
Abstract
Long non-coding RNAs (lncRNAs) interact with the nuclear architecture and are involved in fundamental biological mechanisms, such as imprinting, histone-code regulation, gene activation, gene repression, lineage determination, and cell proliferation, all by regulating gene expression. Understanding the lncRNA regulation of transcriptional or post-transcriptional gene regulation expands our knowledge of disease. Several associations between altered lncRNA function and gene expression have been linked to clinical disease phenotypes. Early advances have been made in developing lncRNAs as biomarkers. Several mouse models reveal that human lncRNAs have very diverse functions. Their involvement in gene and genome regulation as well as disease underscores the importance of lncRNA-mediated regulatory networks. Because of their tissue-specific expression potential, their function as activators or repressors, and their selective targeting of genes, lncRNAs are of potential therapeutic interest. We review the regulatory mechanisms of lncRNAs, their major functional principles, and discuss their role in Mendelian disorders, cancer, cardiovascular disease, and neurological disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Gene regulation
The principle of gene expression is based on epigenetic, transcriptional, and post-transcriptional regulation. Conrad Hal Waddington coined the term “epigenotype or epigenetics” to summarize the complex developmental processes existing between phenotype and genotype in 1942 [1]. Today, we define epigenetics as the study of heritable changes in gene activity, which are not caused by changes in the DNA sequence. The genomic architecture and intra or interchromosomal communication are key mechanisms for accurate gene regulation [2]. Post-transcriptional processes such as alternative splicing, RNA editing, and microRNA-mediated regulation are reviewed elsewhere [3, 4].
Genomic regulators localized on one chromosome that act on the same chromosome are termed cis-regulatory elements. Elements regulating in-trans are interchromosomal regulators that communicate either between homologous or non-homologous chromosomes. Enhancer elements activate gene expression, in contrast to silencers that suppress expression. Insulators determine barriers between different chromatin states (Fig. 1a) and affect expression secondarily [5]. The gene-regulatory elements can exhibit high conservation; however, tissue-specific expression can differ greatly between species [6, 7]. The distance between the down or upstream-located regulator and the target gene ranges several kilobases to 1.5 megabases (Mb) [8]. Functional protein complexes, namely, transcription factors (TFs) and co-activators, bind with chromatin-modifying proteins at DNA consensus motifs. These motif complexes influence the gene expression. The nucleosome state is changed through ATP-dependent remodeling complexes of the SWitch/sucrose nonfermentable (SWI/SNF) families (first found in yeast) or through histone modifiers, such as histone acetyl or methyl transferases [9, 10]. The tissue-specific expression is based on the chromatin state and the bound TF. Histones, the nucleoproteins around which the DNA is wrapped, and histone marks characterize the chromatin state. More than 60 different reversible and irreversible histone modifications are known. They vary between tissues and species and determine the transcriptional active euchromatin or inactive heterochromatin states [11]. The histone modifications, especially during chromatin looping,Footnote 1 determine quantitatively the gene regulation.

a Scheme of gene-regulatory elements influencing gene expression. Enhancer, silencer, and insulator elements can be localized up to 1.5 Mb upstream or downstream of the transcription start site. Response elements within gene promoters bind transcription factors and co-activators to maintain a tissue-specific gene expression. The regulation is dependent on the histone modifications. b Chromatin-looping between a cis-regulatory element (CRE) and its target gene promoter. Recruited modifying, mediating, activating, or repressing proteins build the transcription factor co-activator complex (TFCC) and interact in physical proximity with the promoter to regulate gene expression
Gene-regulatory elements (Fig. 1b) are then in physical proximity to gene promoters to drive transcription [12]. Methylated alleles or gene clusters of either the maternal or paternal alleles in genomic imprinting processes and during X inactivation to compensate gene dosage become manifested in early embryogenesis [13]. The selection of activating or repressing histone marks was found to be operated by a sequential and combinatorial epigenetic code or language depending on the histone modifications involved, the DNA-binding proteins, and non-coding RNAs (ncRNAs), thereby assuring tissue-specific gene expression and epigenetic modifications [14, 15]. The combinatorial diversity is also due to the variety of functional gene regulators, gene promoters, gene homologs or pseudo genes, and embryonic and tissue-specific developmental stages. As inherited, framework or environmental epigenetic conditions determine systemically each functional element supporting proper gene regulation. Alterations of the complex interactions are often clinically apparent in numeric or structural aberrant karyotypes [16]. The physical dissociation of regulator and gene can cause positional effects leading to differentially expressed genes [17].
