
Abstract
Psoriasis is a chronic inflammatory disease of the skin that affects about 2–3% of the population and greatly impairs the quality of life of affected individuals. Psoriatic skin is characterized by excessive proliferation and aberrant differentiation of keratinocytes, as well as redness caused by increased dilation of the dermal blood vessels and infiltration of immune cells. Although the pathogenesis of psoriasis has not yet been completely elucidated, it is generally believed to arise from a complex interplay between hyperproliferating keratinocytes and infiltrating, activated immune cells. So far, the exact triggers that elicit this disease are still enigmatic, yet, it is clear that genetic predisposition significantly contributes to the development of psoriasis. In this review, we summarize current knowledge of important cellular and molecular mechanisms driving the initiation and amplification stages of psoriasis development, with a particular focus on cytokines and emerging evidence illustrating keratinocyte-intrinsic defects as key drivers of inflammation. We also discuss mouse models that have contributed to a better understanding of psoriasis pathogenesis and the preclinical development of novel therapeutics, including monoclonal antibodies against specific cytokines or cytokine receptors that have revolutionized the treatment of psoriasis. Future perspectives that may have the potential to push basic research and open up new avenues for therapeutic intervention are provided.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The skin is the largest organ serving as an interface between the host and the outside world. It provides a barrier that contains the body and prevents dehydration, as well as protects the body from external threats such as microbial agents and from physical dangers such as damage by daily wear and tear and UV light. In addition, the skin serves as a sensory organ and aids in regulating heat. Next to a physical barrier, the skin also acts as an immunological barrier that is important for the defense against invading pathogens. For instance, keratinocytes not only serve as structural components of the epidermis but also as important innate immune sensors. They contain several pattern recognition receptors that sense the presence of pathogen- or danger-associated molecular patterns and through the production of cytokines and proinflammatory mediators they participate in immune responses [1, 2]. Furthermore, keratinocytes are also an important source of antimicrobial proteins (AMPs) that can control microbial colonization [2, 3]. In addition, the skin hosts various resident immune cells that guard the skin and initiate immune responses. Langerhans cells are antigen presenting cells that populate the epidermis and that are tightly connected through their dendrites with keratinocytes. The exact functions of Langerhans cells are still under debate but by continuous sensing of the environment they instruct adaptive immune responses or induce tolerance to environmental antigens [4, 5]. A second type of immune cells that are abundantly present in both the epidermis and the dermis of healthy skin are tissue-resident memory T cells [2, 6, 7]. These adaptive immune cells establish memory after an initial infection and provide long term protection against commonly encountered pathogens [2, 8]. Compared to the epidermis, the dermis contains a more elaborate repertoire of immune cells under steady state including dendritic cells (DCs), macrophages, mast cells, innate lymphoid cells (ILCs), γδ T cells and conventional αβ T cells [6]. Because of this intricate and diverse immune network, the skin serves as an immune sentinel that can efficiently mount an immune response against invading pathogens. Moreover, immune cells and keratinocytes also contribute to the repair of the disturbed epithelial barrier to reestablish homeostasis [2]. However, when immune responses in the skin are targeted to harmless self-antigens or if they spin out of control, they contribute to the development of inflammatory skin diseases such as psoriasis.
Psoriasis is a common chronic inflammatory skin disease that affects 2–3% of the world's population and greatly impairs the quality of life of affected individuals. Psoriasis vulgaris or plaque psoriasis, the most prevalent disease type, is characterized by well-demarcated, red, scaly plaques. However, psoriasis represents a clinical spectrum including more rare subtypes such as pustular, erythrodermic and guttate psoriasis [9], which can be distinguished based on the clinical morphology of the plaques. For instance, pustular psoriasis is characterized by the presence of multiple tender sterile pustules [9]. Psoriasis-affected skin has a thickened epidermis with scaly patches, due to excessive proliferation and aberrant differentiation of keratinocytes, and redness that is caused by increased dilatation of the dermal blood vessels, and infiltration of immune cells [10]. Histologically this is characterized by thickening of the epidermis, also known as acanthosis, and thickening of the stratum corneum, also known as hyperkeratosis. Furthermore, because of the abnormal differentiation, the granular layer is lost, while cells in the stratum corneum retain their nuclei (parakeratosis). Finally, another prominent hallmark of psoriasis is immune cell infiltration, such as the accumulation of neutrophils in the epidermis and in the stratum corneum, also known as the pustules of Kogoj and Munro abscesses, respectively [10].
In certain cases, the cutaneous immune response becomes no longer restricted to the skin and results in systemic inflammation leading to the development of comorbidities. Thus, psoriasis is associated with an increased risk for cardiovascular disease and almost 30% of patients suffer from psoriatic arthritis [11, 12]. Similarly, patients with psoriasis or psoriatic arthritis exhibit increased rates of inflammatory and functional gastrointestinal disorders [13, 14]. Therefore, understanding the molecular and cellular mechanisms driving psoriasis development and ‘run away’ inflammation, is necessary to improve the current treatment options and help in disease prevention. In this review we will focus on the inflammatory pathways and the immune response driving psoriasis pathology.
Etiology and genetic background of psoriasis
Even though the exact etiology of psoriasis is still largely unknown, the concordance rate of psoriasis in monozygotic twins of approximately 70% illustrates that there is a strong genetic component. Through linkage disequilibrium studies in psoriasis-affected families, several psoriasis susceptibility (PSORS) loci have been identified [10]. However, most of the genes responsible for this observed susceptibility remain unknown [10, 15]. Moreover, only for PSORS1, -2 and -4 the linkage association could be replicated in independent studies [16]. PSORS1 is the first discovered susceptibility locus and is one of the strongest heritable risk factors for psoriasis, which lies in the major histocompatibility complex (MHC) region on chromosome 6p21. Although several possible candidate genes for PSORS1 have been identified, there is general consensus that HLA-Cw6 is the allele responsible for psoriasis susceptibility [17]. Since HLA-Cw6 encodes an MHC class I molecule that presents cellular antigens to CD8+ T cells, it is hypothesized that HLA-Cw6 can bind potential self-antigens and in this way links antigen presentation to psoriasis [15, 16]. So far, only LL37 and ADAMTSL5 have been identified as possible autoantigens that bind HLA-Cw6 [18, 19]. In 2012, Jordan et al. showed that CARD14, a scaffolding protein involved in NF-κB activation, is the gene responsible for psoriasis susceptibility in PSORS2 [20]. Psoriasis-associated mutations in CARD14 result in increased NF-κB activation and expression of proinflammatory mediators in primary human keratinocytes [21, 22]. PSORS4 maps to chromosome 1q21, where the epidermal differentiation cluster is located. This cluster is involved in terminal keratinocyte differentiation, which links psoriasis to skin barrier function [16]. The exact genes in PSORS4 responsible for psoriasis association are not yet known, but deletion of LCE3B and LCE3C was shown to be a risk factor for psoriasis [23].
Recently, analysis of single nucleotide polymorphisms (SNP) in genome-wide association studies, exome-wide association studies and immune chips have revealed several new loci linked to psoriasis susceptibility, with at least 63 different psoriasis susceptibility loci being currently identified [16]. Candidate genes often belong to molecular pathways implicated in psoriasis pathology, such as innate and adaptive immune responses or skin barrier function. For instance, several SNPs in genes linked to the IL-23/IL-17 axis, such as IL12Bp40, IL23Ap19, IL23R and TRAF3IP2 have been discovered. Besides, there is a genetic association with genes involved in antigen presentation (HLA-C, ERAP1 and ERAP2) as well. Pathways involved in innate immunity, such as antiviral signaling (IFIH1, DDX58 and RNF114) and NF-κB signaling (CARD14, TNFAIP3, TNIP1 and NFKBIA) also appear to be linked with psoriasis susceptibility [15]. Finally, changes in copy number variance have been detected in psoriasis patients. Beside the deletion of LCE3B/C, an increase in the copy number of β-defensin is associated with psoriasis susceptibility [24]. Together, these genetic links with psoriasis can help to point out and reveal the key pathways implicated in psoriasis pathogenesis.
Not only genetics but also environmental triggers play a role in the occurrence of psoriasis. For instance, psoriasis can be triggered by physical trauma also known as the Koebner phenomenon. Psoriasis lesions often occur on parts of the body that endure more mechanical force such as elbows and knees, and several provoking factors such as physical trauma, friction, tattoos and burns have been described [25]. Therefore, mechanotransduction in the skin might be involved in the onset of psoriasis [26]. Psoriasis prevalence also seems to be influenced by geographical distribution with the highest frequency in populations of northern Europe and the lowest in populations of eastern Asia [27]. Furthermore, also stress can serve as a trigger for the onset and frequency of psoriasis flares [28]. Finally, psoriasis has been associated with the exposure to certain microbiota. In particular, streptococcal infections have been linked to psoriasis occurrence. In addition, T cells recognizing streptococcal protein M can cross-react to keratin and are often found in peripheral blood of psoriasis patients [29]. Several reports described microbiota changes in psoriatic lesions compared to healthy skin that may contribute to differential T cell polarization [30, 31]. However, the available data is often controversial, possibly due to the differences in applied protocols or microbial variability across human subjects. Also changes in the intestinal microbiome in psoriasis patients have been described, with consistent increase in Firmicutes/Bacteroidetes ratio [32]. Furthermore, mice bred in a germ-free facility or treated with broad-spectrum antibiotics (either topically or systemically) are partially protected from imiquimod-induced skin inflammation, accompanied by reduced frequencies of Th17 and Th22 cells [31, 33, 34], suggesting that the microbiome regulates psoriasis-associated immune responses. However, it is still unclear whether these changes are instrumental to the development of psoriasis or are secondary to the skin pathology. In conclusion, the interplay between the genetic background and the environment governs the development of psoriasis and makes it a multi-factorial disease. However, more research is needed to elucidate the exact mechanisms that serve as the tipping point for disease initiation.
Key inflammatory cytokines that drive psoriatic inflammation
IL-23
Although originally thought to be a Th1-driven disease, the discovery of Th17 cells shifted the view on psoriasis as an IL-23/IL-17-dependent pathology [35]. IL-23 belongs to the IL-12 family of cytokines and consists of two subunits: p19 which is unique for IL-23 in man (and shared with IL-39 in mice) and p40 which is shared with IL-12. Initially, the key pathogenic role in psoriasis was attributed to IL-12, since administration of anti-IL-12 antibodies protected mice from the development of psoriasis-like skin inflammation, surprisingly in an IFNγ-independent manner [36]. However, the antibody that was used targets the p40 subunit and, thus, also neutralizes IL-23. The increased p19 and p40 expression but not IL-12-specific p35 expression in psoriatic lesions provided the first evidence that not IL-12, but IL-23 is a pivotal player in psoriasis [37]. Furthermore, intradermal injection of IL-23 in mice is sufficient to induce skin inflammation [38], while genetic deletion of IL-23 protects from imiquimod-induced psoriasis-like inflammation [39]. Finally, the remarkable efficacy of anti-IL-23 therapeutics (discussed below) further illustrates the central role of IL-23 in the pathogenesis of psoriasis [35].
IL-23 is mainly produced by DCs and macrophages, but also keratinocytes were identified as a source of IL-23 [40]. Binding of IL-23 to its receptor, a heterodimeric complex composed of IL23R and IL12Rβ1 subunits, leads to activation of receptor-associated Jak kinase family members, Jak2 and Tyk2. Activated kinases in turn phosphorylate tyrosine residues in the cytoplasmic tail of IL23R facilitating recruitment and activation of STAT3, a key transcription factor which mediates gene expression downstream of IL-23 receptor activation. IL-23 is mainly notorious for inducing IL-17 production in responder cells, such as Th17, which is often referred to as the IL-23/IL-17 axis [35].
