
Abstract
Preservation of mitochondrial quality is paramount for cellular homeostasis. The integrity of mitochondria is guarded by the balanced interplay between anabolic and catabolic mechanisms. The removal of bio-energetically flawed mitochondria is mediated by the process of mitophagy; the impairment of which leads to the accumulation of defective mitochondria which signal the activation of compensatory mechanisms to the nucleus. This process is known as the mitochondrial retrograde response (MRR) and is enacted by Reactive Oxygen Species (ROS), Calcium (Ca2+), ATP, as well as imbalanced lipid and proteostasis. Central to this mitochondria-to-nucleus signalling are the transcription factors (e.g. the nuclear factor kappa-light-chain-enhancer of activated B cells, NF-κB) which drive the expression of genes to adapt the cell to the compromised homeostasis. An increased degree of cellular proliferation is among the consequences of the MRR and as such, engagement of mitochondrial-nuclear communication is frequently observed in cancer. Mitophagy and the MRR are therefore interlinked processes framed to, respectively, prevent or compensate for mitochondrial defects.
In this review, we discuss the available knowledge on the interdependency of these processes and their contribution to cell signalling in cancer.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mitochondria are organelles surrounded by double membranes that are present in every eukaryotic cell. Pivotal for the maintenance of cellular energy metabolism, signalling and death [1], they benefit from a two-way route of interaction with the nucleus: the anterograde, directed from the nucleus to mitochondria, and the retrograde, which goes from mitochondria to the nucleus also known as the mitochondrial retrograde response (MRR) (Fig. 1). The anterograde response is indispensable to build the mitochondria and the retrograde response to signal deficiencies in their quality control (Fig. 1). Mitochondrial quality control is the synthesis between (i) biogenesis of mitochondria and (ii) removal of the defective mitochondria by targeted autophagy (hereafter referred to as mitophagy) [2, 3].

The interplay between anabolic and catabolic mechanisms. The diagram depicts the interplay between anterograde and retrograde signalling. The former is required to build mitochondria and the latter to signalling aberration to their homeostasis. The loss of mitophagy efficiency and hence the impaired control of mitochondrial quality (depicted by the dashed outer mitochondrial membrane, OMM) activates signalling mechanisms that communicate the ongoing dysfunction to the nucleus. This process goes under the name of the mitochondrial retrograde response (MRR) and results in the expression of target genes
The biogenesis of mitochondria is the result of efficient anterograde communication from the nucleus to mitochondria, in which a specific subset of proteins is produced and transferred.
The nuclear DNA (nDNA)-encoded polypeptides are the subunits of mitochondrial enzyme complexes necessary to exert the oxidative phosphorylation (OXPHOS) from where the greater portion of adenosine triphosphate (ATP) originates [4, 5]. The most important regulator of mitochondrial biogenesis is PGC-1α, a member of the peroxisome proliferator-activated receptor γ (PGC) family of transcriptional co-activators [6]. PGC-1α co-activates nuclear respiratory factors 1 and 2 (NRF1/2) that in turn, regulates the expression of many nuclear-encoded mitochondrial genes, such as mitochondrial transcription factor A (TFAM) [7]. TFAM is responsible for transcribing mitochondrially encoded proteins that are involved in mitochondrial DNA (mtDNA) transcription, translation and repair [7]. Therefore, biogenetic mechanisms are essential for the development of mitochondria but insufficient to assure their quality.
Mitophagy removes additional or defective mitochondria thereby regulating the content of the mitochondrial network [8, 9] (Fig. 2). This event is indispensable for cellular health, as it prevents the accumulation of dysfunctional mitochondria [8, 9]. However, when it fails, though, the lost quality of mitochondria triggers a route of communication with the nucleus to produce genes aimed at protecting the cell from demise driven by faulty mitochondria [10, 11].

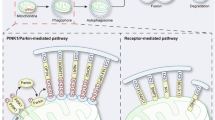
Schematic diagram of the mitochondrial autophagy (mitophagy) mechanisms. a depicts the Parkin-dependent mitophagy in which the accumulation of PINK1 on the mitochondrial surface recruits Parkin to ubiquitylate proteins of the outer mitochondrial membrane (OMM). Consequently, the autophagosome, via the adapter proteins (p62, OPTN and NBR1) is fused on the disposable mitochondria (represented by dashed OMM). In the Parkin-independent pathway of mitophagy (b) autophagy receptor proteins (e.g. FUNDC1 and NIX) or ubiquitin-protein ligases other than Parkin (e.g. SMURF1) accumulate on the bio-energetically impaired mitochondria to drive their recycling via the autophagosomes
Thus, MRR promotes the stabilization of transcriptional factors such as the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κβ) [11], the G-Protein Pathway Suppressor 2 (GPS2) [12] and the forkhead box proteins (FOXOs)] [13]) responsible for driving the metabolic rewiring and resistance to cellular demise driven by faulty mitochondria [11, 14,15,16] (Fig. 3).