lncRNAs influence gene and genome regulation
The Encyclopedia of DNA Elements (ENCODE) consortium was founded in 2003 to characterize and annotate functional genomic elements of the human genome and of several transcriptomes. ENCODE determined that protein-coding genes are not the only major units of the genome that is nearly fully transcribed. Only <3 % of the transcripts originate from protein-coding genes [18]. MicroRNAs (miRNAs), small interfering RNAs (siRNAs), and PIWI-interactingFootnote 2 RNAs (piRNAs) describe the class of short ncRNAs. In contrast, lncRNAs have more than 200 nucleotides, are intra or intergenic, with or without a poly-A signal, oftentimes exhibit low expression levels, and can be highly tissue-specifically expressed and conserved [19, 20]. Linear ncRNAs and circular RNAs (circRNAs) have no protein-coding potential and can exist as mono- or multi-exonic sense and antisense transcripts [21, 22]. The circRNAs are post-transcriptional regulators that are formed by head-to-tail splicing. The first detailed investigation of the circular cerebellar degeneration-related protein 1 transcript (CDR1as) determined antagonistic actions on miRNA [22]. Chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-seq) experiments revealed that H3K4me1, H3K36me3, H3K27ac, and p300 characterize gene-activating enhancers and also lncRNAs loci [20, 23]. RNA sequencing (RNA-seq) of 24 human tissues identified more than 8,000 lncRNAs that are typically co-expressed with their neighboring genes [19]. lncRNAs are tools for the gene and genome regulation within the nucleus [24]. Key roles of lncRNAs have been attributed to the biological processes, chromatin remodeling [25], X-chromosome inactivation [26], embryonic stem cell pluripotency [27], embryogenesis, and development [28] as well as imprinting of genomic loci [13].
Since 1991, the X-inactive specific transcript (Xist) has been investigated to gain insight into the mechanisms of X-chromosomal inactivation (XCI) [26]. In 2013, Xist was shown to exploit the three-dimensional structure of the X chromosome to spread from its transcription site to loci with high gene density and transcription that are in physical nuclear proximity. After the recruitment of the chromatin-modifying polycomb group (PcG) proteins, Xist pulls up further in cis localized X-chromosomal regions to pursue inactivation through the formation of a transcriptionally silent H3K27me3 nuclear compartment and spreads dependently on its internal A-repeat domain across the entire X chromosome [29]. Another project explored the spreading mechanism of Xist on the 150 Mb of the X chromosome [30]. Simon and colleagues described a two-step inactivation mechanism. During the XCI establishment in early embryonic cells, Xist targets gene-rich domains before spreading to intervening gene-poor domains. The mechanism seems to persist as epigenetic memory for a facilitated and more efficient XCI during somatic proliferation and maintenance [30]. The active counterpart of Xist is Tsix, a gene that functions as an antisense to Xist to support the active X chromosome. Furthermore, the additional antagonistic relationship of Xist to Jpx, a lncRNA Xist activator, demonstrates that lncRNA-antisense transcripts regulate lncRNA [31, 32]. Also in 2013, Sun and colleagues showed that the lncRNA, Jpx, displaces the chromatin remodeling and RNA-binding protein, CTCF,Footnote 3 from one X chromosome to regulate the Xist expression in a titration-dependent antagonistic mechanism. Prior to XCI, CTCF normally inhibits the Xist expression. However, in the absence of CTCF, Jpx activates the Xist promoter [33]. In summary, these results broaden the classic understanding of how genes or chromosomes are regulated. The process of chromatin looping to establish gene expression functions inter alia through the actions of lncRNA and their associated proteins.
A second well-studied epigenetically acting lncRNA is the HOX antisense intergenic RNA (HOTAIR). HOTAIR is transcribed from the HOXC gene cluster on chromosome 12 and acts in a repressing manner on 40 kilobases of the HOXD cluster on chromosome 2 in trans through H3K27 trimethylation [34]. The lncRNAs Air and KCNQ1OT1 are both localized on imprinted paternal alleles. They recruit the polycomb repressive complex 2 (PRC2) and a histone methylase mediating the enrichment of the histone modification H3K9me3 to silence the genes KCNQ1 and IGFR2 [35, 36]. In contrast to lncRNA-silencing genes, several activating lncRNAs have been described that promote gene expression of in cis target loci with protein-coding genes [37]. HOXA transcript at the distal tip (HOTTIP) directly interacts in cis with the WDR5/MLL complex ending up in the activation of gene transcription from the HOXA gene cluster through enrichment of the euchromatin characteristic H3K4me3 flag [38]. The cis- and trans-chromosomal interaction lncRNA (CISTR-ACT) is a chondrogenic regulator that interacts in cis and in trans with essential developmental genes determining the cartilage. The CISTR-ACT lncRNA is transcribed from an enhancer that loops to a 24.4 Mb distant chromosome 12 position to induce PTHLH expression. In addition, CISTR-ACT pinpoints SOX9 on the non-homologous chromosome 17 in trans [39]. The knowledge about HOTAIR and CISTR-ACT extend the gene-regulatory background regarding in cis interactions. In in trans communications of homologous or non-homologous chromosomes, the nuclear architecture is a major participating element.
