00395-016-0549-2
Rodriguez M, Lucchesi BR, Schaper J (2002) Apoptosis in myocardial infarction. Ann Med 34:470–479. https://doi.org/10.1080/078538902321012414
Chen L, Zhang D, Yu L, Dong H (2019) Targeting MIAT reduces apoptosis of cardiomyocytes after ischemia/reperfusion injury. Bioengineered 10:121–132. https://doi.org/10.1080/21655979.2019.1605812
Cong L, Su Y, Wei D, Qian L, Xing D, Pan J, Chen Y, Huang M (2020) Catechin relieves hypoxia/reoxygenation-induced myocardial cell apoptosis via down-regulating lncRNA MIAT. J Cell Mol Med 24:2356–2368. https://doi.org/10.1111/jcmm.14919
Mercer JR (2014) Mitochondrial bioenergetics and therapeutic intervention in cardiovascular disease. Pharmacol Ther 141:13–20. https://doi.org/10.1016/j.pharmthera.2013.07.011
Suen DF, Norris KL, Youle RJ (2008) Mitochondrial dynamics and apoptosis. Genes Dev 22:1577–1590. https://doi.org/10.1101/gad.1658508
Wallace DC (1999) Mitochondrial diseases in man and mouse. Science 283:1482–1488. https://doi.org/10.1126/science.283.5407.1482
Gatliff J, Campanella M (2012) The 18 kDa translocator protein (TSPO): a new perspective in mitochondrial biology. Curr Mol Med 12:356–368. https://doi.org/10.2174/1566524011207040356
Morin D, Musman J, Pons S, Berdeaux A, Ghaleh B (2016) Mitochondrial translocator protein (TSPO): from physiology to cardioprotection. Biochem Pharmacol 105:1–13. https://doi.org/10.1016/j.bcp.2015.12.003
Motloch LJ, Hu J, Akar FG (2015) The mitochondrial translocator protein and arrhythmogenesis in ischemic heart disease. Oxid Med Cell Longev 2015:234104. https://doi.org/10.1155/2015/234104
Veenman L, Papadopoulos V, Gavish M (2007) Channel-like functions of the 18-kDa translocator protein (TSPO): regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr Pharm Des 13:2385–2405. https://doi.org/10.2174/138161207781368710
Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2:647–656. https://doi.org/10.1038/nrc883
Kugler W, Veenman L, Shandalov Y, Leschiner S, Spanier I, Lakomek M, Gavish M (2008) Ligands of the mitochondrial 18 kDa translocator protein attenuate apoptosis of human glioblastoma cells exposed to erucylphosphohomocholine. Cell Oncol 30:435–450. https://doi.org/10.3233/clo-2008-0431
Mendonca-Torres MC, Roberts SS (2013) The translocator protein (TSPO) ligand PK11195 induces apoptosis and cell cycle arrest and sensitizes to chemotherapy treatment in pre- and post-relapse neuroblastoma cell lines. Cancer Biol Ther 14:319–326. https://doi.org/10.4161/cbt.23613
Bai X, Yang C, Jiao L, Diao H, Meng Z, Wang L, Cui H, Sun L, Zhang Y, Yang B (2021) LncRNA MIAT impairs cardiac contractile function by acting on mitochondrial translocator protein TSPO in a mouse model of myocardial infarction. Signal Transduct Target Ther 6:172. https://doi.org/10.1038/s41392-021-00538-y
Bujak M, Frangogiannis NG (2007) The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74:184–195. https://doi.org/10.1016/j.cardiores.2006.10.002
Dean RG, Balding LC, Candido R, Burns WC, Cao Z, Twigg SM, Burrell LM (2005) Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem 53:1245–1256. https://doi.org/10.1369/jhc.4A6560.2005
Frangogiannis NG (2020) Cardiac fibrosis. Cardiovasc Res. https://doi.org/10.1093/cvr/cvaa324
Chuang TD, Ansari A, Yu C, Sakurai R, Harb A, Liu J, Khorram O, Rehan VK (2020) Mechanism underlying increased cardiac extracellular matrix deposition in perinatal nicotine-exposed offspring. Am J Physiol Heart Circ Physiol 319:H651–H660. https://doi.org/10.1152/ajpheart.00021.2020
Xing PC, An P, Hu GY, Wang DL, Zhou MJ (2020) LncRNA MIAT promotes inflammation and oxidative stress in sepsis-induced cardiac injury by targeting miR-330-5p/TRAF6/NF-kappaB Axis. Biochem Genet 58:783–800. https://doi.org/10.1007/s10528-020-09976-9