Schematic representation of the mitochondrial retrograde response (MRR) in yeast and mammals. a In yeast, mitochondrial dysfunction (read by reduction of ATP production and drop in ΔΨm) activates the transcription factors Rtg1p and Rtg3p which translocate to the nucleus following dephosphorylation. Mammals do not recapitulate the same pathway of signalling (b); the MRR is instead triggered by alterations in the second messengers [e.g. Ca2+ and Reactive Oxygen Species (ROS)] in response to defective mitochondria and executed by the nuclear relocation of a subset of transcriptional factors (e.g. NF-κB, CREB, NFAT, CHOP and GPS2). As above the dashed OMM is used to flag unhealthy mitochondria which initiates the retrograde route of signalling
MRR is engaged by the presence of defective mitochondria spared by mitophagy [8, 17] which stands as the limiting step in this signalling route [18]. Even though a convergence between mitophagy, remodelling of the mitochondrial network and the MRR has been postulated [19], this mechanism is not clear, leaving several unanswered questions. One of these is the localization of mitochondria driving the retrograde communication and whether this establishes microdomains of signalling.
Cancer cells are known to exploit both mitophagy and the MRR representing therefore the ideal model to comprehend hierarchy and cross-regulation between the remodelling of mitochondria (due to the block of mitophagy) and their retrograde signalling to the nucleus [5].
This review will debate this with the ambition to picture a link between the failed clearance of mitochondria and pathogenic cell signalling [5].
The role of mitophagy in the quality control of mitochondria
Mitophagy is mediated by many different molecules [8,9,10,11], but is principally subdivided into Parkin-dependent and Parkin-independent [20]. In the Parkin-dependent pathway (Fig. 2a), the depolarization of the mitochondrial membrane potential (ΔΨm) induces the activation of E3 ubiquitin ligase Parkin (PARK2) by phosphorylation on serine 65 via the PTEN-induced kinase 1 protein (PINK1) (Fig. 2a) [21]. Consequently, PARK2 is translocated from the cytosol to outer mitochondria membrane (OMM) to promote the ubiquitination and degradation of OMM proteins via proteasomes to allow binding for Optineurin (OPTN) [22], the BRCA1 gene 1 (NBR1), [23] and sequestosome-1 (SQSTM1/p62) (Fig. 2a) [24]. In turn, they directly interact with microtubule-associated proteins 1A/1B light chain 3B (LC3) protein, stimulating the localization of autophagosomes on the mitochondria (Fig. 2a) [24]. Furthermore, PINK1, which accumulates at the OMM upon mitochondrial depolarization, directly recruits the autophagy receptors OPTN or NDP52, proposing a model-mechanism within which PARK2 amplifies the PINK1-initiated mitophagy signalling [25]. The mitochondria-localized PARK2 can also interact with the activating molecule in Beclin-1-regulated autophagy (AMBRA1) to localize it at the OMM (Fig. 2a) and hence activate the phosphoinositide 3-kinase (P13K) complex to facilitate the selectivity of mitophagy [20].
In contrast, in the Parkin-independent pathway (Fig. 2b), dysfunctional mitochondria have high levels of LC3-interacting region (LIR)-containing receptors including FUN14 Domain Containing 1(FUNDC1) (i) [26], BCL2/adenovirus E1B 19 kDa interacting protein 3 (Bnip3) (ii) [27], its homologue Bnip3L/Nix (iii) [28] and BCL2-like 13 (Bcl2L13) (iv) [29] which leads to the recruitment of the autophagosomal membranes by direct interaction with LC3 (Fig. 2b).
Following dissipation of the ΔΨm, the SMAD-specific E3 ubiquitin-protein ligase (SMURF1) can also promote mitophagy through the ubiquitination of mitochondrial proteins (Fig. 2b) [30]. Furthermore, the heat shock protein 90 (Hsp90) and Hsp90 co-chaperone (Cdc37) complex (Hsp90-Cdc37) stabilize and activate the serine/threonine protein kinase ULK1. Consequently, ULK1 phosphorylates the Autophagy-related protein 13 (Atg13) (Fig. 2b) forming a complex [consisting of ULK1 itself, Atg13, focal adhesion kinase family interacting protein of 200 kDa (FIP200), and Atg101] for the formation of the autophagosome [31]. This pathway of mitophagy is negatively regulated by the mammalian target of rapamycin (mTOR) [32]. The latter is repressed by the activation of the AMP-activated protein kinase (AMPK) following the reduction of intracellular ATP thus allowing formation of the ULK complex [33] and deacetylation of ATG proteins by sirtuins [34].
Mitophagy is a fundamental process to retain the quality of mitochondria which is conserved across eukaryotic systems; once lost, the growing population of damaged mitochondria drives pathogenic signalling [12, 19, 35].