As far back as 1904, Theodor Boveri coined the term of chromosomal territories [40]. Chromosomes are not randomly distributed within the nucleus, although mitotic conformational changes occur [41]. Transcriptional active euchromatin is predominantly located in the nuclear center; the compact heterochromatin resides in the outer nucleus. The positioning of chromosomes and their interaction is not predetermined, but rather is a result of a stochastically calculated process, including chromosome looping [42]. In the molecular genetic high-throughput techniques, a modification of chromosome conformation capture (3C) termed Hi-C and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) are methods that allow proximal chromosomal contacts to be identified. The experimental analysis determined the spatial proximity of gene-rich regions and chromosomes and showed the segregation of euchromatin and heterochromatin genomic areas [43, 44]. The topological nuclear domains can determine tissue-specific gene regulation through different sizes and attributes.
Some lncRNAs have been intensively investigated during the last two decades (Table 1). The data yielded insight into the highly organized structure of the nucleus. The complex interplay between chromosome territories, chromatin state, and lncRNAs affects the tissue-specific gene regulation to control developmental stages or to maintain tissue perpetuation [41]. The lncRNAs can reflect nuclear addresses acting in a local, locus-specific, or allele-specific manner for the control of gene expression, genome organization, or regulation. The initial lncRNA transcription signals in cis or in trans and recruits chromatin-modifying complexes or basic factors of the transcription machinery. Moreover, the lncRNA transcription can be involved in the formation of topological nuclear domains, thereby working secondarily on gene regulation [45]. The described lncRNA-mediated regulation scenarios are maintained from different classes of lcnRNAs that belong to the families of competing endogenous lncRNAs (ceRNAs), activating or enhancer-like lncRNAs, natural antisense transcript (NAT) lncRNAs, and small nucleolar RNAs (snoRNAs, Table 1). Table 1 lists the lncRNAs with defined biological functions or with a proven association with human diseases.
Functional insights in lncRNA-mediated gene regulation
We are merely at the beginning in our understanding of the functional processes and the biological width of lncRNA-mediated gene regulation. One controversial question is how lncRNAs can specifically regulate their target genes. Do scaffolds between lncRNAs and mediating protein complexes exist to guarantee the in cis and in trans regulations? Are triple helices of lncRNAs:DNA:DNA formed, or do lncRNAs bind transcription factors and mediate gene expression themselves? During chromatin looping, are lncRNAs dropped from their transcription site for a specific and direct lncRNAs:DNA binding at regulatory sites within a coding gene locus? Do lncRNAs bind the mRNA transcript for post-transcriptional modifications?
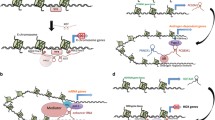
The features of lncRNAs for biologically relevant regulatory processes are their keys to success. Several lncRNAs function as decoys to trap regulatory proteins. DNA damage mediated the induction of the lncRNA PANDA that interacted with the transcription factor NF-YA, to prevent apoptosis by titrating the NF-YA away from target genes. The expression control of pro-apoptotic genes can be a general feature of genes that drive mitosis and which promoters harbor lncRNAs [46] (Fig. 2a). LncRNAs can provide the service as scaffolding adaptors to bind protein complexes for further gene targeting (Fig. 2b). The lncRNA, TERC, serves as scaffold to transport the telomerase complex [47]. Promoter-associated RNA (pRNA) associate with the chromatin-remodeling complex NoRC/TIP5 to induce transcriptional silencing through DNA methylation of rRNA genes [48]. The DNA methylase, DNMT3b, then recognizes the triple helix of lncRNA at the DNA binding site for the transcription factor TTF-1 [49]. For HOTTIP, Wang et al. showed that chromosomal looping with spatial proximity within the HOXA gene cluster is necessary to drive the transcription of several 5′ HOXA genes through direct binding of the co-activating HOTTIP lncRNA with the adaptor protein WDR5 [38]. Some lncRNAs seem to translate higher spatial chromosome structures and processes such as looping into defined chromatin modifications and domains to control gene expression. The same mechanism could be subject to CISTR-ACT and other enhancer-encoded lncRNAs [37, 39] (Fig. 2c). The physical proximity in chromatin loops enables the transformation of higher order genome conformation into biochemical histone modifications and transcription factor recruitment. The lincRNA-p21, HOTAIR, XIST, AIR, and other lncRNAs can bind RNA-binding or chromatin-remodeling proteins to support guiding functions to conduct further remodeling complexes or co-activators or repressors to specific genomic loci [50, 51] (Table 1, Fig. 2d). Xist can stack the transcription factor YY1 that is capable to bind RNA and DNA, thereby attaching Xist to the X chromosome. They form the nucleation center together with PRC2 and squelch gene expression by competing with the transcription machinery [52].