IL-17
IL-17A, often referred to as IL-17, is the best characterized IL-17 family member. It is considered as the most potent mediator of psoriatic inflammation. IL-17A expression is increased in psoriatic lesions and in the blood of patients with psoriasis [41]. IL-23-induced epidermal hyperplasia is reduced in IL-17A-deficient mice or upon IL-17A neutralization [42]. Although Th17 cells have been considered for a long time to be a major source of IL-17, it is expressed by a wider range of cells in psoriatic lesions, such as γδ T cells, ILC3s, neutrophils and mast cells, which also express IL-23R [43, 44]. Therefore, psoriasis is now no longer considered as a strictly Th17-mediated disease but more as an IL-17-driven disease. IL-17 binds to a heterodimeric receptor consisting of IL17RC and IL17RA and not only induces Act1-mediated NF-κB activation, but also activates the C/EBP transcription factor family [45, 46] and enhances the stability of specific mRNAs (Fig. 1). In addition, IL-17A (as well as IL-36) stimulation was shown to induce expression of IκBζ, a co-factor of NF-κB, followed by inflammatory gene expression in keratinocytes [47, 48], while keratinocyte-specific deletion of IκBζ protects from imiquimod- or IL-36-induced skin inflammation [49]. Consequently, IL-17 receptor activation in keratinocytes induces expression of several proinflammatory cytokines, chemokines and AMPs, which sustain and perpetuate the inflammatory reactions during psoriasis [43, 45, 46]. Next to IL-17A, also IL-17F and IL-17C play a role in psoriatic skin inflammation. IL-17A and IL-17F are often co-expressed, bind the same receptor and act in a similar way, while IL-17C is produced by epithelial cells and can thus signal in an autocrine manner [50, 51]. Importantly, IL-17 family members might have different contributions to psoriasis development. Thus, inhibition of IL-17A alone was insufficient to inhibit the development of skin inflammation in IL-36α or STAT3 transgenic mice treated with phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) [52,53,54]. However, antibody-mediated blockade of the IL-17 receptor efficiently inhibited psoriasis-like pathology [54]. Similarly, IL-17 receptor-deficient mice were resistant to imiquimod-induced skin inflammation, comparably to IL-23 knockout mice [39]. Finally, several IL-17A- and IL-17 RA-neutralizing biologics have shown high efficacy in psoriasis patients (discussed below).

Major inflammatory signaling pathways in keratinocytes driving psoriasis. Hyperactivating CARD14 mutations drive the formation of a CARD14-BCL10-MALT1 complex, leading to downstream NF-κB and MAPK signaling and the initiation of pro-inflammatory gene expression, including IL-23, TNF and IL-36. These cytokines can then act in an auto- or paracrine manner and, in turn, initiate NF-κB and MAPK signaling in keratinocytes and other cells. In addition, IL-17 (produced by T cells and ILC3) binding to its receptor can also induce the NF-κB, as well as C/EBPβ. The latter mediates expression of Arginase 1, leading to increased production of polyamines that can stabilize self-RNA and facilitate its uptake by dendritic cells, thus contributing to downstream inflammatory responses. TNF has also been shown to trigger N-WASP phosphorylation in keratinocytes, preventing it from repressing IL-23 expression by controlling H3K9 methylation of the IL-23 promoter
TNF
TNF is another inflammatory cytokine that has been implicated in psoriasis pathology since a long time. As such, high levels of TNF, TNFR1 and TNFR2 are present in lesional skin [55]. Furthermore, TNF was the first cytokine target in psoriasis whose inhibition showed good therapeutic efficacy [43]. TNF is a versatile cytokine that not only shapes inflammatory immune responses, but is also involved in cell death, cell cycling and tissue remodeling [56, 57]. It is a homotrimeric cytokine that can induce NF-κB and MAP kinase activation [58] (Fig. 1) either in a soluble or membrane-bound form that is expressed by a plethora of cell types such as macrophages, DCs, neutrophils, T and B cells and keratinocytes [35]. However, the exact roles of TNF in psoriasis pathogenesis are not completely understood yet. TNF might be involved in both the initiation phase and the chronic phase of the disease. For instance, by stimulating the production of IL-23 by DCs, it can promote disease development [43, 59]. The therapeutic effects of TNF inhibition are also linked to a strong reduction in IL-17-dependent genes [60]. In line with this, TNF potentiates IL-17 signaling by enhancing proinflammatory gene expression in keratinocytes in a synergistic way [61]. Furthermore, it has been recently shown that TNF also induces IL-23 expression in keratinocytes [62]. More specifically, TNF induces the phosphorylation and inactivation of the neural Wiskott-Aldrich syndrome protein (N-WASP) that was in turn shown to repress IL-23 expression by regulating histone methylation. TNF-induced phosphorylation of N-WASP reduced chromatin association of the latter and increased IL-23 expression in keratinocytes [62] (Fig. 1). This shows that TNF can amplify the psoriatic mechanisms that are at play and is, therefore, regarded as a regulator of the IL-23/IL-17 axis.
IL-36
In addition, the IL-1 family cytokines IL-36α, -β and -γ (also known as IL1F6, IL1F8 and IL1F9, respectively) are upregulated in lesional skin of psoriasis patients and implicated in psoriasis development. Thus, IL-36α deficiency protects from imiquimod-induced skin inflammation [63], while loss-of-function mutations in IL36RN, a natural antagonist of IL-36, have been identified as the genetic basis in several cases of generalized pustular psoriasis [64, 65]. Expression of another IL-36 subfamily member with anti-inflammatory properties, IL-38, is reduced in the epidermis of lesional skin in patients with psoriasis, as well as other neutrophil-driven skin diseases [66, 67]. IL-38 deficiency in mice exacerbates imiquimod-driven skin inflammation, possibly via suppressing IL-17 production by γδ T cells, while treatment with recombinant IL-38 ameliorates disease progression [67, 68]. Specific deletion of the IL36R in keratinocytes results in similar levels of protection from psoriasiform inflammation observed in mice lacking IL36R globally [69], demonstrating that keratinocytes are the primary cells that orchestrate dermal inflammation in response to IL-36. However, the specific biological role of IL-36 signaling during psoriasis is not entirely clear yet [70]. IL-36 cytokines produced by keratinocytes after IL-17 and TNF stimulation can in turn further enhance proinflammatory cytokine and AMP expression by keratinocytes [71] (Fig. 1). Recently, IL-36R signaling in keratinocytes was shown to play a major role in the induction of imiquimod-induced psoriasis-like dermatitis by triggering the production of IL-23/IL-17/IL-22 cytokines and neutrophil infiltration [72]. Moreover, IL-36 is also implicated in the cross-talk between immune cells and keratinocytes by promoting DC and macrophage activation [73, 74]. Finally, IL36γ might also promote angiogenesis by inducing endothelial branching [75].
Other cytokines
Several other cytokines are also implicated in psoriasis development, many of which are in one or another way linked to the IL-23/IL-17 axis. For example, IL-22 was shown to induce tissue remodeling in psoriatic lesions by affecting keratinocyte migration, differentiation and gene expression [76,77,78]. IL-22 is expressed in response to IL-23 by Th17 cells [79], but can also be produced by other T cell subsets and innate immune cells. Psoriatic-like lesions in the imiquimod model were strongly reduced in IL-22-deficient mice, suggesting an important role of this cytokine in psoriasis [80]. However, an IL-22 receptor antagonist was discontinued because of a lack of efficacy [81], questioning the driving role of IL-22 in psoriasis in humans.
The development of psoriatic lesions in patients that are treated with the type I interferons IFNα or -β for viral infections or multiple sclerosis provided the first evidence that type I interferons are involved in psoriasis pathogenesis [82,83,84,85]. Furthermore, neutralization of IFNα could prevent the development of psoriatic lesions upon xenotransplantation of non-lesional skin from psoriasis patients to mice, which points to a major role for type I interferons during the initiation phases of psoriasis [86]. Remarkably, therapeutic targeting of IFNα by MEDI-545, an anti-IFNα monoclonal antibody, did not show clinical improvements in psoriasis patients, though this might reflect the initiating role of type I interferons in psoriasis [87].
Lesional skin and serum of psoriasis patients also contains increased levels of IFNγ and these levels were also shown to correlate with disease severity [88]. Furthermore, even in healthy volunteers, a single intradermal injection of IFNγ could induce transcriptomic changes that are overlapping with those observed in lesional skin [89]. Therefore, IFNγ was considered a Th1-mediated driver of psoriasis for a long time. However, after the discovery of the cardinal IL-23/IL-17 axis and because of the poor therapeutic efficacy of IFNγ-neutralizing antibodies, IFNγ is no longer regarded as the central driver of psoriasis [90]. Still, IFNγ might be involved in the early phases through activation of antigen-presenting cells and it may also affect inflammation by synergizing with other cytokines such as IL-17 [43, 91].
Finally, it should be mentioned that also IL-9, IL-18, IL-19, IL-20, IL-21, IL-24, Il-27, IL-29 and IL-33 have been linked with psoriasis [92,93,94,95,96]. However, their specific importance and the mechanisms by which they influence psoriatic disease still require further research.
Current model of psoriasis pathogenesis
Although the pathogenesis of psoriasis is not completely elucidated, substantial advances have been made over the last decade. Research on psoriasis has long been fuelled by the paradigm that psoriasis is driven by T cell-mediated immune responses targeting keratinocytes. However, as described above, psoriasis cannot be explained solely on the basis of T-cell activation. Instead, a complex interplay between infiltrating, activated immune cells and hyperproliferating keratinocytes is now believed to be at the base of the disease (Fig. 2).

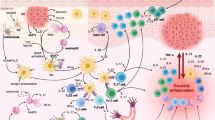
Mechanistic model depicting the inflammatory response in psoriasis. Insults that damage keratinocytes drive the release of self-nucleotides and antimicrobial peptides (AMPs) such as LL37, which together with ADAMTSL5 and neo-lipids are suggested to activate autoreactive T cells. Furthermore, AMPs form complexes with self-nucleic acids that stimulate plasmacytoid dendritic cells (pDCs) to produce IFNα and -β, which enhance myeloid DC (mDC) cell differentiation and secretion of IL-23, TNF and IL-6. These cytokines can activate T cells and type 3 innate lymphoid cells (ILC3s), which, in turn, produce IL-17 and other proinflammatory cytokines such as TNF, IL-17F, IL-22 and IFNγ. This cytokine cocktail activates keratinocytes to produce several AMPs, cytokines and chemokines acting on T cell, mDCs, neutrophils and keratinocytes. In addition, keratinocyte-intrinsic defects, such as hyperactivating CARD14 mutations, can induce this proinflammatory cascade culminating in the recruitment and activation of IL-17-producing cells, which further propagate and amplify the inflammatory response
Role of keratinocytes in disease initiation
The exact triggers that initiate psoriatic disease are not yet known. However, the association with the Koebner phenomenon suggests that trauma can be a trigger. Furthermore, also microbial agents and certain medicines (e.g., lithium, type I IFN blockers) have been correlated with psoriasis onset [10]. Because some of these triggers are encountered regularly, genetic predisposition probably plays a considerable role in shaping the right microenvironment for the pathogenic events to occur. According to the current model for psoriasis pathogenesis, trauma or infectious agents can trigger keratinocytes which respond by releasing self-nucleic acids, AMPs, and alarmins like the cytokine IL-36 (Fig. 2) [43]. Certain AMPs such as LL37, hBD2, hBD3 and lysozyme form complexes with self-nucleic acids. By inducing TLR7/9 signaling in plasmacytoid DCs (pDCs) these AMP-nucleotide complexes breach the innate tolerance to self-DNA [97,98,99]. An elegant study by Lou and colleagues have recently shown that IL-17 stimulation of keratinocytes can rewire the metabolic urea cycle leading to the production of polyamines that protect released self-RNA [100] (Fig. 1). More specifically, IL-17 stimulation results in the downregulation of protein phosphatase 6 enabling phosphorylation and activation of the transcription factor C/EBPβ, which in turn leads to increased expression of Arginase 1 and production of polyamines [100]. Upon TLR7/9 stimulation, pDCs quickly respond by producing type I interferons. IFNα and -β, in turn, activate myeloid DCs (mDCs) which become inflammatory mDCs known as Tip-DCs [43] (Fig. 2). Furthermore, self-RNA complexed with LL37 can also directly activate mDCs by engaging TLR7/8 [97, 100]. Noteworthy, IL-17-induced polyamines were shown to facilitate self-RNA uptake by myeloid DC [100]. The dermal inflammatory mDCs release IL-6, NO, TNF, IL-12, IL-20 and IL-23, which seems to be dependent on TNF [59, 101] and ultimately initiate a chronic IL-17-mediated inflammatory response.