In cancer, the crosstalk between anabolism and catabolism of mitochondria is impaired and with the abundance of dysfunctional mitochondria, the consumption of ATP rises as well as the accumulation of ROS [36, 37]. The intracellular accumulation of defective mitochondria is capitalized by cancer cells to enable oncogenic development [19, 38]. As forcing mitophagy is not a suitable strategy to arrest such a pattern, it is crucial to comprehend where the dysfunctional mitochondria accumulate when mitophagy is impaired. This could determine where the MRR begins and therefore offer a discrete target to revert the process. Recently, we showed that remodelling of the mitochondrial network, following evasion of mitophagy, is a determinant in the interaction with the nucleus by expediting the cross-organelles communication via specific contact sites. The aggressiveness of cancer could be therefore favoured not only by the accumulation of unhealthy mitochondria but also by their repositioning within the cell.
The mitochondrial retrograde response (MRR)
Transcription factors, stress response mechanisms and mitochondrial activity are all profoundly altered in the pathogenesis of uncontrolled cellular proliferation. Cancer cells exploit MRR, which is primed by upregulation of ROS and deregulation Ca2+ [15, 16], for their survival capacity [5, 39, 40]. During conditions that could endanger their biochemical and structural integrity, mitochondria crosstalk with the nucleus to sustain their reprogramming and adapt to the perturbed environment [10, 11]. The altered metabolism in cancer cells is characterized by the high-consumption rate of glucose that is degraded through glycolysis to obtain ATP. In this scenario, the mitochondrial tricarboxylic acid (TCA) cycle is reduced causing low OXPHOS activity and increased mitochondrial ROS (mtROS) [41]. The inefficient mitochondrial respiration [42] and the parallel accumulation of ROS further compromise organelle integrity by damaging mtDNA [43]. In addition, loading of mitochondrial cholesterol is also a factor in the mitochondrial aetiology of tumour cells [41]. By shielding mitochondrial membranes from pro-apoptotic protein-mediated permeabilization, the excess of cholesterol may delay the commitment of the organelle to apoptosis resulting in pro-longed survival of cells with unhealthy mitochondria [41].
Even though MRR is linked with the pathogenesis of cancer, the most detailed information into this route of signalling is available in a non-mammalian model: the yeast [11]. In S. cerevisiae, the MRR depends on the basic helix-loop-helix/leucine zipper (bHLH/LeuZip) transcription factors which act as a nuclear sensor of mitochondrial dysfunction (manifested by a reduction in ATP content) for the cellular reprogramming to occur [44]. When activated, the retrograde regulation protein (Rtg1/3p) complex translocates from the cytoplasm to the nucleus, where it controls the expression of genes that encode mitochondrial proteins (Fig. 3a) [5, 44]. Inhibition of retrograde signalling is mediated by a cytosolic phosphoprotein, Mks1p, that when activated by phosphorylation, binds to the Bmh-sensitive protein (Bmh1/2p) and prevents dephosphorylation of Rtg3p (Fig. 3a). Rtg2p, an activator of this pathway, binds to Mks1p—a negative regulator of the RTG pathway, limiting its interaction with Bmh1/2p and thus allowing partial dephosphorylation of Rtg3p and Rtg1/3p translocation to the nucleus (Fig. 3a) [44, 45]. The MRR, even though executed in the cytosol-nucleus portion of the cell, is triggered by a decrease in ATP concentration which sees the Mks1p released from Bmh1/2p and then bound with Rtg2p (Fig. 3a) [46].
The lack of homology with the yeast Rtg1/2/3 system in mammals [4] reverted the attention towards cytosolic elements, capable of shuttling into the nucleus to prime the expression of a specific subset of genes.