a lncRNA can serve as decoys to control the actions of DNA-binding proteins, i.e., PANDA. b Scaffold structures of lncRNA with protein partners display functional units for gene regulation, TERC. c Enhancer-encoded lncRNA act specifically on their target genes through chromatin loops, i.e., CISTR-ACT and HOTTIP. d Either DNA-bound adaptor proteins bind lncRNA or DNA-bound lncRNA serves as guides for further functional processes, i.e., Xist or HOTAIR. Triple-helix formations of lncRNA with the DNA double helix are possible
lncRNAs in development and disease
In addition to epigenetic functions during X-chromosome inactivation, imprinting and co-activation, or repression of genes, lncRNAs have been attributed to various functions in cellular homeostasis, during development, and in pathogenesis of diseases. The half-STAU1-binding site RNA (½-sbsRNA) co-activate the STAU1-mediated mRNA decay by dsRNA formation to regulate the degradation of translationally active mRNAs [53]. The terminal differentiation-induced lncRNA (TINCR) regulates the somatic tissue differentiation through binding to differentiation-mediating mRNAs for proper translation [54]. TINCR directly binds to the STAU1 protein, thereby stabilizing differentiating mRNAs. Braveheart (Bvht) was identified as lncRNA responsible for the establishment of the cardiovascular lineage determination and the maintenance of the cardiac fate [55]. Bvht conducts its functions through interaction with SUZ12, an important subunit of PRC2. EGO (eosinophil granule ontogeny) plays a role during eosinophil development [56]. Fendrr is a lateral mesoderm-specific lncRNA controlling mesodermal differentiation and its developing derivatives heart and body wall through binding to the histone-remodeling complexes PRC2 and TrxG/MLL [57]. Fendrr-lacking embryos showed dysregulation of mesoderm-specific transcription factors and reduction of PRC2 enrichment at their loci. Gomafu is involved in neuronal and retinal development. Its downregulation, the binding of splicing factors and the resulting altered splicing patterns, was associated with schizophrenia [58]. The muscle-specific lncRNA linc-MD1 is a competing endogenous RNA (ceRNA) that controls muscle differentiation via sponging of miRNA. Moreover, linc-MD1 seems to be involved in the pathogenesis of Duchenne muscle dystrophy [59]. The lncRNA, FMR4, triggers the ratio of proliferation and apoptosis and was silenced in patients with the fragile X syndrome [60]. CISTR-ACT was dysregulated due to chromosomal translocations in two different families with the autosomal-dominant Mendelian disorder of the chondrodysplasia brachydactyly type E (BDE). Chromosome 12 translocations physically disrupted CISTR-ACT from the major chondrogenic morphogene, PTHLH, and caused dysregulation of the coding gene and lncRNA [39]. These data underscore the important interface between genome conformation and gene-lncRNA regulation. The suppression of UBE3A-ATS can activate UBE3A in patients with the neurogenetic disorder of Angelman’s syndrome [61, 62]. The severe phenotypes of leukemia, myelofibrosis, sarcoma, and vasculitis were detected in Xist-depleted mice and suggest Xist-mediated in vivo cancer repression. The loss of Xist seemed to reactivate the X chromosome leading finally through aberrant hematopoietic stem cells to cancer [63]. The metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) that is very abundantly expressed co-localizes with splicing factors in the nuclear speckles and regulates alternative splicing of pre-mRNAs [64]. Its association with migration, metastasis, and tumor growth in lung adenocarcinoma has been shown [65, 66]. MALAT1 is required for mitotic proliferation and seems to mediate its activity positively through activated p53 and B-MYB, an oncogenic transcription factor. Thus, the dysregulation of splicing factors and alternative splicing led to the dysregulation of cell-cycle-regulated transcription factors promoting cellular proliferation [66]. For the non-polyadenylated MALAT1, a 3′ triple helix formation that served as translational enhancer was found, was inhibited by miRNAs, and was argued for a major role in regulation and stabilization of MALAT1 [67]. HOTAIR was proposed as a diagnostic marker in breast and colorectal cancer. Its depletion resulted in reduced invasiveness, and its expression level correlated with differentially regulated genes of the PRC2 complex [25, 68]. Currently, upregulated HOTTIP and HOXA13 expressions were associated with the prognosis and progression of the hepatocellular carcinoma (HCC) [69]. The highly upregulated lncRNA HULC in liver cancer was found in the blood of HCC patients, promising a potential biomarker [70]. HULC sponges several miRNA such as miR-372, leading to transcriptional inhibition of target genes, i.e., the transcription factor CREB. The CREB motif within the HULC promoter supports CREB-mediated upregulation in liver cancer through an autoregulatory mechanism blocking the miR-372 function [71]. Moreover, HULC correlated with upregulated hepatitis B virus X protein (HBx) that importantly contributes to HCC and that was able to promote HULC expression. The HULC-mediated downregulation of the tumor suppressor p18 supported the HCC proliferation [72]. The expression of BACE1 antisense transcript (BACE1-AS) was linked to increased amyloid-β 1–42 in patients with Alzheimer’s disease and gave rise for a stabilizing function of the lncRNA [73]. Aberrant ANRIL transcripts and mutations were associated with cardiovascular disease and cancer [74, 75]. The existence of linear and circular ANRIL transcripts was found in patients with atherosclerosis [74]. The prostate cancer-associated ncRNA transcript 1 lncRNA PCAT-1 [76, 77], SchlAP1 (second chromosome locus associated with prostate-1) [78], and CTBP1-AS [79] indicate cancer cell invasiveness and metastasis in prostate cancer progression. SchlAP1 antagonizes the tumor-suppressing functions of the SWI/SNF chromatin-remodeling complex [78, 80], and CTBP1-AS represses CTBP not only by interacting with histone deacetylases and the transcriptional repressor PSF but also by inhibiting tumor-suppressor genes in a general manner [79].