Various factors (e.g., chemicals, viral or endogenous nucleic acids, mutations) may cause damage and even cell death of keratinocytes. Dysregulation of necroptotic cell death markers has been documented in the epidermis of human psoriasis lesions as well as in imiquimod-induced psoriasiform skin of mice [102]. Moreover, inhibition of necroptotic cell death was shown to reduce imiquimod-induced inflammatory responses in the skin, indicating a proinflammatory effect of necrotic cell death in the pathogenesis of psoriasis [102]. Recently, several studies reported an important role for endogenous Z-DNA sensing by Z-DNA-binding protein 1 (ZBP1) in keratinocytes in which necroptosis and skin inflammation was initiated by disruption of the kinase RIPK1 [103, 104]. However, the causal role of necroptotic cell death in psoriasis, as well as the underlying molecular mechanisms, in more physiological conditions remain to be determined.
Intrinsic defects in cytokine and growth factor signalling in keratinocytes may be responsible for their aberrant hyperproliferation and differentiation, as well as inflammatory cell infiltration and activation. For instance, enhanced expression of IL-23 specifically in keratinocytes (IL-23 transgene or deletion of N-WASP, a negative regulator of IL-23 expression) leads to the development of psoriasis-like skin inflammation and psoriatic arthritis in mice [62, 105]. Recent evidence based on the imiquimod-induced psoriasis model also suggests that IL-36R signaling in keratinocytes mediates early expression of IL-23 and is indispensable for the induction of neutrophil infiltration but not keratinocyte hyperproliferation [72]. In addition, hyperactivating mutations in the CARD14 gene, which is specifically expressed in keratinocytes and mucosal tissues, have been linked to psoriasis-susceptibility in human patients [20]. Specific CARD14 mutations enable its interaction with the downstream signaling proteins, BCL10 and MALT1, which induces NF-κB and p38/JNK MAP kinase signaling and expression of several proinflammatory mediators in keratinocytes [21, 106] (Figs. 1, 2). Importantly, keratinocyte-specific expression of the psoriasis-associated mutant CARD14 (E138A) was recently shown to be sufficient for driving the development of psoriasis-like skin inflammation in mice [107, 108]. Collectively, these studies highlight the instrumental role that keratinocytes are playing in initiating inflammatory responses in psoriasis.
Role of T cells in the initiation and amplification of inflammation
Autoreactive T cells producing IFNγ and IL-17 might be another, alternative pathogenic mechanism that can initiate psoriasis [43]. Moreover, the strong predisposition linked to the HLA-Cw6 allele favors an autoimmune disease mechanism. So far, LL37, ADAMTSL5 and neo-lipids have been identified as self-antigens (Fig. 2), evidenced by the existence of LL37- and ADAMTSL5-specific autoreactive T cells in psoriasis patients [18, 19, 109, 110]. However, deep sequencing of the T cell repertoire in lesional skin showed that the αβ and γδ T cell populations are highly polyclonal [111]. Therefore, the exact way of how these self-antigens and autoreactive T cells might initiate psoriasis is not entirely clear and still requires further research.
By the production of IL-23, activated inflammatory mDCs can steer and sustain T cell responses [112]. The essential role of T cells in the disease mechanism was first illustrated by the therapeutic efficacy of drugs that deplete or inhibit activated T cells such as alefacept and denileukin diftitox [10]. Furthermore, both CD4+ T helper (Th) cells and CD8+ cytotoxic T (Tc) cells were shown to be crucial for the development of skin lesions, and both CD4+ and CD8+ T cell numbers are increased in lesions and peripheral blood of psoriasis patients [43]. So far, Th1, Th17 and Th22 (a distinct T cell population that only produces IL-22) cells and their CD8+ counterparts Tc1, Tc17 and Tc22 have been implicated in the disease [113, 114]. However, because of the strong clinical impact of anti-IL-17 therapeutics but not of IFNγ and IL-22 inhibitory drugs, Th17/Tc17 cells seem to play a more prominent role. Upon activation, these T cell populations produce a cytokine cocktail with IL-17 as the most potent ingredient. Next to ‘conventional’ T cells, also γδ T cells and ILC3s are enriched in lesional skin and serve as additional sources of IL-17 upon IL-23 stimulation [115,116,117,118] (Fig. 2). ILC3s, an innate immune system’s counterpart of Th17 cells, secrete IL-17, IL-22 and GM-CSF in response to antigen-independent activation by microbial products or inflammatory cytokines, thus modulating cross-talk between innate and adaptive immune systems [119]. Importantly, intradermal injection of ILC3s was sufficient to drive the development of psoriatic lesions in healthy human skin transplants implanted in SCID mice, even in the absence of Th17 cells [120].
Psoriasis recurrence is often observed in healed skin upon cessation of treatment, suggesting that a certain predisposition facilitating recurrence remains in the tissue. Indeed, engraftment of non-lesional skin from psoriatic patients onto immunocompromised mice resulted in the development of psoriatic lesions that was dependent on local T cell proliferation [121]. It was subsequently shown that resolved psoriatic lesions contain psoriasis-specific tissue-resident memory T cells that produce IL-17 and IL-22 [122, 123]. Furthermore, even never-lesional skin from patients with psoriasis harbors T cells capable of inducing psoriasis-associated responses further underscoring the pathogenic role that T cells play in the initiation of psoriasis development [124].
Role of keratinocytes in the amplification of inflammation
Several of the cytokines produced by T cells and ILC3s act on keratinocytes and will thus further amplify the inflammatory response in psoriasis. Deletion of IL-17RA in T cells or myeloid cells had no effect on the development of imiquimod-induced skin inflammation, while deletion of IL-17RA in keratinocytes inhibited neutrophil infiltration and disease development, highlighting the instrumental role of keratinocyte-derived signals in psoriasis pathogenesis [125]. TNF, IL-22 and IFNγ have synergistic effects on IL-17 signaling and thus enhance the effects of IL-17 on keratinocytes [43]. In response to stimulation with these cytokines, keratinocytes become a source of AMPs, cytokines and chemokines, which have various effector functions (Fig. 2) [43]. In psoriatic plaques, keratinocytes produce excessive amounts of AMPs including LL37, S100A proteins (S100A7, S100A8 and S100A9), β-defensins, RNase7, REG3A and lipocalin-2 [3]. These AMPs are small proteins that have anti-bacterial, -fungal, -protozoal and -viral activities and can prevent microbial colonization. In addition, they also have immunomodulatory functions that further feed the inflammatory pathways at play. For instance, next to its capacity to activate TLR7/8/9 signaling, LL37 might serve as a self-antigen and can regulate the expression of cytokines such as IL-36γ by keratinocytes [110, 126]. Furthermore, AMPs are involved in a wide range of psoriatic processes that contribute to the formation of psoriatic plaques such as chemotaxis of immune cells, increased proliferation and aberrant differentiation of keratinocytes and angiogenesis [3, 127]. Next to AMPs, psoriatic keratinocytes also produce a vast amount of cytokines including TNF, IL-1 family members, IL-6, IL-8, IL-17C, IL-19 and IL-36γ [43], which can induce both paracrine and autocrine signaling. For instance, IL-19 and IL-20 act on keratinocytes and induce hyperproliferation leading to epidermal hyperplasia, while IL-1β is an important mediator of Th17 differentiation [128, 129]. Notably, keratinocytes can also be a source of IL-23 and in this way might shape a favorable microenvironment for IL-17-producing cells [40, 62]. Finally, upon stimulation with psoriatic cytokines, keratinocytes also produce several chemokines that serve as attractants for immune cells. CXCL1, -2, -3 and -8 are strongly upregulated by psoriatic keratinocytes and attract neutrophils to psoriatic lesions, which further contribute to the production of AMPs and proinflammatory mediators, in particular IL-17 [44, 130]. In addition, CCL20, a chemokine that recruits CCR6-positive cells, is markedly induced by keratinocytes in psoriatic plaques. Because most psoriatic IL-17-producing cells such as Th17 cells and γδ T cells bear a CCR6 receptor, this again induces higher IL-17 signals in the skin [43]. Together, the inflammatory mediators that are produced by keratinocytes upon IL-17 stimulation establish a feedforward loop that fuels IL-17 production by immune cells resulting in a vicious cycle of inflammation in psoriasis.
Mouse models of psoriasis
Our improved understanding of psoriasis pathogenesis stems in a large part from studies that rely on the use of mice as a disease model. However, it is important to remember that although mouse and human skin share similar features, also significant differences exist. Mouse skin is thinner, has more densely distributed hair follicles [131] and also contains a cutaneous muscular layer called panniculus carnosus, which is usually absent in human skin. On the other hand, human epidermis contains more distinct cell layers compared to murine epidermis [132]. The skin transcriptomes of mice and human only partially overlap [132] and also skin-resident immune cells show some differences. For instance, murine epidermis contains special dendritic epidermal γδ T cells that are involved in the defense against infections and in wound healing [133], while T cells in the human skin predominantly express an αβ T cell receptor [132, 134]. Furthermore, spontaneous development of psoriasis seems to be unique to humans and mouse models fail to exactly reproduce psoriasis pathology. For instance, comparative transcriptomics between several mouse models shows that these mouse models share multiple gene expression patterns with human psoriasis but cannot completely mirror all involved pathways [135, 136]. Notwithstanding these differences, several mouse models, which closely resemble psoriasis pathology, serve as valuable tools for preclinical research and drug development. Ideally, psoriasis models should meet three criteria: they should have histological hallmarks that are similar to those in psoriasis, a similar disease mechanism and respond to therapeutics that have shown to be effective in psoriasis patients [131, 137]. Different strategies to induce psoriasis-like disease in mice have been employed and can be subdivided in spontaneous models, genetically engineered models, induced models and xenograft models [134, 138], which reproduce typical characteristics of human psoriasis to a varying degree (Table 1).
Spontaneous models
The first psoriasis-like mouse models that have been described were caused by homozygous spontaneous mutations. For instance, mice with asebia (Scd1ab⁄Scd1ab), chronic proliferative dermatitis (Sharpincpdm/Sharpincpdm) and flaky skin (Ttc7fsn/Ttc7fsn) mutations develop skin inflammation with histological features that resemble those in psoriasis [139,140,141]. However, the role of T cells in these models is limited and they don’t respond to treatment with anti-psoriatic drugs such as cyclosporine A [134]. Therefore, psoriasis models caused by spontaneous mutations are considered to have limited applications.
Genetically engineered mouse models
Over the last two decades, several genetically engineered mouse strains that develop psoriasis-like inflammation have been established. These involve both transgenic animals that overexpress certain genes and animals with a (conditional) deletion in certain genes. Transgenic (over)expression of genes induced by the K5 and K14 promotor leads to epidermal-specific expression and is often employed to obtain mouse lines that develop psoriasis-like inflammation. Among these, several mouse lines that overexpress cytokines and mediators associated with psoriasis have been generated. For example, K14-IL-23, K5-IL-17C and K14-IL-17ind/+ mouse lines all develop skin inflammation that resembles psoriasis to a greater or lesser extent [105, 142, 143]. Furthermore, expression of a constitutively active STAT3 (Stat3C), a transcription factor that mediates IL-23 signaling, induces psoriasis-like inflammation in K5-Stat3C mice [144]. The most recent mouse model involves expression of human psoriasis-associated mutant CARD14(E138A) [107, 108, 145, 146]. Inducible keratinocyte-specific expression of CARD14(E138A) was shown to be sufficient for the development of skin inflammation in the ears with several characteristics of psoriasis such as acanthosis and parakeratosis, neutrophilic infiltration and a TNF/IL-23/IL-17 cascade-related cytokine profile [108], illustrating the important role of keratinocytes in the initiation of psoriasis. Mutant CARD14-driven skin inflammation can be reduced upon treatment with IL-23-, TNF- and (to some extent) IL-17-neutralizing antibodies [107, 145, 146]. Importantly, CARD14(E138A) expression still induces psoriasiform skin inflammation in RAG-1-deficient mice that lack T and B cells, suggesting that inflammation caused by keratinocyte-intrinsic hyperactive CARD14-driven signaling is independent of adaptive immunity ([107]; our own unpublished observations).
Finally, also mouse strains that are deficient for certain genes have been described to develop psoriasiform inflammation. For instance, mice lacking JunB and c-Jun transcription factors in epidermal cells develop skin inflammation that includes many hallmarks of psoriasis [147]. In general, genetically engineered mouse models allow to investigate the effect of a single gene on skin inflammation in vivo and in this way help to elucidate the events and key mediators that promote psoriasis. However, because psoriasis is a multifactorial disease, single genetic alterations might not completely represent the complex phenotype observed in psoriasis.