The same pathway shown in Fig. 3a is triggered following depolarization of the mitochondrial membrane (ΔΨm) [47, 48]. Even though, compared to yeast, in mammals, the molecular coordination between mitochondria and the nucleus remains poorly understood [5, 49], the recent findings on GPS2, a functional homologue of Rtg2, have unveiled a selective interconnection between the two organelles (Fig. 3b) [12]. GPS2 acts as a transcriptional activator of the nuclear-encoded mitochondrial genes in mammals. In response to the loss of the ΔΨm, GPS2 translocates to the nucleus to activate the transcription of stress response genes (Fig. 3b) by activating the Histone H3 Lysine 9 (H3K9) demethylation and RNA polymerase II (RNA POL2), through inhibition of the Ubiquitin-conjugating enzyme E2 13 (Ubc13) [12]. In addition, the disruption of ΔΨm impairs mitochondrial Ca2+ uptake, causing an increase in free Ca2+ within the cytoplasm [45]. This activates a plethora of Ca2+-dependent effectors such as protein kinase C (PKC) (i), calcium/calmodulin-dependent protein kinase type IV (CamKIV) (ii), c-Jun N-terminal kinases (JNK) (iii), and mitogen-activated protein kinase (MAPK) (iv) (Fig. 3b). These proteins engage transcription factors such as activating transcription factor 2 (ATF2), CCAAT/enhancer-binding protein delta (CEBP/δ), CCAAT/enhancer-binding protein delta (CREB), Early growth response protein 1 (Egr-1), and CCAAT-enhancer-binding protein homologous protein (CHOP) that process nuclear events for the transcription of genes [11]. Moreover, the increased levels of Ca2+ activates the Ca2+-dependent serine-threonine phosphatase Calcineurin which in turn induces the nuclear translocation of Nuclear factor of activated T-cells (NFAT) and NF-κB (Fig. 3b) [45] to promote the expression of anti-apoptotic genes, cytokines, immunoreceptors and adhesion molecules. In addition, NF-κB is promptly engaged by the increased redox stress [43]. The production of mtROS triggers the MRR too and is implicit in many pathophysiological conditions including hypoxia, ischemia/reperfusion injury, chemical stress and drug treatment [50, 51]. The increased redox stress (standard in cancer cells) depicts a natural feedback loop between MRR and mitophagy. By studying the protein SKN-1, the nematode homologue of Nuclear factor erythroid 2-like 2 (NFE2L2) [2, 19], it was indeed possible to delineate that the accumulation of ROS, originated from defective mitochondria, promotes the expression of genes involved in mitochondrial biogenesis and mitophagy [12, 19, 35, 52, 53] thus suggesting that mitophagy is naturally engaged to control MRR. In physiology, the crosstalk between mitochondrial biogenesis and mitophagy preserves mitochondrial quality allowing the cells to adjust their mitochondrial content.
In pathology, and still using cancer as an example, mitophagy is unable to remove damaged mitochondria resulting in a negative feedback on mitochondrial biogenesis thus leading to a progressive accumulation of unhealthy mitochondria which drives the MRR further.
Beyond transcription factors, other molecules take part in the MRR [45, 54]. Alterations of OXPHOS function decrease the NAD+/NADH ratio creating a deficit in mitochondria that leads to cytosolic NAD+/NADH imbalance [55]. The respiratory compromised cells redirect pyruvate towards lactate production which triggers MRR [56]. In addition, NAD+ is a co-substrate for the deacetylases sirtuins which are involved in the regulation of various cellular pathways such as histone modification and modulation of FOXO3, NF-κB and PGC-1α [57].
The tricarboxylic acid (TCA) cycle precursor metabolites including succinate [38], fumarate [58, 59] and 2-ketoglutarate (2-kg) [38, 60] accumulate in both mitochondria and cytosol during mitochondrial dysfunctions [38] representing a cue for cellular re-adaptation.
The mitochondrial unfolded protein response (UPRmt) is also considered an MRR trigger [61]. Its activation, which results in a reduction of nuclear and mtDNA-encoded OXPHOS transcripts (thereby operating as counteractor of the anterograde signalling) establishes an efficient pathway of retro-communication with the nucleus via sequential activation of the c-Jun N-terminal kinase (a component of AP-1 transcription factor c-Jun) and the transcription factors C/EBPβ and C/EBP homologous protein (CHOP) [62].
All this implies that the unresolved complexity of the MRR demands more systematic investigation to integrate the diverse aspects of this route of communication. Below, we illustrate one which regards the remodelling mitochondria that holds the potential to inform and better interpret the dynamics of this compensatory signaling pathway.
The mitochondrial space occupancy in cancer cells and the increased interaction with the nucleus
The shape of mitochondria is tightly linked to their function [63] same as the interaction they endure with other intracellular structures [64]. The transitions of mitochondrial morphology are defined as mitochondrial dynamics (fusion and fission events) and it take an active part in both physiological and pathological responses [64].
In highly OXPHOS active cells, mitochondria appear elongated; while in cells relying more on glycolysis, mitochondria present a reduced size and a more fragmented phenotype [65, 66]. Tumour cells are able to modify the shape of their mitochondrial network according to their needs thereby altering space occupancy within the cell [63] and exploiting this to evade apoptosis [63]. The molecular machinery regulating mitochondrial dynamics is therefore indispensable for driving the adaptability of cancer cells within a changing environment.
Mitochondrial fusion entails both dynamic-like guanosine triphosphatases (GTPase), such as mitofusin 1 and 2 (MFN1 and MFN2), and dynamin-related protein optic atrophy 1 (OPA1), responsible for the maintenance of a reticular mitochondrial network inside the cell via OMM and IMM, respectively [66]. In contrast, mitochondrial fragmentation or fission is mediated by a cytosolic GTPase Dynamin-related Protein 1 (Drp1) that binds to its receptor on the OMM [66].