Current research directions
The idea that lncRNAs are nuclear addresses or addressors respectively plays a major role in gene and genome regulation. The regulation of the regulators is not clear. The lncRNAs also need to be directed. One hypothesis is that the interplay between the marginally expressed lncRNAs, tissue-specific TF, and histone modifications ensure tissue-specific gene expression. In which extent, is stochastical coincidence present? In the human genome, 46 chromosomes containing ~3 × 109 bp communicate while underlying dramatic conformational changes during mitosis. However, the cellular and nuclear infrastructures remain to fulfill the particularized cellular tasks.
The lncRNAs are often highly tissue specific. Despite the barely understood mechanisms of their specific target-gene regulation, lncRNAs have a potential therapeutic value. To date, most of the therapeutic agents serve an inhibitory function. Blocking lncRNAs could lead to the upregulation of genes and have a stimulatory effect. The subclass of natural antisense transcripts (NATs), shown in Table 1, can be degraded or inhibited in binding their target mRNA through single-stranded antagonistic oligo-nucleotides (antagoNATs) [81]. The endogenous de-repression of genes could be the key in various haplo-insufficiencies. Moreover, upregulated lncRNAs in cancer that normally exhibit decoy functions could be also targets for antagoNATs [81]. Previously, antisense oligo-nucleotides (ASOs) were successfully applied to silence the RNA gain-of-function effect in the hereditary degenerative disease myotonic dystrophy type 1 (DM1), and a performed myogenic long-term Malat1 knockdown was effective [82]. Currently, siRNAs are being introduced therapeutically in patients.
Detailed system biological approaches are needed to locate, to annotate, and to characterize lncRNAs in development and disease. Various genetic model systems have to be established to understand the functional roles of lncRNAs:protein interactions that modulate chromatin remodeling complexes, gene, and genome regulation to investigate lncRNA-associated pathogenesis of disease or developmental defects. In two different projects generating Malat1 knockout mice, any apparent phenotype or alteration of the murine development was observed [83, 84]. Only in cis genes of Malat1 were differentially expressed [84]. The lncRNA NEAT1 is highly expressed in the mammal-specific nuclear paraspeckles Footnote 4. Interestingly, the NEAT1-depleted mice had no phenotypes, suggesting environmentally provoked nuclear structures [85]. In Malat1-depleted mice, showing no phenotype again, Neat1 was downregulated in several tissues lacking Malat1, indicating its dispensability in mice paraspeckles [86]. These data indicate that human-specific lncRNAs that do not exert their human functions in animal models may exist. In contrast, a Hotair deletion in mice was associated with malformation of the spine and metacarpals and a general, non-selective de-repression of genes [87]. In the latest study of 18 knockout models for approved lncRNAs, only five displayed apparent phenotypes [88]. The results indicate that long-term and more precise phenotypization could reveal additional subtle and highly tissue-specific behavioral phenotypes. The lncRNAs that have been associated with clinically apparent phenotypes are only the tip of the iceberg. The detailed molecular analysis of lncRNAs will augment the understanding of the nuclear regulation networks and discover more pathogenic lncRNAs or circRNAs that can serve as clinically relevant prognostic or diagnostic biomarker or as therapeutic targets.
Notes
Chromatin looping: Studying the structural properties and spatial organization of chromosomes is important for the understanding and evaluation of the regulation of gene expression, DNA replication and repair, and recombination. One example of chromosomal interaction is chromosome looping in which a chromosomal region can fold in order to bring an enhancer and associated TF within close proximity to a gene.
PIWI: The piwi or PIWI, originally P-element-induced wimpy testis in Drosophila, class of genes was originally identified as encoding regulatory proteins responsible for maintaining incomplete differentiation in stem cells. PIWI proteins are highly conserved nucleic acid-binding proteins.
CTCF: Transcriptional repressor CTCF, also known as 11-zinc finger protein or CCCTC-binding factor, is a transcription factor that is involved in many cellular processes, including transcriptional regulation, insulator activity, and regulation of chromatin architecture.
Paraspeckle: A paraspeckle is an irregularly shaped cell compartment, approximately 0.2–1 μm in size, found in the nuclear interchromatin space.