Topical application and intradermal injection models
As an alternative approach to genetic models, topical application of imiquimod cream or intradermal injection of IL-23 has been used. In 2009, Van der Fits et al. described that topical application of imiquimod-containing cream (Aldara®) could induce psoriasis-like inflammation in mice [39]. Originally, Aldara cream has been used as a treatment for genital warts caused by human papilloma virus and (pre)cancerous skin lesions. Remarkably, an exacerbation of psoriatic lesions in patients was noticed as a side effect of this treatment [39]. Imiquimod is a TLR7/8 agonist and a potent immune activator that can induce IL-23 production by dendritic cells and in this way drive psoriasiform changes [148]. However, it has been shown that also other components of the Aldara cream such as isostearic acid contribute to the psoriasiform inflammation through inflammasome activation in a TLR7-independent way [149]. The Aldara-induced skin inflammation is an IL-23/IL-17-driven model, partially dependent on T cells presenting several histological hallmarks of psoriasis such as acanthosis, parakeratosis and angiogenesis [39, 138]. For this reason and because of the easy applicability, this model has been widely used to study the impact of several genes and chemical substances on psoriasis-like inflammation. Nonetheless, similar to other mouse models, there are some limitations to the use of the imiquimod model. This model mainly represents the acute phase of psoriasis-like inflammation but does not correspond to its chronic nature. Moreover, several systemic effects such as splenomegaly and dehydration occur during the treatment, probably caused by accidental ingestion during grooming and effects on other organ systems such as the intestine. In this way the observed skin inflammation is affected by mechanisms that are not completely relevant to psoriasis pathology [150].
Intradermal injection of IL-23 is another way to model psoriatic inflammation. Injection of IL-23 induces erythema and ear swelling, which is characterized by acanthosis, parakeratosis and infiltrating immune cells such as CD4+ T cells, dendritic cells, neutrophils and macrophages [38, 151]. IL-23-induced psoriasis was shown to be mediated by both IL-22 and IL-17 [42, 79]. Hedrick et al. described two phases in this model, an early T cell-independent phase, followed by a second, more chronic, T cell-dependent phase [151]. Furthermore, analysis of the gene expression profile showed significant overlap between this model and human psoriasis [135]. Next to IL-23 injections, also intradermal IL-17, IL-21 and IL-22 injections have been shown to induce psoriasiform skin inflammation, although IL-23 injections mirror psoriasis pathology more closely [151,152,153].
Xenograft models
Finally, in an attempt to better preserve immunologic and genotypic characteristics of human skin, xenograft models were developed. Here, skin explants from lesional or non-lesional psoriatic skin together with infiltrating immune cells are transplanted onto immunocompromised mice, such as nude mice, SCID (Prkdcscid) mice and AGR129 mice [121, 154, 155]. Transplantation in each of these strains shows a T cell-dependent disease course, illustrating the essential role of T cells in psoriasis pathogenesis. Interestingly, non-lesional psoriatic skin transplanted onto AGR129 mice (Rag-2-deficient mice lacking IFN I and II receptors) leads to the spontaneous development of psoriatic lesions, suggesting that all necessary immune ‘components’ to develop psoriatic lesions are also present in non-lesional skin [121, 138]. Unfortunately, xenograft models are technically difficult and require large skin fragments. In addition, because skin fragments are transplanted, these models cannot recapitulate more systemic co-morbidities of psoriasis such as psoriatic arthritis [134]. Nevertheless, these humanized mouse models offer the highest resemblance to human psoriasis serving as good models for preclinical studies and drug development.
In a further attempt to circumvent the restrictions of mouse models and reconstruct human epidermis in vitro, several three-dimensional (3D) skin models have been established and are now commercially available [156]. These human skin equivalents rely on the co-culture of primary cells (e.g., keratinocytes, fibroblasts) supported by extracellular matrix components. However, to more faithfully reproduce the intricated interplay of skin keratinocytes and resident immune cells, 3D skin models require exogenous addition of immune cells or artificial substitution with cytokine stimulation. A further layer of complexity is added by the need to simulate infiltration of the skin models with non-resident immune cells, such as neutrophils or macrophages. Shin et al. have recently recapitulated T cell migration into reconstructed epidermis by supplementing human skin constructs with in vitro polarized T cells derived from psoriasis patients [157]. These skin constructs showed increased epidermal proliferation and psoriasis-associated cytokine profile and responded to psoriasis treatments [157]. However, although promising, 3D skin models are technologically complex, heterogenous and still currently inadequate to fully reproduce the complex multi-factorial in vivo environment necessary to study psoriasis pathology. Nevertheless, despite these limitations, 3D skin models have been used to study the role of cytokine signaling in cutaneous responses or for drug testing [156] and can nicely complement existing mouse models.
Current therapies for the treatment of psoriasis
So far, there is no cure for psoriasis, since the current therapeutics manage the disease by controlling symptoms. The psoriasis area and severity index (PASI) is often used to score the severity of psoriasis and as a measure to follow up the efficacy of therapeutics. PASI is determined as a combined score for erythema (redness), induration (thickness) and scaling and it also takes into account the body surface area that is involved. A treatment is often considered successful if the PASI score is reduced with 75% compared to baseline, also termed PASI75 [10]. Psoriasis patients with mild psoriasis are usually treated with topical treatments containing coal tar, salicylic acid, vitamin D3, anthralin, retinoids or corticosteroids. Patients that do not respond to topical treatments or patients that suffer from moderate to severe psoriasis can benefit from phototherapy with UV-B radiation or UV-A radiation in combination with psoralen [158]. Furthermore, also systemic drugs including cyclosporin, methotrexate and acitetrin are used for moderate to severe psoriasis. However, these systemic drugs have immune-suppressive capacities and can lead to toxicity when administered chronically [159].
The development of biologics, most often monoclonal antibodies, was a great breakthrough in psoriasis treatment. These biological drugs specifically target key players in psoriasis pathogenesis. Therefore, they have greater efficacy and a better safety profile compared to traditional systemic drugs [160]. Table 2 gives an overview of currently used biologics for the treatment of psoriasis and psoriatic arthritis.
The first approved biologics for psoriasis were biologics that interfered with the activation of T cells. Efalizumab and alefacept both block co-stimulation of T cells by antigen presenting cells. However, because of severe side effects or lower effectiveness compared to the newer biologics, these drugs were withdrawn from the market [160]. Abatacept (trade name Orensia), is a fusion protein composed of the Fc region of the immunoglobulin IgG1 fused to the extracellular domain of CTLA-4 that binds CD80/CD86 and in this way prevents co-stimulation of T cells. It is used for the treatment of rheumatoid arthritis and was in 2017 approved by the FDA for treatment of psoriatic arthritis.
TNF inhibitors, another class of biologics, interfere with TNF function and in this way reduce inflammatory signaling. Several anti-TNF biologics are currently used to treat psoriasis. Etanercept was the first approved drug and is a human TNF-receptor fusion protein. Next, also other TNF inhibitors were developed: infliximab, adalimumab and golimumab are monoclonal antibodies, while certolizumab consist of an antibody Fab fragment conjugated to poly-ethylene glycol. They all bind both to soluble and membrane-bound TNF [159, 160]. Nowadays, also biosimilars for etanercept, infliximab and adalimumab are approved by the FDA. These biosimilar agents are highly similar to the already approved biologics, however, because of the biologic origin of these products, it is almost impossible to manufacture an exact generic copy [159]. TNF inhibitors generally show an improved efficacy compared to traditional systemic drugs; ~ 50–80% of patients reached PASI75 [35]. However, they are, like almost any drug, linked with side-effects. For instance, infusion reactions are observed in 4% of patients and because of the immunosuppressive properties, patients can be more sensitive to infections [159]. Furthermore, loss of efficacy can also occur through time due to the development of anti-drug antibodies or because of adaptations in the pathogenic mechanisms. Surprisingly, TNF inhibitors have also been linked to the development of paradoxical skin lesions that had psoriasiform and eczematous-like characteristics [35].
Next to TNF inhibitors, also biologics that directly target the IL-17/IL-23 axis have been developed. Ustekinumab targets the p40 subunit that is shared by IL-12 and IL-23 and in this way inhibits both Th1 and Th17 pathways in psoriasis. 50 to 80 percent of patients showed an improvement of 75 percent in the PASI score in phase III clinical trials with ustekinumab [35]. In addition, guselkumab, tildrakizumab and risankizumab have recently been approved for treatment of psoriasis. They are IL-23-specific inhibitors that target the p19 subunit of IL-23. Comparative testing with ustekinumab and etanercept, showed that these IL-23-specific therapeutics are superior and can reach a PASI90 in more than 50 percent of patients [35, 161]. In addition, also another IL-23-specific inhibitor, mirikizumab, is being developed and undergoes clinical testing. Finally, also IL-17 inhibitors have shown great promise for the treatment of psoriasis. So far, there are three biologics that target IL-17 on the market. Secukinumab, ixekizumab and netakimab are monoclonal antibodies that target IL-17A directly. Recently, superiority to Secukinumab in achieving complete psoriasis skin clearance in a Phase 3b study was reported for Bimekizumab, which is a monoclonal antibody that targets both IL-17A and IL-17F. Brodalumab is an IL-17RA inhibitor and thus inhibits signaling by several of the IL-17 family members: IL-17A, IL-17C, IL-17F and heterodimers of IL-17A and F, as well as IL-17E (IL-25). Compared to ustekinumab and etanercept, these IL-17 inhibitors showed better efficacy and they could even reduce the PASI score with 100% in 30 to 60 percent of psoriasis patients [35, 159]. Unfortunately, similar to anti-TNF biologics, also anti-IL-23 and anti-IL-17 biologics are accompanied with side effects such as increased susceptibility for infections. For instance, because of its biological functions, blocking of IL-17 might sensitize to fungal infections such as Candida infections [162]. Nevertheless, anti-IL-23 and anti-IL-17 biologics show great efficacy with relatively good safety profiles compared to the traditional systemic therapies.
Even though biologics revolutionized the treatment of psoriasis, the perfect therapy does not exist yet. The benefits require ongoing administration of treatment and there are currently no known therapies for psoriasis that can induce long-lasting tolerance. Moreover, there is still a subset of patients that does not respond to these novel biologic therapies [35]. Furthermore, treatment with biologics is very expensive and might not always be an affordable option. Therefore, novel, more targeted drugs to treat psoriasis are being developed using small molecule inhibitors. For instance, in 2014, the FDA approved the use of Aprimelast (Otelza®), a phosphodiesterase 4 (PDE4) inhibitor to treat psoriasis [159]. Inhibition of PDE4 increases intracellular cAMP levels and this reduces the production of proinflammatory cytokines and promotes anti-inflammatory signaling [163]. In addition, other small molecule inhibitors such as Tofacitinib and Ruxolitinib that target Janus kinases in the JAK/STAT signaling pathways, are being tested for the treatment of psoriasis [164].