Mahtta D, Sudhakar D, Koneru S, Silva GV, Alam M, Virani SS, Jneid H (2020) Targeting inflammation after myocardial infarction. Curr Cardiol Rep 22:110. https://doi.org/10.1007/s11886-020-01358-2
Ong SB, Hernandez-Resendiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek XY, Cabrera-Fuentes HA, Hausenloy DJ (2018) Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther 186:73–87. https://doi.org/10.1016/j.pharmthera.2018.01.001
Liu W, Liu Y, Zhang Y, Zhu X, Zhang R, Guan L, Tang Q, Jiang H, Huang C, Huang H (2015) MicroRNA-150 protects against pressure overload-induced cardiac hypertrophy. J Cell Biochem 116:2166–2176. https://doi.org/10.1002/jcb.25057
Li Y, Wang J, Sun L, Zhu S (2018) LncRNA myocardial infarction-associated transcript (MIAT) contributed to cardiac hypertrophy by regulating TLR4 via miR-93. Eur J Pharmacol 818:508–517. https://doi.org/10.1016/j.ejphar.2017.11.031
Li Z, Liu Y, Guo X, Sun G, Ma Q, Dai Y, Zhu G, Sun Y (2018) Long noncoding RNA myocardial infarctionassociated transcript is associated with the microRNA1505p/P300 pathway in cardiac hypertrophy. Int J Mol Med 42:1265–1272. https://doi.org/10.3892/ijmm.2018.3700
Wei JQ, Shehadeh LA, Mitrani JM, Pessanha M, Slepak TI, Webster KA, Bishopric NH (2008) Quantitative control of adaptive cardiac hypertrophy by acetyltransferase p300. Circulation 118:934–946. https://doi.org/10.1161/CIRCULATIONAHA.107.760488
Zeng Z, Pan Y, Wu W, Li L, Wu Z, Zhang Y, Deng B, Wei S, Zhang W, Lin F, Song Y (2019) Myocardial hypertrophy is improved with berberine treatment via long non-coding RNA MIAT-mediated autophagy. J Pharm Pharmacol 71:1822–1831. https://doi.org/10.1111/jphp.13170
Beesley SJ, Weber G, Sarge T, Nikravan S, Grissom CK, Lanspa MJ, Shahul S, Brown SM (2018) Septic cardiomyopathy. Crit Care Med 46:625–634. https://doi.org/10.1097/CCM.0000000000002851
Brieler J, Breeden MA, Tucker J (2017) Cardiomyopathy: an overview. Am Fam Physician 96:640–646
Bugger H, Abel ED (2014) Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 57:660–671. https://doi.org/10.1007/s00125-014-3171-6
Jia G, DeMarco VG, Sowers JR (2016) Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 12:144–153. https://doi.org/10.1038/nrendo.2015.216
Bern C (2015) Chagas’ disease. N Engl J Med 373:456–466. https://doi.org/10.1056/NEJMra1410150
Perez-Molina JA, Molina I (2018) Chagas disease. Lancet 391:82–94. https://doi.org/10.1016/S0140-6736(17)31612-4
Frade AF, Laugier L, Ferreira LR, Baron MA, Benvenuti LA, Teixeira PC, Navarro IC, Cabantous S, Ferreira FM, da Silva CD, Gaiotto FA, Bacal F, Pomerantzeff P, Santos RH, Kalil J, Cunha-Neto E, Chevillard C (2016) Myocardial infarction-associated transcript, a long noncoding RNA, is overexpressed during dilated cardiomyopathy due to chronic Chagas disease. J Infect Dis 214:161–165. https://doi.org/10.1093/infdis/jiw095
Cimolai MC, Alvarez S, Bode C, Bugger H (2015) Mitochondrial mechanisms in septic cardiomyopathy. Int J Mol Sci 16:17763–17778. https://doi.org/10.3390/ijms160817763
Ehrman RR, Sullivan AN, Favot MJ, Sherwin RL, Reynolds CA, Abidov A, Levy PD (2018) Pathophysiology, echocardiographic evaluation, biomarker findings, and prognostic implications of septic cardiomyopathy: a review of the literature. Crit Care 22:112. https://doi.org/10.1186/s13054-018-2043-8
Nattel S, Maguy A, Le Bouter S, Yeh YH (2007) Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 87:425–456. https://doi.org/10.1152/physrev.00014.2006