References
Waddington CH (2012) The epigenotype. 1942. Int J Epidemiol 41:10–13
Mercer TR, Mattick JS (2013) Understanding the regulatory and transcriptional complexity of the genome through structure. Genome Res 23:1081–1088
Chen K, Rajewsky N (2007) The evolution of gene regulation by transcription factors and microRNAs. Nat Rev Genet 8:93–103
Poulos MG, Batra R, Charizanis K, Swanson MS (2011) Developments in RNA splicing and disease. Cold Spring Harb Perspect Biol 3:a000778
Phillips-Cremins JE, Corces VG (2013) Chromatin insulators: linking genome organization to cellular function. Mol Cell 50:461–474
Odom DT et al (2007) Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet 39:730–732
Vavouri T, Walter K, Gilks WR, Lehner B, Elgar G (2007) Parallel evolution of conserved non-coding elements that target a common set of developmental regulatory genes from worms to humans. Genome Biol 8:R15
Vavouri T, McEwen GK, Woolfe A, Gilks WR, Elgar G (2006) Defining a genomic radius for long-range enhancer action: duplicated conserved non-coding elements hold the key. Trends Genet 22:5–10
Schubeler D, Francastel C, Cimbora DM, Reik A, Martin DI, Groudine M (2000) Nuclear localization and histone acetylation: a pathway for chromatin opening and transcriptional activation of the human beta-globin locus. Genes Dev 14:940–950
Vignali M, Hassan AH, Neely KE, Workman JL (2000) ATP-dependent chromatin-remodeling complexes. Mol Cell Biol 20:1899–1910
Kouzarides T (2007) Chromatin modifications and their function. Cell 128:693–705
Blackwood EM, Kadonaga JT (1998) Going the distance: a current view of enhancer action. Science 281:60–63
Lee JT, Bartolomei MS (2013) X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152:1308–1323
Berger SL (2002) Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12:142–148
Lee JS, Smith E, Shilatifard A (2010) The language of histone crosstalk. Cell 142:682–685
Kleinjan DA, van Heyningen V (2005) Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet 76:8–32
Wijchers PJ, de Laat W (2011) Genome organization influences partner selection for chromosomal rearrangements. Trends Genet 27:63–71
Maher B (2012) ENCODE: the human encyclopaedia. Nature 489:46–48
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927
Guttman M et al (2009) Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458:223–227
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J (2013) Natural RNA circles function as efficient microRNA sponges. Nature 495:384–388
Memczak S et al (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495:333–338
Khalil AM et al (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 106:11667–11672
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166
Kogo R et al (2011) Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res 71:6320–6326
Brown CJ, Ballabio A, Rupert JL, Lafreniere RG, Grompe M, Tonlorenzi R, Willard HF (1991) A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349:38–44
Dinger ME et al (2008) Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res 18:1433–1445
Pauli A, Rinn JL, Schier AF (2011) Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet 12:136–149
Engreitz JM et al (2013) The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341:1237973
Simon MD et al (2013) High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 504:465–469
Lee JT, Davidow LS, Warshawsky D (1999) Tsix, a gene antisense to Xist at the X-inactivation centre. Nat Genet 21:400–404
Tian D, Sun S, Lee JT (2010) The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 143:390–403
Sun S, Del Rosario BC, Szanto A, Ogawa Y, Jeon Y, Lee JT (2013) Jpx RNA activates Xist by evicting CTCF. Cell 153:1537–1551
Rinn JL et al (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129:1311–1323
Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P (2008) The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322:1717–1720
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32:232–246
Orom UA et al (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58
Wang KC et al (2011) A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472:120–124
Maass PG et al (2012) A misplaced lncRNA causes brachydactyly in humans. J Clin Invest 122:3990–4002
Strickfaden H, Zunhammer A, van Koningsbruggen S, Kohler D, Cremer T (2010) 4D chromatin dynamics in cycling cells: Theodor Boveri’s hypotheses revisited. Nucleus 1:284–297
Cremer T, Cremer M (2010) Chromosome territories. Cold Spring Harb Perspect Biol 2:a003889
Bickmore WA (2013) The spatial organization of the human genome. Annu Rev Genomics Hum Genet 14:67–84
Fullwood MJ et al (2009) An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462:58–64
Lieberman-Aiden E et al (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293
Batista PJ, Chang HY (2013) Long noncoding RNAs: cellular address codes in development and disease. Cell 152:1298–1307
Hung T et al (2011) Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet 43:621–629
Zappulla DC, Cech TR (2006) RNA as a flexible scaffold for proteins: yeast telomerase and beyond. Cold Spring Harb Symp Quant Biol 71:217–224
Mayer C, Schmitz KM, Li J, Grummt I, Santoro R (2006) Intergenic transcripts regulate the epigenetic state of rRNA genes. Mol Cell 22:351–361