To maintain migratory potential, breast, thyroid and glioblastoma cancer cells the mitochondrial morphology towards fission [67, 68] which is mirrored by the high degree of DRP1 expression and reduced Mfn 1/2 presence [63, 68]. This pattern, however, has not yet been recapitulated in the conditions which drive the essential reprogramming of cells required for the transition to cancer and resistance to chemotherapeutics.
The role of mitochondrial dynamics in the activation and execution of MRR remains substantially unknown. Quite interestingly, an association between pro-fusion elements (e.g. OPA1, Mfn1/2) and this pathway of communication has been already drawn by the characterization of the molecular physiology of the mitochondrial F1Fo-ATPase Inhibitory Factor 1 (IF1) [69].
The over-expression of the latter promotes MRR by mediating cyto-protective resistance to organelle disassembly and facilitating the adaptive cellular reprogramming required for tumour development [50]. IF1, which is the most characterized regulator of the F1Fo-ATPsynthase [70, 71], protects cells from ATP depletion by inhibiting the hydrolytic activity of the enzyme [70]. Upregulated in many human carcinomas [72], IF1 remodels mitochondrial cristae via the stabilization of pro-fusion OPA1 [73]. Mechanistically, IF1 promotes cell survival by repressing apoptosis and preventing mitochondrial recruitment of Drp1, thus hindering fission and permeabilization of mitochondria [74]. The morphological transitions of mitochondria (from elongated to fragmented, and vice-versa) modify and regulate mitochondrial function and hence the retrograde signalling [64].
Among the molecules which populate the outer mitochondrial membrane of the mitochondria, the mitochondrial 18kD Translocator Protein (TSPO)[75], is the one on which we have devoted great attention to decrypt the interplay between physical presence of the mitochondrial network and MRR (Fig. 4). TSPO stabilizes the mitochondrial network by preventing its mitophagy-mediated removal [76], making this an indispensable promoter and facilitator of MRR during cell pathology (Fig. 4). We have demonstrated that in cancerous cells of the mammary gland, TSPO by impairing intracellular cell signalling promotes the translocation of NF-κB into the nucleus onto which cholesterol accumulates to promote the translation of cyto-protective genes and interaction between the two organelles. Namely, we have demonstrated that TSPO recruits on mitochondria the holoenzyme Protein Kinase A (PKA) and the A-kinase anchoring protein Acyl-CoA Binding Domain Containing 3 (ACBD3) which complexes with the nucleus via the A-kinase-anchoring protein AKAP9525 [18] (Fig. 4). It allows the redistribution of cholesterol which sustains the pro-survival response. Therefore, TSPO could be required for the formation of the Nucleus Associated Mitochondria (NAM) [18] thus fulfilling a role beyond the manipulation of Ca2+ and ROS cell signalling [76] at the basis of MRR [4] (Fig. 4).

Mito-nuclear contacts as catalysts of the mitochondrial retrograde response. Dysfunctional mitochondria that escape the quality control by mitophagy enlarge the size of the mitochondrial network, increasing proximity with the nuclear envelope. Such a gain in space establishes contacts between dysfunctional mitochondria (represented by dashed OMM) and the nucleus to facilitate MRR and hence sustain pathogenic signalling. The outer mitochondrial membrane protein TSPO is required for the formation of the nucleus–mitochondria tethering complex together with ABCD3, PKA and AKAP95. Notably, TSPO when accumulated on the mitochondria prevents Parkin-mediated mitophagy thus emerging as a valuable tool to inform the link between declined mitophagy and increased MRR
The recent piece of evidence shows the expression of mitophagy genes during MRR [19]. This makes us speculate on the existence of a threshold level beyond which the segregation of mitochondria becomes irrelevant for the MRR. The repositioning of mitochondria on the nucleus [73, 76] could be indicative of this and therefore the contacts between mitochondria and nucleus a mechanism to evade the autophagy-mediated selection of defective mitochondria and so a prerequisite to drive reprogramming.
Concluding remarks
Mitochondria are highly dynamic organelles undergoing coordinated cycles of fission and fusion indispensable for their shape, location and size. Their transient and rapid morphological adaptation is crucial for many cellular processes. Mutations in the core machinery components and defects in mitochondrial autophagy have been therefore associated with numerous human diseases. In this review, we focused our attention on the ill-defined interplay between accumulation of defective mitochondria within the cytosol and associated cell survival mechanisms in cancer cells driven by the retrograde response pathway. Elucidating how these events are regulated, from a molecular but also biological point of view, represents a crucial step to the understanding of chronic diseases. The discovery of new components that regulate these events is in constant development and must continue in the following years with the support of technologies to aid this task.
We thus feel necessary to better understand why and how dysfunctional mitochondria move and occupy different portions of the cell. And critical is their movements towards the nucleus which drives reprogramming of mammalian cells to evade demise and promote proliferation. In vivo and in vitro studies on defective mitochondria have been fundamental for shedding light on the crucial role of these organelles in the cell fate decision. However, the interrelation between mitochondrial space occupancy and genetic redesign in cancer remains ill-defined limiting our capacity to disentangle the complexity of the disease.