Atienza F, Almendral J, Moreno J, Vaidyanathan R, Talkachou A, Kalifa J, Arenal A, Villacastin JP, Torrecilla EG, Sanchez A, Ploutz-Snyder R, Jalife J, Berenfeld O (2006) Activation of inward rectifier potassium channels accelerates atrial fibrillation in humans: evidence for a reentrant mechanism. Circulation 114:2434–2442. https://doi.org/10.1161/CIRCULATIONAHA.106.633735
Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Zhang Y, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B (2010) MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 122:2378–2387. https://doi.org/10.1161/CIRCULATIONAHA.110.958967
Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, Lin H, Xiao L, Maguy A, Qi XY, Li Y, Gao X, Dong D, Zhang Y, Bai Y, Ai J, Sun L, Lu H, Luo XY, Wang Z, Lu Y, Yang B, Nattel S (2013) MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. J Clin Invest 123:1939–1951. https://doi.org/10.1172/JCI62185
Schotten U, Ausma J, Stellbrink C, Sabatschus I, Vogel M, Frechen D, Schoendube F, Hanrath P, Allessie MA (2001) Cellular mechanisms of depressed atrial contractility in patients with chronic atrial fibrillation. Circulation 103:691–698. https://doi.org/10.1161/01.cir.103.5.691
Workman AJ, Kane KA, Rankin AC (2001) The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res 52:226–235. https://doi.org/10.1016/s0008-6363(01)00380-7
Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S (1997) Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res 81:512–525. https://doi.org/10.1161/01.res.81.4.512
Zhang H, Garratt CJ, Zhu J, Holden AV (2005) Role of up-regulation of IK1 in action potential shortening associated with atrial fibrillation in humans. Cardiovasc Res 66:493–502. https://doi.org/10.1016/j.cardiores.2005.01.020
Wang Z, Lu Y, Yang B (2011) MicroRNAs and atrial fibrillation: new fundamentals. Cardiovasc Res 89:710–721. https://doi.org/10.1093/cvr/cvq350
Wang Z, Yue L, White M, Pelletier G, Nattel S (1998) Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation 98:2422–2428. https://doi.org/10.1161/01.cir.98.22.2422
Burstein B, Nattel S (2008) Atrial structural remodeling as an antiarrhythmic target. J Cardiovasc Pharmacol 52:4–10. https://doi.org/10.1097/FJC.0b013e3181668057
Dzeshka MS, Lip GY, Snezhitskiy V, Shantsila E (2015) Cardiac fibrosis in patients with atrial fibrillation: mechanisms and clinical implications. J Am Coll Cardiol 66:943–959. https://doi.org/10.1016/j.jacc.2015.06.1313
Lau DH, Linz D, Schotten U, Mahajan R, Sanders P, Kalman JM (2017) Pathophysiology of paroxysmal and persistent atrial fibrillation: rotors, foci and fibrosis. Heart Lung Circ 26:887–893. https://doi.org/10.1016/j.hlc.2017.05.119
Yao L, Zhou B, You L, Hu H, Xie R (2020) LncRNA MIAT/miR-133a-3p axis regulates atrial fibrillation and atrial fibrillation-induced myocardial fibrosis. Mol Biol Rep 47:2605–2617. https://doi.org/10.1007/s11033-020-05347-0
Zhang LS JY, Liu YY, Yu YH, Sun X, Tian H, Xu J, Yue E, Lv Y, Dong CR, Wang XY, Liu GQ, Zhang DY, Wang ZG, Bai YL, Yang BF (2020) Long non-coding RNA MIAT governs atrial fibrillation via dual mechanisms as a competitive endogenous RNA and an antisense RNA. Basic Res Cardiol (unpublished data)
Jayprokas CSM (2017) Cancer and noncoding RNAs: antisense RNA and cancer. Elsevier Publisher, Amsterdam
Garcia-Padilla C, Dominguez JN, Aranega AE, Franco D (2019) Differential chamber-specific expression and regulation of long non-coding RNAs during cardiac development. Biochim Biophys Acta Gene Regul Mech 1862:194435. https://doi.org/10.1016/j.bbagrm.2019.194435
Farsangi SJ, Rostamzadeh F, Sheikholeslami M, Jafari E, Karimzadeh M (2020) Modulation of the expression of long non-coding RNAs H19, GAS5, and MIAT by endurance exercise in the hearts of rats with myocardial infarction. Cardiovasc Toxicol. https://doi.org/10.1007/s12012-020-09607-0