Schmitz KM, Mayer C, Postepska A, Grummt I (2010) Interaction of noncoding RNA with the rDNA promoter mediates recruitment of DNMT3b and silencing of rRNA genes. Genes Dev 24:2264–2269
Huarte M et al (2010) A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 142:409–419
Yoon JH et al (2012) LincRNA-p21 suppresses target mRNA translation. Mol Cell 47:648–655
Jeon Y, Lee JT (2011) YY1 tethers Xist RNA to the inactive X nucleation center. Cell 146:119–133
Gong C, Maquat LE (2011) lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 470:284–288
Kretz M et al (2013) Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 493:231–235
Klattenhoff CA et al (2013) Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152:570–583
Wagner LA, Christensen CJ, Dunn DM, Spangrude GJ, Georgelas A, Kelley L, Esplin MS, Weiss RB, Gleich GJ (2007) EGO, a novel, noncoding RNA gene, regulates eosinophil granule protein transcript expression. Blood 109:5191–5198
Grote P et al (2013) The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell 24:206–214
Barry G et al (2013) The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry. doi:10.1038/mp.2013.45
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147:358–369
Khalil AM, Faghihi MA, Modarresi F, Brothers SP, Wahlestedt C (2008) A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS ONE 3:e1486
Landers M, Calciano MA, Colosi D, Glatt-Deeley H, Wagstaff J, Lalande M (2005) Maternal disruption of Ube3a leads to increased expression of Ube3a-ATS in trans. Nucleic Acids Res 33:3976–3984
Meng L, Person RE, Beaudet AL (2012) Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet 21:3001–3012
Yildirim E, Kirby JE, Brown DE, Mercier FE, Sadreyev RI, Scadden DT, Lee JT (2013) Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 152:727–742
Tripathi V et al (2010) The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39:925–938
Schmidt LH et al (2011) The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol 6:1984–1992
Tripathi V et al (2013) Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet 9:e1003368
Wilusz JE, JnBaptiste CK, Lu LY, Kuhn CD, Joshua-Tor L, Sharp PA (2012) A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26:2392–2407
Gupta RA et al (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464:1071–1076
Quagliata L et al (2013) lncRNA HOTTIP/HOXA13 expression is associated with disease progression and predicts outcome in hepatocellular carcinoma patients. Hepatology. doi:10.1002/hep.26740
Panzitt K et al (2007) Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 132:330–342
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q (2010) CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res 38:5366–5383
Du Y, Kong G, You X, Zhang S, Zhang T, Gao Y, Ye L, Zhang X (2012) Elevation of highly up-regulated in liver cancer (HULC) by hepatitis B virus X protein promotes hepatoma cell proliferation via down-regulating p18. J Biol Chem 287:26302–26311
Faghihi MA et al (2008) Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med 14:723–730
Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE (2010) Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet 6:e1001233
Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I (2007) Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res 67:3963–3969
Ge X, Chen Y, Liao X, Liu D, Li F, Ruan H, Jia W (2013) Overexpression of long noncoding RNA PCAT-1 is a novel biomarker of poor prognosis in patients with colorectal cancer. Med Oncol 30:588
Prensner JR et al (2011) Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 29:742–749
Prensner JR et al (2013) The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet 45:1392–1398
Takayama K et al (2013) Androgen-responsive long noncoding RNA CTBP1-AS promotes prostate cancer. Embo J 32:1665–1680
Lu P, Roberts CW (2013) The SWI/SNF tumor suppressor complex: regulation of promoter nucleosomes and beyond. Nucleus 4:374–378
Wahlestedt C (2013) Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat Rev Drug Discov 12:433–446
Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA (2012) Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature 488:111–115
Eissmann M et al (2012) Loss of the abundant nuclear non-coding RNA MALAT1 is compatible with life and development. RNA Biol 9:1076–1087
Zhang B et al (2012) The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep 2:111–123
Nakagawa S, Naganuma T, Shioi G, Hirose T (2011) Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J Cell Biol 193:31–39
Nakagawa S, Ip JY, Shioi G, Tripathi V, Zong X, Hirose T, Prasanth KV (2012) Malat1 is not an essential component of nuclear speckles in mice. RNA 18:1487–1499
Li L et al (2013) Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep 5:3–12
Sauvageau M et al (2013) Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2:e01749
Zhang X et al (2009) A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood 113:2526–2534
Hu W, Yuan B, Flygare J, Lodish HF (2011) Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes Dev 25:2573–2578
Onoguchi M, Hirabayashi Y, Koseki H, Gotoh Y (2012) A noncoding RNA regulates the neurogenin1 gene locus during mouse neocortical development. Proc Natl Acad Sci U S A 109:16939–16944
Navarro P et al (2010) Molecular coupling of Tsix regulation and pluripotency. Nature 468:457–460