This warrants a more structured investigation to insight the architecture of the cross-organelles communication to advance our current capacity to predict, mark and curb malignant progression.
References
Lu H, Li G, Liu L et al (2013) Regulation and function of mitophagy in development and cancer. Autophagy 9:1720–1736. https://doi.org/10.4161/auto.26550
Palikaras K, Tavernarakis N (2014) Mitochondrial homeostasis: the interplay between mitophagy and mitochondrial biogenesis. Exp Gerontol 56:182–188. https://doi.org/10.1016/j.exger.2014.01.021
Herst PM, Rowe MR, Carson GM, Berridge MV (2017) Functional mitochondria in health and disease. Front Endocrinol 8:296
Quirós PM, Mottis A, Auwerx J (2016) Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol
Yang D, Kim J (2019) Mitochondrial Retrograde Signalling and Metabolic Alterations in the Tumour Microenvironment. Cells 8:. https://doi.org/https://doi.org/10.3390/cells8030275
Sanchis-Gomar F, García-Giménez JL, Gómez-Cabrera MC, Pallardó FV (2014) Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr Pharm Des 20:5619–5633. https://doi.org/10.2174/1381612820666140306095106
Johri A, Chandra A, Flint Beal M (2013) PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic Biol Med 62:37–46. https://doi.org/10.1016/j.freeradbiomed.2013.04.016
Chourasia AH, Boland ML, Macleod KF (2015) Mitophagy and Cancer. Cancer Metab 3:4. https://doi.org/10.1186/s40170-015-0130-8
Youle RJ, Narendra DP (2011) Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12:9–14. https://doi.org/10.1038/nrm3028
Guha M, Srinivasan S, Ruthel G et al (2014) Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene 33:5238–5250. https://doi.org/10.1038/onc.2013.467
Butow RA, Avadhani NG (2004) Mitochondrial signaling: the retrograde response. Mol Cell 14:1–15. https://doi.org/10.1016/s1097-2765(04)00179-0
Cardamone MD, Tanasa B, Cederquist CT et al (2018) Mitochondrial retrograde signaling in mammals is mediated by the transcriptional cofactor GPS2 via direct mitochondria-to-nucleus translocation. Mol Cell 69:757-772.e7. https://doi.org/10.1016/j.molcel.2018.01.037
Kim S, Koh H (2017) Role of FOXO transcription factors in crosstalk between mitochondria and the nucleus. J Bioenerg Biomembr 49:. https://doi.org/https://doi.org/10.1007/s10863-017-9705-0
Liu Z, Butow RA (2006) Mitochondrial retrograde signaling. Annu Rev Genet 40:159–185. https://doi.org/10.1146/annurev.genet.40.110405.090613
Carden T, Singh B, Mooga V et al (2017) Epigenetic modification of miR-663 controls mitochondria-to-nucleus retrograde signaling and tumor progression. J Biol Chem 292:20694–20706. https://doi.org/10.1074/jbc.M117.797001
Feske S, Okamura H, Hogan PG, Rao A (2003) Ca2+/calcineurin signalling in cells of the immune system. Biochem Biophys Res Commun 311:1117–1132. https://doi.org/10.1016/j.bbrc.2003.09.174
Amuthan G, Biswas G, Zhang SY et al (2001) Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J 20:1910–1920. https://doi.org/10.1093/emboj/20.8.1910
Desai R, East DA, Hardy L et al (2020) Mitochondria form contact sites with the nucleus to couple prosurvival retrograde response. Sci Adv 6(51):eabc9955: https://doi.org/10.1126/sciadv.abc9955
Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521:525–528. https://doi.org/10.1038/nature14300
Matic I, Strobbe D, Di Guglielmo F, Campanella M (2017) Molecular biology digest of cell mitophagy. In: International Review of Cell and Molecular Biology
Ordureau A, Sarraf SA, Duda DM et al (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56:360–375. https://doi.org/10.1016/j.molcel.2014.09.007
Wong YC, Holzbaur ELF (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci 111:E4439 LP-E4448. https://doi.org/https://doi.org/10.1073/pnas.1405752111
Hamacher-Brady A, Brady NR (2016) Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci 73:775–795. https://doi.org/10.1007/s00018-015-2087-8
Narendra D, Tanaka A, Suen D-F, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183:795–803. https://doi.org/10.1083/jcb.200809125
Lazarou M, Sliter DA, Kane LA et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. https://doi.org/10.1038/nature14893
Liu L, Feng D, Chen G et al (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14:177–185. https://doi.org/10.1038/ncb2422
Zhu Y, Massen S, Terenzio M et al (2013) Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem 288:1099–1113. https://doi.org/10.1074/jbc.M112.399345