Jae N, Dimmeler S (2015) Long noncoding RNAs in diabetic retinopathy. Circ Res 116:1104–1106. https://doi.org/10.1161/CIRCRESAHA.115.306051
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146:353–358. https://doi.org/10.1016/j.cell.2011.07.014
Bosson AD, Zamudio JR, Sharp PA (2014) Endogenous miRNA and target concentrations determine susceptibility to potential ceRNA competition. Mol Cell 56:347–359. https://doi.org/10.1016/j.molcel.2014.09.018
Broderick JA, Zamore PD (2014) Competitive endogenous RNAs cannot alter microRNA function in vivo. Mol Cell 54:711–713. https://doi.org/10.1016/j.molcel.2014.05.023
Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M (2014) Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell 54:766–776. https://doi.org/10.1016/j.molcel.2014.03.045
Jens M, Rajewsky N (2015) Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet 16:113–126. https://doi.org/10.1038/nrg3853
Thomson DW, Dinger ME (2016) Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet 17:272–283. https://doi.org/10.1038/nrg.2016.20
Cai B, Ma W, Ding F, Zhang L, Huang Q, Wang X, Hua B, Xu J, Li J, Bi C, Guo S, Yang F, Han Z, Li Y, Yan G, Yu Y, Bao Z, Yu M, Li F, Tian Y, Pan Z, Yang B (2018) The long noncoding RNA CAREL controls cardiac regeneration. J Am Coll Cardiol 72:534–550. https://doi.org/10.1016/j.jacc.2018.04.085
Zhang Y, Jiao L, Sun L, Li Y, Gao Y, Xu C, Shao Y, Li M, Li C, Lu Y, Pan Z, Xuan L, Zhang Y, Li Q, Yang R, Zhuang Y, Zhang Y, Yang B (2018) LncRNA ZFAS1 as a SERCA2a inhibitor to cause intracellular Ca(2+) overload and contractile dysfunction in a mouse model of myocardial infarction. Circ Res 122:1354–1368. https://doi.org/10.1161/CIRCRESAHA.117.312117
Delihas N, Rokita SE, Zheng P (1997) Natural antisense RNA/target RNA interactions: possible models for antisense oligonucleotide drug design. Nat Biotechnol 15:751–753. https://doi.org/10.1038/nbt0897-751
Dolnick BJ (1997) Naturally occurring antisense RNA. Pharmacol Ther 75:179–184. https://doi.org/10.1016/s0163-7258(97)00050-8
Knowling S, Morris KV (2011) Non-coding RNA and antisense RNA. Nature’s trash or treasure? Biochimie 93:1922–1927. https://doi.org/10.1016/j.biochi.2011.07.031
Cai B, Ma W, Wang X, Sukhareva N, Hua B, Zhang L, Xu J, Li X, Li S, Liu S, Yu M, Xu Y, Song R, Xu B, Yang F, Han Z, Ding F, Huang Q, Yu Y, Zhao Y, Wang J, Bamba D, Zagidullin N, Li F, Tian Y, Pan Z, Yang B (2020) Targeting LncDACH1 promotes cardiac repair and regeneration after myocardium infarction. Cell Death Differ 27:2158–2175. https://doi.org/10.1038/s41418-020-0492-5
Acknowledgements
The authors greatly thank Prof. Zhiguo Wang for continued support and valuable advice of this manuscript.
Funding
This work was supported by the Funds for National Key Research and Development Program of China (2017YFC1307403 to Baofeng Yang, 2017YFC1702003 to Yong Zhang); the Key Program of National Natural Science Foundation of China (81730012 to Yong Zhang); the National Natural Science Foundation of China (81861128022, 81570301, 81570399 to Yong Zhang); and Qiqihar Academy of Medical Sciences Project (QMSI2020B-04 to Chao Yang).
Author information
Authors and Affiliations
Contributions
BY conceived idea of the article. CY performed the literature search and wrote the manuscript. YZ critically revised the work. All authors approved the final version of the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, C., Zhang, Y. & Yang, B. MIAT, a potent CVD-promoting lncRNA. Cell. Mol. Life Sci. 79, 43 (2022). https://doi.org/10.1007/s00018-021-04046-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-021-04046-8