Ogawa Y, Lee JT (2003) Xite, X-inactivation intergenic transcription elements that regulate the probability of choice. Mol Cell 11:731–743
van Dijk M et al (2012) HELLP babies link a novel lincRNA to the trophoblast cell cycle. J Clin Invest 122:4003–4011
Sonkoly E et al (2005) Identification and characterization of a novel, psoriasis susceptibility-related noncoding RNA gene, PRINS. J Biol Chem 280:24159–24167
Beckedorff FC, Ayupe AC, Crocci-Souza R, Amaral MS, Nakaya HI, Soltys DT, Menck CF, Reis EM, Verjovski-Almeida S (2013) The intronic long noncoding RNA ANRASSF1 recruits PRC2 to the RASSF1A promoter, reducing the expression of RASSF1A and increasing cell proliferation. PLoS Genet 9:e1003705
Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B (2010) Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with anril expression. PLoS Genet 6:e1000899
Kotake Y, Nakagawa T, Kitagawa K, Suzuki S, Liu N, Kitagawa M, Xiong Y (2011) Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30:1956–1962
Pasmant E, Sabbagh A, Vidaud M, Bieche I (2011) ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J 25:444–448
Yap KL, Li S, Munoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, Gil J, Walsh MJ, Zhou MM (2010) Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 38:662–674
Arita T et al (2013) Circulating long non-coding RNAs in plasma of patients with gastric cancer. Anticancer Res 33:3185–3193
Kallen AN et al (2013) The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell 52:101–112
Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A, Galun E (2007) The H19 non-coding RNA is essential for human tumor growth. PLoS ONE 2:e845
Thorvaldsen JL, Duran KL, Bartolomei MS (1998) Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev 12:3693–3702
Yang Z, Zhou L, Wu LM, Lai MC, Xie HY, Zhang F, Zheng SS (2011) Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann Surg Oncol 18:1243–1250
Liu Y et al (2012) A genetic variant in long non-coding RNA HULC contributes to risk of HBV-related hepatocellular carcinoma in a Chinese population. PLoS ONE 7:e35145
Golding MC, Magri LS, Zhang L, Lalone SA, Higgins MJ, Mann MR (2011) Depletion of Kcnq1ot1 non-coding RNA does not affect imprinting maintenance in stem cells. Development 138:3667–3678
Nakano S et al (2006) Expression profile of LIT1/KCNQ1OT1 and epigenetic status at the KvDMR1 in colorectal cancers. Cancer Sci 97:1147–1154
Gutschner T et al (2013) The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res 73:1180–1189
Xu C, Yang M, Tian J, Wang X, Li Z (2011) MALAT-1: a long non-coding RNA and its important 3′ end functional motif in colorectal cancer metastasis. Int J Oncol 39:169–175
Lee GL, Dobi A, Srivastava S (2011) Prostate cancer: diagnostic performance of the PCA3 urine test. Nat Rev Urol 8:123–124
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP (2010) A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465:1033–1038
Colley SM, Leedman PJ (2011) Steroid receptor RNA activator—a nuclear receptor coregulator with multiple partners: Insights and challenges. Biochimie 93:1966–1972
Cooper C, Guo J, Yan Y, Chooniedass-Kothari S, Hube F, Hamedani MK, Murphy LC, Myal Y, Leygue E (2009) Increasing the relative expression of endogenous non-coding steroid receptor RNA activator (SRA) in human breast cancer cells using modified oligonucleotides. Nucleic Acids Res 37:4518–4531
Redon S, Zemp I, Lingner J (2013) A three-state model for the regulation of telomerase by TERRA and hnRNPA1. Nucleic Acids Res 41:9117–9128
Ritter O, Haase H, Schulte HD, Lange PE, Morano I (1999) Remodeling of the hypertrophied human myocardium by cardiac bHLH transcription factors. J Cell Biochem 74:551–561
Ishii N et al (2006) Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet 51:1087–1099
Rapicavoli NA, Poth EM, Blackshaw S (2010) The long noncoding RNA RNCR2 directs mouse retinal cell specification. BMC Dev Biol 10:49
Haddad F, Bodell PW, Qin AX, Giger JM, Baldwin KM (2003) Role of antisense RNA in coordinating cardiac myosin heavy chain gene switching. J Biol Chem 278:37132–37138
Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK (2008) The DISC locus in psychiatric illness. Mol Psychiatry 13:36–64
Scheele C et al (2007) Altered regulation of the PINK1 locus: a link between type 2 diabetes and neurodegeneration? FASEB J 21:3653–3665
Chen WL, Lin JW, Huang HJ, Wang SM, Su MT, Lee-Chen GJ, Chen CM, Hsieh-Li HM (2008) SCA8 mRNA expression suggests an antisense regulation of KLHL1 and correlates to SCA8 pathology. Brain Res 1233:176–184
Acknowledgements
We apologize to all scientists whose important work of lncRNAs could not be cited in this review. The German Research Foundation (DFG MA-5028/1–3, LU-435/15-1) supports our work. We also receive support from the ECRC, a joint cooperation between the Max-Delbrück-Center for Molecular Medicine (MDC) and the Medical Faculty of the Charité.
Conflict of interest
The authors declare no interest conflicts.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Maass, P.G., Luft, F.C. & Bähring, S. Long non-coding RNA in health and disease. J Mol Med 92, 337–346 (2014). https://doi.org/10.1007/s00109-014-1131-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-014-1131-8