Novak I, Dikic I (2011) Autophagy receptors in developmental clearance of mitochondria. Autophagy 7:301–303. https://doi.org/10.4161/auto.7.3.14509
Murakawa T, Yamaguchi O, Hashimoto A et al (2015) Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun 6:7527. https://doi.org/10.1038/ncomms8527
Koganti P, Levy-Cohen G, Blank M (2018) Smurfs in Protein Homeostasis, Signaling, and Cancer. Front Oncol 8:295. https://doi.org/10.3389/fonc.2018.00295
Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24:24–41. https://doi.org/10.1038/cr.2013.168
Roca-Agujetas V, de Dios C, Lestón L et al (2019) Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxid Med Cell Longev 2019:3809308. https://doi.org/10.1155/2019/3809308
Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19:121–135. https://doi.org/10.1038/nrm.2017.95
Tang BL (2016) Sirt1 and the Mitochondria. Mol Cells 39:87–95. https://doi.org/https://doi.org/10.14348/molcells.2016.2318
Ploumi C, Daskalaki I, Tavernarakis N (2017) Mitochondrial biogenesis and clearance: a balancing act. FEBS J 284:183–195. https://doi.org/10.1111/febs.13820
Yan C, Li T-S (2018) Dual Role of Mitophagy in Cancer Drug Resistance. Anticancer Res 38:617–621. https://doi.org/10.21873/anticanres.12266
Weigl S, Paradiso A, Tommasi S (2013) Mitochondria and familial predisposition to breast cancer. Curr Genomics 14:195–203. https://doi.org/10.2174/1389202911314030005
Zong W-X, Rabinowitz JD, White E (2016) Mitochondria and Cancer. Mol Cell 61:667–676. https://doi.org/10.1016/j.molcel.2016.02.011
Sullivan LB, Chandel NS (2014) Mitochondrial reactive oxygen species and cancer. Cancer Metab 2:17. https://doi.org/10.1186/2049-3002-2-17
Hsu C-C, Tseng L-M, Lee H-C (2016) Role of mitochondrial dysfunction in cancer progression. Exp Biol Med (Maywood) 241:1281–1295. https://doi.org/10.1177/1535370216641787
Ribas V, García-Ruiz C, Fernández-Checa JC (2016) Mitochondria, cholesterol and cancer cell metabolism. Clin Transl Med 5:22. https://doi.org/10.1186/s40169-016-0106-5
Kirkinezos IG, Moraes CT (2001) Reactive oxygen species and mitochondrial diseases. Semin Cell Dev Biol 12:449–457. https://doi.org/10.1006/scdb.2001.0282
Yakes FM, Van Houten B (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA 94:514–519. https://doi.org/10.1073/pnas.94.2.514
Sekito T, Thornton J, Butow RA (2000) Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol Biol Cell 11:2103–2115. https://doi.org/10.1091/mbc.11.6.2103
da Cunha FM, Torelli NQ, Kowaltowski AJ (2015) Mitochondrial retrograde signaling: triggers, pathways, and outcomes. Oxid Med Cell Longev 2015:482582. https://doi.org/10.1155/2015/482582
Zhang F, Pracheil T, Thornton J, Liu Z (2013) Adenosine triphosphate (ATP) is a candidate signaling molecule in the mitochondria-to-nucleus retrograde response pathway. Genes (Basel) 4:86–100. https://doi.org/10.3390/genes4010086
Borghouts C, Benguria A, Wawryn J, Jazwinski SM (2004) Rtg2 protein links metabolism and genome stability in yeast longevity. Genetics 166:765–777. https://doi.org/10.1534/genetics.166.2.765
Miceli MV, Jiang JC, Tiwari A et al (2011) Loss of mitochondrial membrane potential triggers the retrograde response extending yeast replicative lifespan. Front Genet 2:102. https://doi.org/10.3389/fgene.2011.00102
Arnould T, Michel S, Renard P (2015) Mitochondria retrograde signaling and the UPR mt: Where are we in mammals? Int J Mol Sci 16:18224–18251. https://doi.org/10.3390/ijms160818224
Formentini L, Sánchez-Aragó M, Sánchez-Cenizo L, Cuezva JM (2012) The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol Cell 45:731–742. https://doi.org/10.1016/j.molcel.2012.01.008
Srinivasan S, Avadhani NG (2012) Cytochrome c oxidase dysfunction in oxidative stress. Free Radic Biol Med 53:1252–1263. https://doi.org/10.1016/j.freeradbiomed.2012.07.021
Ma Q (2013) Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Yun J, Finkel T (2014) Mitohormesis. Cell Metab 19:757–766. https://doi.org/10.1016/j.cmet.2014.01.011
Bohovych I, Khalimonchuk O (2016) Sending out an SOS: mitochondria as a signaling hub. Front cell Dev Biol 4:109. https://doi.org/10.3389/fcell.2016.00109
Sullivan LB, Gui DY, Hosios AM et al (2015) Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162:552–563. https://doi.org/10.1016/j.cell.2015.07.017
DiMauro S, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348:2656–2668. https://doi.org/10.1056/NEJMra022567