Future perspectives
Our knowledge about the disease mechanisms that lead to psoriasis has greatly increased over the last years. However, also considerable questions remain. For instance, next to the above-mentioned cellular players, also several other cell types such as mast cells, macrophages and NK cells are involved in psoriasis pathogenesis; however, their exact roles are not fully understood. Furthermore, both deregulated innate immune responses and autoimmune responses seem to be at the base of this disease, but how these responses interact and feed into each other to initiate the disease is still unclear. Finally, also the breaks that keep inflammatory processes in check and restore homeostasis in the skin are enigmatic. For instance, regulatory T cells (Tregs) are a subset of T cells responsible for suppressing immune responses, but their role in psoriasis still needs clarification. The numbers of Tregs are elevated in lesional skin of psoriasis patients [165], but some reports have indicated that Tregs are functionally impaired and might even acquire Th17-like proinflammatory characteristics in response to IL-23 [166,167,168,169,170]. Similarly, IL-10-producing regulatory B cells of psoriasis patients were reduced in number and showed decreased IL-10 production [171, 172]. Genetic association studies are also indicating novel therapeutic targets. For example, psoriasis patients harboring hyperactivating CARD14 mutations that are associated with psoriasis might benefit from CARD14 signaling inhibitors. In this regard, we have recently reported that a small molecule inhibitor of MALT1, a protease that is activated by CARD14 and that mediates signaling leading to increased psoriasis-associated gene expression (e.g., TNF, IL-23), attenuates the development of psoriatic-like dermatitis in a newly developed CARD14(E138A) transgenic mouse model [108]. Moreover, genetic associations also point to a role for defects in negative regulators of proinflammatory signaling pathways in psoriasis. For instance, SNPs in inhibitors of NF-κB signaling (TNFAIP3, TNIP1 and NFKBIA) or JAK-STAT signaling (SOCS1)), can predispose to psoriasis [15, 173]. Such observations indicate that a better knowledge of the link between psoriasis susceptibility genes and the response to certain treatments might allow to stratify patients and thus enable personalized medicine. For instance, SNPs in TNFAIP3 have been linked to a better response to anti-TNF therapy [174]. Furthermore, also natural cytokine antagonists might prevent excessive skin inflammation as evidenced by the close connection of loss-of-function mutations in IL36RN with generalized pustular psoriasis [64, 65]. The association of these negative regulators with psoriasis indicates that this disease is not merely caused by an excessive inflammatory response, but also by a failure to adequately dampen inflammatory response and to restore homeostasis. In addition, ever growing evidence supporting the role of keratinocytes as initiators of psoriatic inflammation might further shift the focus to topical therapies rather than systemic biologics. Clearly, further studies are needed to obtain better insights in the etiology and inflammatory signals that drive psoriasis, which would ultimately lead to the development of more targeted and more effective therapies that will also benefit refractory patients. Ideally, such treatments would also have an effect on comorbidities of psoriasis such as psoriatic arthritis, cardiovascular disease, inflammatory bowel disease and other immune-related disorders.
References
Pasparakis M, Haase I, Nestle FO (2014) Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol 14:289–301. https://doi.org/10.1038/nri3646
Eyerich S, Eyerich K, Traidl-Hoffmann C, Biedermann T (2018) Cutaneous barriers and skin immunity: differentiating a connected network. Trends Immunol 39:315–327. https://doi.org/10.1016/j.it.2018.02.004
Niyonsaba F, Kiatsurayanon C, Chieosilapatham P, Ogawa H (2017) Friends or Foes? Host defense (antimicrobial) peptides and proteins in human skin diseases. Exp Dermatol 26:989–998. https://doi.org/10.1111/exd.13314
Deckers J, Hammad H, Hoste E (2018) Langerhans cells: sensing the environment in health and disease. Front Immunol 9:93. https://doi.org/10.3389/fimmu.2018.00093
West HC, Bennett CL (2017) Redefining the role of langerhans cells as immune regulators within the skin. Front Immunol 8:1941. https://doi.org/10.3389/fimmu.2017.01941
Heath WR, Carbone FR (2013) The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol 14:978–985. https://doi.org/10.1038/ni.2680
Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, Elco CP, Huang V, Matos TR, Kupper TS, Clark RA (2015) Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci Transl Med 7:279ra239. https://doi.org/10.1126/scitranslmed.3010302
Mueller SN, Zaid A, Carbone FR (2014) Tissue-resident T cells: dynamic players in skin immunity. Front Immunol 5:332. https://doi.org/10.3389/fimmu.2014.00332
Raychaudhuri SK, Maverakis E, Raychaudhuri SP (2014) Diagnosis and classification of psoriasis. Autoimmun Rev 13:490–495. https://doi.org/10.1016/j.autrev.2014.01.008
Lowes MA, Suarez-Farinas M, Krueger JG (2014) Immunology of psoriasis. Annu Rev Immunol 32:227–255. https://doi.org/10.1146/annurev-immunol-032713-120225
Hu SC, Lan CE (2017) Psoriasis and cardiovascular comorbidities: focusing on severe vascular events, cardiovascular risk factors and implications for treatment. Int J Mol Sci 18:2211. https://doi.org/10.3390/ijms18102211
Mease PJ, Gladman DD, Papp KA, Khraishi MM, Thaci D, Behrens F, Northington R, Fuiman J, Bananis E, Boggs R, Alvarez D (2013) Prevalence of rheumatologist-diagnosed psoriatic arthritis in patients with psoriasis in European/North American dermatology clinics. J Am Acad Dermatol 69:729–735. https://doi.org/10.1016/j.jaad.2013.07.023
Egeberg A, Mallbris L, Warren RB, Bachelez H, Gislason GH, Hansen PR, Skov L (2016) Association between psoriasis and inflammatory bowel disease: a Danish nationwide cohort study. Br J Dermatol 175:487–492. https://doi.org/10.1111/bjd.14528
Zohar A, Cohen AD, Bitterman H, Feldhamer I, Greenberg-Dotan S, Lavi I, Comanesther D, Batat E, Zisman D (2016) Gastrointestinal comorbidities in patients with psoriatic arthritis. Clin Rheumatol 35:2679–2684. https://doi.org/10.1007/s10067-016-3374-y
Harden JL, Krueger JG, Bowcock AM (2015) The immunogenetics of psoriasis: a comprehensive review. J Autoimmun 64:66–73. https://doi.org/10.1016/j.jaut.2015.07.008
Capon F (2017) The genetic basis of psoriasis. Int J Mol Sci 18:2526. https://doi.org/10.3390/ijms18122526
Elder JT (2006) PSORS1: linking genetics and immunology. J Invest Dermatol 126:1205–1206. https://doi.org/10.1038/sj.jid.5700357
Arakawa A, Siewert K, Stohr J, Besgen P, Kim SM, Ruhl G, Nickel J, Vollmer S, Thomas P, Krebs S, Pinkert S, Spannagl M, Held K, Kammerbauer C, Besch R, Dornmair K, Prinz JC (2015) Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med 212:2203–2212. https://doi.org/10.1084/jem.20151093
Mabuchi T, Hirayama N (2016) Binding affinity and interaction of LL-37 with HLA-C*06:02 in psoriasis. J Invest Dermatol 136:1901–1903. https://doi.org/10.1016/j.jid.2016.04.033
Jordan CT, Cao L, Roberson ED, Pierson KC, Yang CF, Joyce CE, Ryan C, Duan S, Helms CA, Liu Y, Chen Y, McBride AA, Hwu WL, Wu JY, Chen YT, Menter A, Goldbach-Mansky R, Lowes MA, Bowcock AM (2012) PSORS2 is due to mutations in CARD14. Am J Hum Genet 90:784–795. https://doi.org/10.1016/j.ajhg.2012.03.012
Afonina IS, Van Nuffel E, Baudelet G, Driege Y, Kreike M, Staal J, Beyaert R (2016) The paracaspase MALT1 mediates CARD14-induced signaling in keratinocytes. EMBO Rep 17:914–927. https://doi.org/10.15252/embr.201642109
Jordan CT, Cao L, Roberson ED, Duan S, Helms CA, Nair RP, Duffin KC, Stuart PE, Goldgar D, Hayashi G, Olfson EH, Feng BJ, Pullinger CR, Kane JP, Wise CA, Goldbach-Mansky R, Lowes MA, Peddle L, Chandran V, Liao W, Rahman P, Krueger GG, Gladman D, Elder JT, Menter A, Bowcock AM (2012) Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet 90:796–808. https://doi.org/10.1016/j.ajhg.2012.03.013
de Cid R, Riveira-Munoz E, Zeeuwen PL, Robarge J, Liao W, Dannhauser EN, Giardina E, Stuart PE, Nair R, Helms C, Escaramis G, Ballana E, Martin-Ezquerra G, den Heijer M, Kamsteeg M, Joosten I, Eichler EE, Lazaro C, Pujol RM, Armengol L, Abecasis G, Elder JT, Novelli G, Armour JA, Kwok PY, Bowcock A, Schalkwijk J, Estivill X (2009) Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet 41:211–215. https://doi.org/10.1038/ng.313
Hollox EJ, Huffmeier U, Zeeuwen PL, Palla R, Lascorz J, Rodijk-Olthuis D, van de Kerkhof PC, Traupe H, de Jongh G, den Heijer M, Reis A, Armour JA, Schalkwijk J (2008) Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet 40:23–25. https://doi.org/10.1038/ng.2007.48
Sagi L, Trau H (2011) The Koebner phenomenon. Clin Dermatol 29:231–236. https://doi.org/10.1016/j.clindermatol.2010.09.014
Malakou LS, Gargalionis AN, Piperi C, Papadavid E, Papavassiliou AG, Basdra EK (2018) Molecular mechanisms of mechanotransduction in psoriasis. Ann Transl Med 6:245. https://doi.org/10.21037/atm.2018.04.09
Parisi R, Symmons DP, Griffiths CE, Ashcroft DM; on behalf of the Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team, Identification, Management of P, Associated ComorbidiTy project t (2013) Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol 133:377–385. https://doi.org/10.1038/jid.2012.339
Rousset L, Halioua B (2018) Stress and psoriasis. Int J Dermatol 57:1165–1172. https://doi.org/10.1111/ijd.14032
Valdimarsson H, Thorleifsdottir RH, Sigurdardottir SL, Gudjonsson JE, Johnston A (2009) Psoriasis—as an autoimmune disease caused by molecular mimicry. Trends Immunol 30:494–501. https://doi.org/10.1016/j.it.2009.07.008
Chang HW, Yan D, Singh R, Liu J, Lu XY, Ucmak D, Lee K, Afifi L, Fadrosh D, Leech J, Vasquez KS, Lowe MM, Rosenblum MD, Scharschmidt TC, Lynch SV, Liao W (2018) Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome 6:154. https://doi.org/10.1186/s40168-018-0533-1
Stehlikova Z, Kostovcikova K, Kverka M, Rossmann P, Dvorak J, Novosadova I, Kostovcik M, Coufal S, Srutkova D, Prochazkova P, Hudcovic T, Kozakova H, Stepankova R, Rob F, Juzlova K, Hercogova J, Tlaskalova-Hogenova H, Zakostelska ZJ (2019) Crucial role of microbiota in experimental psoriasis revealed by a gnotobiotic mouse model. Front Microbiol 10:236. https://doi.org/10.3389/fmicb.2019.00236
Myers B, Brownstone N, Reddy V, Chan S, Thibodeaux Q, Truong A, Bhutani T, Chang HW, Liao W (2019) The gut microbiome in psoriasis and psoriatic arthritis. Best Pract Res Cl Rh 33:101494. https://doi.org/10.1016/j.berh.2020.101494
Zakostelska Z, Malkova J, Klimesova K, Rossmann P, Hornova M, Novosadova I, Stehlikova Z, Kostovcik M, Hudcovic T, Stepankova R, Juzlova K, Hercogova J, Tlaskalova-Hogenova H, Kverka M (2016) Intestinal microbiota promotes psoriasis-like skin inflammation by enhancing Th17 response. PLoS ONE 11:e0159539. https://doi.org/10.1371/journal.pone.0159539