Cantó C, Menzies KJ, Auwerx J (2015) NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab 22:31–53. https://doi.org/10.1016/j.cmet.2015.05.023
Adam J, Yang M, Soga T, Pollard PJ (2014) Rare insights into cancer biology. Oncogene 33:2547–2556. https://doi.org/10.1038/onc.2013.222
Sullivan LB, Martinez-Garcia E, Nguyen H et al (2013) The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell 51:236–248. https://doi.org/10.1016/j.molcel.2013.05.003
Losman J-A, Kaelin WGJ (2013) What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 27:836–852. https://doi.org/10.1101/gad.217406.113
Nargund AM, Pellegrino MW, Fiorese CJ et al (2012) Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337:587–590. https://doi.org/10.1126/science.1223560
Zhang Z, Tan M, Xie Z et al (2011) Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol 7:58–63. https://doi.org/10.1038/nchembio.495
Trotta AP, Chipuk JE (2017) Mitochondrial dynamics as regulators of cancer biology. Cell Mol Life Sci 74:1999–2017. https://doi.org/10.1007/s00018-016-2451-3
Picard M, Shirihai OS, Gentil BJ, Burelle Y (2013) Mitochondrial morphology transitions and functions: Implications for retrograde signaling? Am J Physiol—Regul Integr Comp Physiol 304:. https://doi.org/https://doi.org/10.1152/ajpregu.00584.2012
Chen H, Chan DC (2017) Mitochondrial dynamics in regulating the unique phenotypes of cancer and stem cells. Cell Metab 26(1):39–48. https://doi.org/10.1016/j.cmet.2017.05.016
Maycotte P, Marín-Hernández A, Goyri-Aguirre M et al (2017) Mitochondrial dynamics and cancer. Tumour Biol J Int Soc Oncodev Biol Med 39:1010428317698391. https://doi.org/10.1177/1010428317698391
Ferreira-da-Silva A, Valacca C, Rios E et al (2015) Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE 10:e0122308. https://doi.org/10.1371/journal.pone.0122308
Zhao J, Zhang J, Yu M et al (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32:4814–4824. https://doi.org/10.1038/onc.2012.494
Sánchez-Aragó M, Formentini L, García-Bermúdez J, Cuezva JM (2012) IF1 reprograms energy metabolism and signals the oncogenic phenotype in cancer. Cell Cycle 11:2963–2964. https://doi.org/10.4161/cc.21387
Faccenda D, Campanella M (2012) Molecular Regulation of the mitochondrial F(1)F(o)-ATPsynthase: physiological and pathological significance of the inhibitory factor 1 (IF(1)). Int J Cell Biol 2012:367934. https://doi.org/10.1155/2012/367934
García-Bermúdez J, Cuezva JM (2016) The ATPase inhibitory factor 1 (IF1): a master regulator of energy metabolism and of cell survival. Biochim Biophys Acta 1857:1167–1182. https://doi.org/10.1016/j.bbabio.2016.02.004
Sánchez-Cenizo L, Formentini L, Aldea M et al (2010) Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem 285:25308–25313. https://doi.org/10.1074/jbc.M110.146480
Faccenda D, Nakamura J, Gorini G et al (2017) Control of mitochondrial remodeling by the ATPase inhibitory factor 1 unveils a pro-survival relay via OPA1. Cell Rep 18:1869–1883. https://doi.org/10.1016/j.celrep.2017.01.070
Faccenda D, Tan CH, Seraphim A et al (2013) IF1 limits the apoptotic-signalling cascade by preventing mitochondrial remodelling. Cell Death Differ 20:686–697. https://doi.org/10.1038/cdd.2012.163
Gatliff J, Campanella M (2016) TSPO: kaleidoscopic 18-kDa amid biochemical pharmacology, control and targeting of mitochondria. Biochem J 473(2):107–121. https://doi.org/10.1042/BJ20150899
Gatliff J, East D, Crosby J et al (2014) TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 10:2279–2296. https://doi.org/10.4161/15548627.2014.991665
Acknowledgements
We would like to express our genuine gratitude to Dr. Previdelli, Miss Jemma Gane and Miss Parmis Vadafar for carefully reading the manuscript and providing feedback. The researches activities lead by M.C. on the topics of this script are supported by: The European Research Council Consolidator Grant COG 2018—819600_FIRM; AIRC-MFAG 21903; The Petplan Charitable Trust; LAM-Bighi Grant Initiative.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no competing interests of any nature to report. This research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Strobbe, D., Sharma, S. & Campanella, M. Links between mitochondrial retrograde response and mitophagy in pathogenic cell signalling. Cell. Mol. Life Sci. 78, 3767–3775 (2021). https://doi.org/10.1007/s00018-021-03770-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-021-03770-5