Zanvit P, Konkel JE, Jiao X, Kasagi S, Zhang DF, Wu RQ, Chia C, Ajami NJ, Smith DP, Petrosino JF, Abbatiello B, Nakatsukasa H, Chen QM, Belkaid Y, Chen ZJ, Chen WJ (2015) Antibiotics in neonatal life increase murine susceptibility to experimental psoriasis. Nat Commun 6:8424. https://doi.org/10.1038/ncomms9424
Hawkes JE, Yan BY, Chan TC, Krueger JG (2018) Discovery of the IL-23/IL-17 signaling pathway and the treatment of psoriasis. J Immunol 201:1605–1613. https://doi.org/10.4049/jimmunol.1800013
Hong K, Chu A, Ludviksson BR, Berg EL, Ehrhardt RO (1999) IL-12, independently of IFN-gamma, plays a crucial role in the pathogenesis of a murine psoriasis-like skin disorder. J Immunol 162:7480–7491
Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, Dhodapkar M, Krueger JG (2004) Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med 199:125–130. https://doi.org/10.1084/jem.20030451
Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, Lucian L, Geissler R, Brodie S, Kimball AB, Gorman DM, Smith K, Malefyt RD, Kastelein RA, McClanahan TK, Bowman EP (2006) IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med 203:2577–2587. https://doi.org/10.1084/jem.20060244
van der Fits L, Mourits S, Voerman JSA, Kant M, Boon L, Laman JD, Cornelissen F, Mus AM, Florencia E, Prens EP, Lubberts E (2009) Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 182:5836–5845. https://doi.org/10.4049/jimmunol.0802999
Piskin G, Sylva-Steenland RMR, Bos JD, Teunissen MBM (2006) In vitro and in situ expression of IL-23 by keratinocytes in healthy skin and psoriasis lesions: enhanced expression in psoriatic skin. J Immunol 176:1908–1915. https://doi.org/10.4049/jimmunol.176.3.1908
Suarez-Farinas M, Li K, Fuentes-Duculan J, Hayden K, Brodmerkel C, Krueger JG (2012) Expanding the psoriasis disease profile: interrogation of the skin and serum of patients with moderate-to-severe psoriasis. J Invest Dermatol 132:2552–2564. https://doi.org/10.1038/jid.2012.184
Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A (2011) IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J Immunol 186:1495–1502. https://doi.org/10.4049/jimmunol.1001001
Chiricozzi A, Romanelli P, Volpe E, Borsellino G, Romanelli M (2018) Scanning the immunopathogenesis of psoriasis. Int J Mol Sci 19:179. https://doi.org/10.3390/ijms19010179
Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT (2011) Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Invest Dermatol 131:S106–S106
Amatya N, Garg AV, Gaffen SL (2017) IL-17 signaling: the yin and the yang. Trends Immunol 38:310–322. https://doi.org/10.1016/j.it.2017.01.006
Chiricozzi A, Nograles KE, Johnson-Huang LM, Fuentes-Duculan J, Cardinale I, Bonifacio KM, Gulati N, Mitsui H, Guttman-Yassky E, Suarez-Farinas M, Krueger JG (2014) IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS ONE 9:e90284. https://doi.org/10.1371/journal.pone.0090284
Johansen C, Mose M, Ommen P, Bertelsen T, Vinter H, Hailfinger S, Lorscheid S, Schulze-Osthoff K, Iversen L (2015) I kappa B zeta is a key driver in the development of psoriasis. Proc Natl Acad Sci 112:E5825–E5833. https://doi.org/10.1073/pnas.1509971112
Muller A, Hennig A, Lorscheid S, Grondona P, Schulze-Osthoff K, Hailfinger S, Kramer D (2018) I kappa B zeta is a key transcriptional regulator of IL-36-driven psoriasis-related gene expression in keratinocytes. Proc Natl Acad Sci 115:10088–10093. https://doi.org/10.1073/pnas.1801377115
Lorscheid S, Muller A, Loffler J, Resch C, Bucher P, Kurschus FC, Waisman A, Schakel K, Hailfinger S, Schulze-Osthoff K, Kramer D (2019) Keratinocyte-derived I kappa B zeta drives psoriasis and associated systemic inflammation. JCI Insight 4:e130835. https://doi.org/10.1172/jci.insight.130835
Brembilla NC, Senra L, Boehncke WH (2018) The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol 9:1682. https://doi.org/10.3389/fimmu.2018.01682
Ramirez-Carrozzi V, Sambandam A, Luis E, Lin ZG, Jeet S, Lesch J, Hackney J, Kim J, Zhou MJ, Lai J, Modrusan Z, Sai T, Lee W, Xu M, Caplazi P, Diehl L, de Voss J, Balazs M, Gonzalez L, Singh H, Ouyang W, Pappu R (2011) IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 12:1159-U1146. https://doi.org/10.1038/ni.2156
Martin DA, Towne JE, Kricorian G, Klekotka P, Gudjonsson JE, Krueger JG, Russell CB (2013) The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. J Invest Dermatol 133:17–26. https://doi.org/10.1038/jid.2012.194
Nakajima K, Kanda T, Takaishi M, Shiga T, Miyoshi K, Nakajima H, Kamijima R, Tarutani M, Benson JM, Elloso MM, Gutshall LL, Naso MF, Iwakura Y, DiGiovanni J, Sano S (2011) Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J Immunol 186:4481–4489. https://doi.org/10.4049/jimmunol.1000148
Towne JE, Bigler J, Zhang Y, Kerkof K, Timour M, Rand H, Klekotka P, Martin DA, Salinger D, Russell CB (2013) Three IL-17 ligands contribute to psoriasis: Blockade of IL-17RA signaling with brodalumab. J Invest Dermatol 133:S45–S45
Ettehadi P, Greaves MW, Wallach D, Aderka D, Camp RDR (1994) Elevated tumor-necrosis-factor-alpha (Tnf-Alpha) biological-activity in psoriatic skin-lesions. Clin Exp Immunol 96:146–151. https://doi.org/10.1111/j.1365-2249.1994.tb06244.x
Banno T, Gazel A, Blumenberg M (2004) Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J Biol Chem 279:32633–32642. https://doi.org/10.1074/jbc.M400642200
Grine L, Dejager L, Libert C, Vandenbroucke RE (2015) An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev 26:25–33. https://doi.org/10.1016/j.cytogfr.2014.10.009
Borghi A, Verstrepen L, Beyaert R (2016) TRAF2 multitasking in TNF receptor-induced signaling to NF-kappa B, MAP kinases and cell death. Biochem Pharmacol 116:1–10. https://doi.org/10.1016/j.bcp.2016.03.009
Zaba LC, Cardinale I, Gilleaudeau P, Sulhvan-Whalen M, Suarez-Farinas M, Fuentes-Duculan J, Novitskaya I, Khatcherian A, Bluth MJ, Lowes MA, Krueger JG (2007) Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med 204:3183–3194. https://doi.org/10.1084/jem.20071094
Zaba LC, Suarez-Farinas M, Fuentes-Duculan J, Nograles KE, Guttman-Yassky E, Cardinale I, Lowes MA, Krueger JG (2009) Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J Allergy Clin Immun 124:1022–1030. https://doi.org/10.1016/j.jaci.2009.08.046
Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, Chimenti S, Krueger JG (2011) Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 131:677–687. https://doi.org/10.1038/jid.2010.340
Li H, Yao Q, Mariscal AG, Wu XD, Hulse J, Pedersen E, Helin K, Waisman A, Vinkel C, Thomsen SF, Avgustinova A, Benitah SA, Lovato P, Norsgaard H, Mortensen MS, Veng L, Rozell B, Brakebusch C (2018) Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat Commun 9:1420. https://doi.org/10.1038/s41467-018-03704-z
Milora KA, Fu HF, Dubaz O, Jensen LE (2015) Unprocessed interleukin-36 alpha regulates psoriasis-like skin inflammation in cooperation with interleukin-1. J Invest Dermatol 135:2992–3000. https://doi.org/10.1038/jid.2015.289
Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, Zribi J, Bal E, Cluzeau C, Chrabieh M, Towne JE, Douangpanya J, Pons C, Mansour S, Serre V, Makni H, Mahfoudh N, Fakhfakh F, Bodemer C, Feingold J, Hadj-Rabia S, Favre M, Genin E, Sahbatou M, Munnich A, Casanova JL, Sims JE, Turki H, Bachelez H, Smahi A (2011) Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. New Engl J Med 365:620–628. https://doi.org/10.1056/NEJMoa1013068
Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, Knight J, Spain SL, Nestle FO, Burden AD, Capon F, Trembath RC, Barker JN (2011) Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet 89:432–437. https://doi.org/10.1016/j.ajhg.2011.07.022
Boutet MA, Bart G, Penhoat M, Amiaud J, Brulin B, Charrier C, Morel F, Lecron JC, Rolli-Derkinderen M, Bourreille A, Vigne S, Gabay C, Palmer G, Le Goff B, Blanchard F (2016) Distinct expression of interleukin (IL)-36alpha, beta and gamma, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid arthritis and Crohn’s disease. Clin Exp Immunol 184:159–173. https://doi.org/10.1111/cei.12761
Mercurio L, Morelli M, Scarponi C, Eisenmesser EZ, Doti N, Pagnanelli G, Gubinelli E, Mazzanti C, Cavani A, Ruvo M, Dinarello CA, Albanesi C, Madonna S (2018) IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis 9:1104. https://doi.org/10.1038/s41419-018-1143-3
Han Y, Mora J, Huard A, da Silva P, Wiechmann S, Putyrski M, Schuster C, Elwakeel E, Lang G, Scholz A, Scholz T, Schmid T, de Bruin N, Billuart P, Sala C, Burkhardt H, Parnham MJ, Ernst A, Brune B, Weigert A (2019) IL-38 ameliorates skin inflammation and limits IL-17 production from gammadelta T Cells. Cell Rep 27(835–846):e835. https://doi.org/10.1016/j.celrep.2019.03.082
Hernandez-Santana YE, Leon G, St Leger D, Fallon PG, Walsh PT (2020) Keratinocyte interleukin-36 receptor expression orchestrates psoriasiform inflammation in mice. Life Sci Alliance 3:e201900586. https://doi.org/10.26508/lsa.201900586
Towne JE, Sims JE (2012) IL-36 in psoriasis. Curr Opin Pharmacol 12:486–490. https://doi.org/10.1016/j.coph.2012.02.009
Carrier Y, Ma HL, Ramon HE, Napierata L, Small C, O’Toole M, Young DA, Fouser LA, Nickerson-Nutter C, Collins M, Dunussi-Joannopoulos K, Medley QG (2011) Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol 131:2428–2437. https://doi.org/10.1038/jid.2011.234
Goldstein JD, Bassoy EY, Caruso A, Palomo J, Rodriguez E, Lemeille S, Gabay C (2020) IL-36 signaling in keratinocytes controls early IL-23 production in psoriasis-like dermatitis. Life Sci Alliance 3:e202000688. https://doi.org/10.26508/lsa.202000688
Foster AM, Baliwag J, Chen CS, Guzman AM, Stoll SW, Gudjonsson JE, Ward NL, Johnston A (2014) IL-36 promotes myeloid cell infiltration, activation, and inflammatory activity in skin. J Immunol 192:6053–6061. https://doi.org/10.4049/jimmunol.1301481
Mutamba S, Allison A, Mahida Y, Barrow P, Foster N (2012) Expression of IL-1Rrp2 by human myelomonocytic cells is unique to DCs and facilitates DC maturation by IL-1F8 and IL-1F9. Eur J Immunol 42:607–617. https://doi.org/10.1002/eji.201142035
Bridgewood C, Fearnley GW, Berekmeri A, Laws P, Macleod T, Ponnambalam S, Stacey M, Graham A, Wittmann M (2018) IL-36 gamma Is a strong inducer of IL-23 in psoriatic cells and activates angiogenesis. Front Immunol 9:200. https://doi.org/10.3389/fimmu.2018.00200
Boniface K, Guignouard E, Pedretti N, Garcia M, Delwail A, Bernard FX, Nau F, Guillet G, Dagregorio G, Yssel H, Lecron JC, Morel F (2007) A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol 150:407–415. https://doi.org/10.1111/j.1365-2249.2007.03511.x
Sa SM, Valdez PA, Wu JF, Jung K, Zhong F, Hall L, Kasman I, Winer J, Modrusan Z, Danilenko DM, Ouyang WJ (2007) The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol 178:2229–2240. https://doi.org/10.4049/jimmunol.178.4.2229
Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R (2006) IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol 36:1309–1323. https://doi.org/10.1002/eji.200535503
Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W (2007) Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445:648–651. https://doi.org/10.1038/nature05505
Van Belle AB, de Heusch M, Lemaire MM, Hendrickx E, Warnier G, Dunussi-Joannopoulos K, Fouser LA, Renauld JC, Dumoutier L (2012) IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J Immunol 188:462–469. https://doi.org/10.4049/jimmunol.1102224
Antoniu SA (2012) Discontinued drugs 2011: pulmonary, allergy, gastrointestinal and arthritis. Expert Opin Investig Drugs 21:1607–1618. https://doi.org/10.1517/13543784.2012.712112
Amschler K, Meyersburg D, Kitze B, Schon MP, Mossner R (2016) Onset of psoriasis upon interferon beta treatment in a multiple sclerosis patient. Eur J Dermatol 26:211–212. https://doi.org/10.1684/ejd.2015.2602
Funk J, Langeland T, Schrumpf E, Hanssen LE (1991) Psoriasis induced by interferon-alpha. Br J Dermatol 125:463–465. https://doi.org/10.1111/j.1365-2133.1991.tb14774.x
La Mantia L, Capsoni F (2010) Psoriasis during interferon beta treatment for multiple sclerosis. Neurol Sci 31:337–339. https://doi.org/10.1007/s10072-009-0184-x
Pauluzzi P, Kokelj F, Perkan V, Pozzato G, Moretti M (1993) Psoriasis exacerbation induced by interferon-alpha. Report of two cases. Acta Derm Venereol 73:395. https://doi.org/10.2340/0001555573395
Nestle FO, Conrad C, Tun-Kyi A, Homey B, Gombert M, Boyman O, Burg G, Liu YJ, Gilliet M (2005) Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med 202:135–143. https://doi.org/10.1084/jem.20050500
Bissonnette R, Papp K, Maari C, Yao Y, Robbie G, White WI, Le C, White B (2010) A randomized, double-blind, placebo-controlled, phase I study of MEDI-545, an anti-interferon-alfa monoclonal antibody, in subjects with chronic psoriasis. J Am Acad Dermatol 62:427–436. https://doi.org/10.1016/j.jaad.2009.05.042
Abdallah MA, Abdel-Hamid MF, Kotb AM, Mabrouk EA (2009) Serum interferon-gamma is a psoriasis severity and prognostic marker. Cutis 84:163–168
Johnson-Huang LM, Suarez-Farinas M, Pierson KC, Fuentes-Duculan J, Cueto I, Lentini T, Sullivan-Whalen M, Gilleaudeau P, Krueger JG, Haider AS, Lowes MA (2012) A single intradermal injection of IFN-gamma induces an inflammatory state in both non-lesional psoriatic and healthy skin. J Invest Dermatol 132:1177–1187. https://doi.org/10.1038/jid.2011.458
Harden JL, Johnson-Huang LM, Chamian MF, Lee E, Pearce T, Leonardi CL, Haider A, Lowes MA, Krueger JG (2015) Humanized anti-IFN-gamma (HuZAF) in the treatment of psoriasis. J Allergy Clin Immunol 135:553–556. https://doi.org/10.1016/j.jaci.2014.05.046
Teunissen MB, Koomen CW, de Waal MR, Wierenga EA, Bos JD (1998) Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol 111:645–649. https://doi.org/10.1046/j.1523-1747.1998.00347.x
Baliwag J, Barnes DH, Johnston A (2015) Cytokines in psoriasis. Cytokine 73:342–350. https://doi.org/10.1016/j.cyto.2014.12.014
Long D, Chen Y, Wu H, Zhao M, Lu Q (2019) Clinical significance and immunobiology of IL-21 in autoimmunity. J Autoimmun 99:1–14. https://doi.org/10.1016/j.jaut.2019.01.013
Mitsui A, Tada Y, Takahashi T, Shibata S, Kamata M, Miyagaki T, Fujita H, Sugaya M, Kadono T, Sato S, Asano Y (2016) Serum IL-33 levels are increased in patients with psoriasis. Clin Exp Dermatol 41:183–189. https://doi.org/10.1111/ced.12670
Ruiz-Romeu E, Ferran M, de Jesus-Gil C, Garcia P, Sagrista M, Casanova JM, Fernandez JM, Chiriac A, Hollo P, Celada A, Pujol RM, Santamaria-Babi LF (2018) Microbe-dependent induction of IL-9 by CLA(+) T cells in psoriasis and relationship with IL-17A. J Invest Dermatol 138:580–587. https://doi.org/10.1016/j.jid.2017.08.048
Wolk K, Witte K, Witte E, Raftery M, Kokolakis G, Philipp S, Schonrich G, Warszawska K, Kirsch S, Prosch S, Sterry W, Volk HD, Sabat R (2013) IL-29 is produced by T(H)17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci Transl Med 5:204ra129. https://doi.org/10.1126/scitranslmed.3006245
Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, Homey B, Barrat FJ, Zal T, Gilliet M (2009) Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med 206:1983–1994. https://doi.org/10.1084/jem.20090480
Lande R, Chamilos G, Ganguly D, Demaria O, Frasca L, Durr S, Conrad C, Schroder J, Gilliet M (2015) Cationic antimicrobial peptides in psoriatic skin cooperate to break innate tolerance to self-DNA. Eur J Immunol 45:203–213. https://doi.org/10.1002/eji.201344277
Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, Zal T, Mellman I, Schroder JM, Liu YJ, Gilliet M (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449:564–569. https://doi.org/10.1038/nature06116
Lou F, Sun Y, Xu Z, Niu L, Wang Z, Deng S, Liu Z, Zhou H, Bai J, Yin Q, Cai X, Sun L, Wang H, Li Q, Wu Z, Chen X, Gu J, Shi YL, Tao W, Ginhoux F, Wang H (2020) Excessive polyamine generation in keratinocytes promotes self-RNA sensing by dendritic cells in psoriasis. Immunity 53:204–216. https://doi.org/10.1016/j.immuni.2020.06.004
Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, Nussbaum R, Novitskaya I, Carbonaro H, Cardinale I, Kikuchi T, Gilleaudeau P, Sullivan-Whalen M, Wittkowski KM, Papp K, Garovoy M, Dummer W, Steinman RM, Krueger JG (2005) Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci 102:19057–19062. https://doi.org/10.1073/pnas.0509736102
Duan X, Liu X, Liu N, Huang Y, Jin Z, Zhang S, Ming Z, Chen H (2020) Inhibition of keratinocyte necroptosis mediated by RIPK1/RIPK3/MLKL provides a protective effect against psoriatic inflammation. Cell Death Dis 11:134. https://doi.org/10.1038/s41419-020-2328-0
Devos M, Tanghe G, Gilbert B, Dierick E, Verheirstraeten M, Nemegeer J, de Reuver R, Lefebvre S, De Munck J, Rehwinkel J, Vandenabeele P, Declercq W, Maelfait J (2020) Sensing of endogenous nucleic acids by ZBP1 induces keratinocyte necroptosis and skin inflammation. J Exp Med 217:e20191913. https://doi.org/10.1084/jem.20191913
Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO, Fisher A, Lane R, Young GR, Kassiotis G, Kaiser WJ, Pasparakis M (2020) Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 580:391–395. https://doi.org/10.1038/s41586-020-2129-8
Chen L, Deshpande M, Grisotto M, Smaldini P, Garcia R, He Z, Gulko PS, Lira SA, Furtado GC (2020) Skin expression of IL-23 drives the development of psoriasis and psoriatic arthritis in mice. Sci Rep 10:8259. https://doi.org/10.1038/s41598-020-65269-6
Howes A, O’Sullivan PA, Breyer F, Ghose A, Cao L, Krappmann D, Bowcock AM, Ley SC (2016) Psoriasis mutations disrupt CARD14 autoinhibition promoting BCL10-MALT1-dependent NF-kappaB activation. Biochem J 473:1759–1768. https://doi.org/10.1042/BCJ20160270
Manils J, Webb LV, Howes A, Janzen J, Boeing S, Bowcock AM, Ley SC (2020) CARD14(E138A) signalling in keratinocytes induces TNF-dependent skin and systemic inflammation. Elife 9:e56720. https://doi.org/10.7554/eLife.56720
Van Nuffel E, Staal J, Baudelet G, Haegman M, Driege Y, Hochepied T, Afonina IS, Beyaert R (2020) MALT1 targeting suppresses CARD14-induced psoriatic dermatitis in mice. EMBO Rep 21:e49237. https://doi.org/10.15252/embr.201949237
Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, Chen YL, Hardman C, Xue L, Cerundolo V, Ogg G (2016) Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med 213:2399–2412. https://doi.org/10.1084/jem.20160258
Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, Chamilos G, Feldmeyer L, Marinari B, Chon S, Vence L, Riccieri V, Guillaume P, Navarini AA, Romero P, Costanzo A, Piccolella E, Gilliet M, Frasca L (2014) The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun 5:5621. https://doi.org/10.1038/ncomms6621
Harden JL, Hamm D, Gulati N, Lowes MA, Krueger JG (2015) Deep sequencing of the T-cell receptor repertoire demonstrates polyclonal T-cell infiltrates in psoriasis. F1000Res 4:460. https://doi.org/10.12688/f1000research.6756.1
Zaba LC, Fuentes-Duculan J, Eungdamrong NJ, Abello MV, Novitskaya I, Pierson KC, Gonzalez J, Krueger JG, Lowes MA (2009) Psoriasis is characterized by accumulation of immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic cells. J Invest Dermatol 129:79–88. https://doi.org/10.1038/jid.2008.194
Kagami S, Rizzo HL, Lee JJ, Koguchi Y, Blauvelt A (2010) Circulating Th17, Th22, and Th1 cells are increased in psoriasis. J Invest Dermatol 130:1373–1383. https://doi.org/10.1038/jid.2009.399
Res PC, Piskin G, de Boer OJ, van der Loos CM, Teeling P, Bos JD, Teunissen MB (2010) Overrepresentation of IL-17A and IL-22 producing CD8 T cells in lesional skin suggests their involvement in the pathogenesis of psoriasis. PLoS ONE 5:e14108. https://doi.org/10.1371/journal.pone.0014108
Cai Y, Shen X, Ding C, Qi C, Li K, Li X, Jala VR, Zhang HG, Wang T, Zheng J, Yan J (2011) Pivotal role of dermal IL-17-producing gammadelta T cells in skin inflammation. Immunity 35:596–610. https://doi.org/10.1016/j.immuni.2011.08.001
Soare A, Weber S, Maul L, Rauber S, Gheorghiu AM, Luber M, Houssni I, Kleyer A, von Pickardt G, Gado M, Simon D, Rech J, Schett G, Distler JHW, Ramming A (2018) Cutting edge: homeostasis of innate lymphoid cells is imbalanced in psoriatic arthritis. J Immunol 200:1249–1254. https://doi.org/10.4049/jimmunol.1700596
Teunissen MBM, Munneke JM, Bernink JH, Spuls PI, Res PCM, Te Velde A, Cheuk S, Brouwer MWD, Menting SP, Eidsmo L, Spits H, Hazenberg MD, Mjosberg J (2014) Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol 134:2351–2360. https://doi.org/10.1038/jid.2014.146
Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, Perera GK, Chapman A, Smith CH, Di Meglio P, Nestle FO (2014) Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol 134:984–991. https://doi.org/10.1038/jid.2013.477
Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F (2019) ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis 10:315. https://doi.org/10.1038/s41419-019-1540-2
Keren A, Shemer A, Ginzburg A, Ullmann Y, Schrum AG, Paus R, Gilhar A (2018) Innate lymphoid cells 3 induce psoriasis in xenotransplanted healthy human skin. J Allergy Clin Immunol 142:305–308. https://doi.org/10.1016/j.jaci.2018.02.015
Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO (2004) Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med 199:731–736. https://doi.org/10.1084/jem.20031482
Cheuk S, Wiken M, Blomqvist L, Nylen S, Talme T, Stahle M, Eidsmo L (2014) Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 192:3111–3120. https://doi.org/10.4049/jimmunol.1302313
Matos TR, O’Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, Kupper TS, Krueger JG, Clark RA (2017) Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest 127:4031–4041. https://doi.org/10.1172/JCI93396
Gallais Serezal I, Hoffer E, Ignatov B, Martini E, Zitti B, Ehrstrom M, Eidsmo L (2019) A skewed pool of resident T cells triggers psoriasis-associated tissue responses in never-lesional skin from patients with psoriasis. J Allergy Clin Immunol 143:1444–1454. https://doi.org/10.1016/j.jaci.2018.08.048
Moos S, Mohebiany AN, Waisman A, Kurschus FC (2019) Imiquimod-induced psoriasis in mice depends on the IL-17 signaling of keratinocytes. J Invest Dermatol 139:1110–1117. https://doi.org/10.1016/j.jid.2019.01.006
Li N, Yamasaki K, Saito R, Fukushi-Takahashi S, Shimada-Omori R, Asano M, Aiba S (2014) Alarmin function of cathelicidin antimicrobial peptide LL37 through IL-36gamma induction in human epidermal keratinocytes. J Immunol 193:5140–5148. https://doi.org/10.4049/jimmunol.1302574
Lai Y, Li D, Li C, Muehleisen B, Radek KA, Park HJ, Jiang Z, Li Z, Lei H, Quan Y, Zhang T, Wu Y, Kotol P, Morizane S, Hata TR, Iwatsuki K, Tang C, Gallo RL (2012) The antimicrobial protein REG3A regulates keratinocyte proliferation and differentiation after skin injury. Immunity 37:74–84. https://doi.org/10.1016/j.immuni.2012.04.010
Witte E, Kokolakis G, Witte K, Philipp S, Doecke WD, Babel N, Wittig BM, Warszawska K, Kurek A, Erdmann-Keding M, Kunz S, Asadullah K, Kadin ME, Volk HD, Sterry W, Wolk K, Sabat R (2014) IL-19 is a component of the pathogenetic IL-23/IL-17 cascade in psoriasis. J Invest Dermatol 134:2757–2767. https://doi.org/10.1038/jid.2014.308
Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, Vom Baur E, Witte K, Warszawska K, Philipp S, Johnson-Leger C, Volk HD, Sterry W, Sabat R (2009) IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J Mol Med (Berl) 87:523–536. https://doi.org/10.1007/s00109-009-0457-0
Schon MP, Broekaert SM, Erpenbeck L (2017) Sexy again: the renaissance of neutrophils in psoriasis. Exp Dermatol 26:305–311. https://doi.org/10.1111/exd.13067
Gudjonsson JE, Johnston A, Dyson M, Valdimarsson H, Elder JT (2007) Mouse models of psoriasis. J Invest Dermatol 127:1292–1308. https://doi.org/10.1038/sj.jid.5700807
Gerber PA, Buhren BA, Schrumpf H, Homey B, Zlotnik A, Hevezi P (2014) The top skin-associated genes: a comparative analysis of human and mouse skin transcriptomes. Biol Chem 395:577–591. https://doi.org/10.1515/hsz-2013-0279
Sutoh Y, Mohamed RH, Kasahara M (2018) Origin and evolution of dendritic epidermal T cells. Front Immunol 9:1059. https://doi.org/10.3389/fimmu.2018.01059
Wagner EF, Schonthaler HB, Guinea-Viniegra J, Tschachler E (2010) Psoriasis: what we have learned from mouse models. Nat Rev Rheumatol 6:704–714. https://doi.org/10.1038/nrrheum.2010.157
Suarez-Farinas M, Arbeit R, Jiang W, Ortenzio FS, Sullivan T, Krueger JG (2013) Suppression of molecular inflammatory pathways by Toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS ONE 8:e84634. https://doi.org/10.1371/journal.pone.0084634
Swindell WR, Johnston A, Carbajal S, Han G, Wohn C, Lu J, Xing X, Nair RP, Voorhees JJ, Elder JT, Wang XJ, Sano S, Prens EP, DiGiovanni J, Pittelkow MR, Ward NL, Gudjonsson JE (2011) Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PLoS ONE 6:e18266. https://doi.org/10.1371/journal.pone.0018266
Danilenko DM (2008) Review paper: preclinical models of psoriasis. Vet Pathol 45:563–575. https://doi.org/10.1354/vp.45-4-563
Chuang SY, Lin CH, Sung CT, Fang JY (2018) Murine models of psoriasis and their usefulness for drug discovery. Expert Opin Drug Discov 13:551–562. https://doi.org/10.1080/17460441.2018.1463214
Brown WR, Hardy MH (1988) A hypothesis on the cause of chronic epidermal hyperproliferation in asebia mice. Clin Exp Dermatol 13:74–77. https://doi.org/10.1111/j.1365-2230.1988.tb00661.x
HogenEsch H, Gijbels MJ, Offerman E, van Hooft J, van Bekkum DW, Zurcher C (1993) A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am J Pathol 143:972–982
Sundberg JP, France M, Boggess D, Sundberg BA, Jenson AB, Beamer WG, Shultz LD (1997) Development and progression of psoriasiform dermatitis and systemic lesions in the flaky skin (fsn) mouse mutant. Pathobiology 65:271–286. https://doi.org/10.1159/000164138
Croxford AL, Karbach S, Kurschus FC, Wortge S, Nikolaev A, Yogev N, Klebow S, Schuler R, Reissig S, Piotrowski C, Brylla E, Bechmann I, Scheller J, Rose-John S, Thomas Wunderlich F, Munzel T, von Stebut E, Waisman A (2014) IL-6 regulates neutrophil microabscess formation in IL-17A-driven psoriasiform lesions. J Invest Dermatol 134:728–735. https://doi.org/10.1038/jid.2013.404
Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, Chen CS, Fu W, Gudjonsson JE, McCormick TS, Ward NL (2013) Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol 190:2252–2262. https://doi.org/10.4049/jimmunol.1201505
Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, Itami S, Nickoloff BJ, DiGiovanni J (2005) Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med 11:43–49. https://doi.org/10.1038/nm1162
Mellett M, Meier B, Mohanan D, Schairer R, Cheng P, Satoh TK, Kiefer B, Ospelt C, Nobbe S, Thome M, Contassot E, French LE (2018) CARD14 gain-of-function mutation alone is sufficient to drive IL-23/IL-17-mediated psoriasiform skin inflammation in vivo. J Invest Dermatol 138:2010–2023. https://doi.org/10.1016/j.jid.2018.03.1525
Wang M, Zhang S, Zheng G, Huang J, Songyang Z, Zhao X, Lin X (2018) Gain-of-function mutation of card14 leads to spontaneous psoriasis-like skin inflammation through enhanced keratinocyte response to IL-17A. Immunity 49(66–79):e65. https://doi.org/10.1016/j.immuni.2018.05.012
Zenz R, Eferl R, Kenner L, Florin L, Hummerich L, Mehic D, Scheuch H, Angel P, Tschachler E, Wagner EF (2005) Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature 437:369–375. https://doi.org/10.1038/nature03963
Wohn C, Ober-Blobaum JL, Haak S, Pantelyushin S, Cheong C, Zahner SP, Onderwater S, Kant M, Weighardt H, Holzmann B, Reizis B, Becher B, Prens EP, Clausen BE (2013) Langerin(neg) conventional dendritic cells produce IL-23 to drive psoriatic plaque formation in mice. Proc Natl Acad Sci 110:10723–10728. https://doi.org/10.1073/pnas.1307569110
Walter A, Schafer M, Cecconi V, Matter C, Urosevic-Maiwald M, Belloni B, Schonewolf N, Dummer R, Bloch W, Werner S, Beer HD, Knuth A, van den Broek M (2013) Aldara activates TLR7-independent immune defence. Nat Commun 4:1560. https://doi.org/10.1038/ncomms2566
Grine L, Steeland S, Van Ryckeghem S, Ballegeer M, Lienenklaus S, Weiss S, Sanders NN, Vandenbroucke RE, Libert C (2016) Topical imiquimod yields systemic effects due to unintended oral uptake. Sci Rep 6:20134. https://doi.org/10.1038/srep20134
Hedrick MN, Lonsdorf AS, Shirakawa AK, Richard Lee CC, Liao F, Singh SP, Zhang HH, Grinberg A, Love PE, Hwang ST, Farber JM (2009) CCR6 is required for IL-23-induced psoriasis-like inflammation in mice. J Clin Invest 119:2317–2329. https://doi.org/10.1172/jci37378
Sarra M, Caruso R, Cupi ML, Monteleone I, Stolfi C, Campione E, Diluvio L, Mazzotta A, Botti E, Chimenti S, Costanzo A, MacDonald TT, Pallone F, Monteleone G (2011) IL-21 promotes skin recruitment of CD4(+) cells and drives IFN-gamma-dependent epidermal hyperplasia. J Immunol 186:5435–5442. https://doi.org/10.4049/jimmunol.1003326
Wu L, Chen X, Zhao J, Martin B, Zepp JA, Ko JS, Gu C, Cai G, Ouyang W, Sen G, Stark GR, Su B, Vines CM, Tournier C, Hamilton TA, Vidimos A, Gastman B, Liu C, Li X (2015) A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J Exp Med 212:1571–1587. https://doi.org/10.1084/jem.20150204
Krueger GG, Chambers DA, Shelby J (1981) Involved and uninvolved skin from psoriatic subjects: are they equally diseased? Assessment by skin transplanted to congenitally athymic (nude) mice. J Clin Invest 68:1548–1557. https://doi.org/10.1172/jci110409
Nickoloff BJ, Kunkel SL, Burdick M, Strieter RM (1995) Severe combined immunodeficiency mouse and human psoriatic skin chimeras. Validation of a new animal model. Am J Pathol 146:580–588
Desmet E, Ramadhas A, Lambert J, Van Gele M (2017) In vitro psoriasis models with focus on reconstructed skin models as promising tools in psoriasis research. Exp Biol Med (Maywood) 242:1158–1169. https://doi.org/10.1177/1535370217710637
Shin JU, Abaci HE, Herron L, Guo Z, Sallee B, Pappalardo A, Jackow J, Wang EHC, Doucet Y, Christiano AM (2020) Recapitulating T cell infiltration in 3D psoriatic skin models for patient-specific drug testing. Sci Rep 10:4123. https://doi.org/10.1038/s41598-020-60275-0
Golbari NM, Porter ML, Kimball AB (2018) Current guidelines for psoriasis treatment: a work in progress. Cutis 101:10–12
Kaushik SB, Lebwohl MG (2019) Review of safety and efficacy of approved systemic psoriasis therapies. Int J Dermatol 58:649–658. https://doi.org/10.1111/ijd.14246
Ronholt K, Iversen L (2017) Old and new biological therapies for psoriasis. Int J Mol Sci 18:2297. https://doi.org/10.3390/ijms18112297
Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, Papp KA, Sofen H, Puig L, Foley P, Ohtsuki M, Flack M, Geng Z, Gu Y, Valdes JM, Thompson EHZ, Bachelez H (2018) Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 392:650–661. https://doi.org/10.1016/S0140-6736(18)31713-6
Saunte DM, Mrowietz U, Puig L, Zachariae C (2017) Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br J Dermatol 177:47–62. https://doi.org/10.1111/bjd.15015
Li H, Zuo J, Tang W (2018) Phosphodiesterase-4 Inhibitors for the treatment of inflammatory diseases. Front Pharmacol 9:1048. https://doi.org/10.3389/fphar.2018.01048
Wcislo-Dziadecka D, Zbiciak-Nylec M, Brzezinska-Wcislo L, Bebenek K, Kazmierczak A (2017) Newer treatments of psoriasis regarding IL-23 inhibitors, phosphodiesterase 4 inhibitors, and Janus kinase inhibitors. Dermatol Ther. https://doi.org/10.1111/dth.12555
Liu Y, Jarjour W, Olsen N, Zheng SG (2020) Traitor or warrior-Treg cells sneaking into the lesions of psoriatic arthritis. Clin Immunol 215:108425. https://doi.org/10.1016/j.clim.2020.108425
Karczewski J, Dobrowolska A, Rychlewska-Hanczewska A, Adamski Z (2016) New insights into the role of T cells in pathogenesis of psoriasis and psoriatic arthritis. Autoimmunity 49:435–450. https://doi.org/10.3109/08916934.2016.1166214
Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, McCormick TS, Cooper KD (2005) Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol 174:164–173. https://doi.org/10.4049/jimmunol.174.1.164
Uttarkar S, Brembilla NC, Boehncke WH (2019) Regulatory cells in the skin: pathophysiologic role and potential targets for anti-inflammatory therapies. J Allergy Clin Immunol 143:1302–1310. https://doi.org/10.1016/j.jaci.2018.12.1011
Yang L, Li B, Dang E, Jin L, Fan X, Wang G (2016) Impaired function of regulatory T cells in patients with psoriasis is mediated by phosphorylation of STAT3. J Dermatol Sci 81:85–92. https://doi.org/10.1016/j.jdermsci.2015.11.007
Kannan AK, Su Z, Gauvin DM, Paulsboe SE, Duggan R, Lasko LM, Honore P, Kort ME, McGaraughty SP, Scott VE, Gauld SB (2019) IL-23 induces regulatory T cell plasticity with implications for inflammatory skin diseases. Sci Rep 9:17675. https://doi.org/10.1038/s41598-019-53240-z
Hayashi M, Yanaba K, Umezawa Y, Yoshihara Y, Kikuchi S, Ishiuji Y, Saeki H, Nakagawa H (2016) IL-10-producing regulatory B cells are decreased in patients with psoriasis. J Dermatol Sci 81:93–100. https://doi.org/10.1016/j.jdermsci.2015.11.003
Mavropoulos A, Varna A, Zafiriou E, Liaskos C, Alexiou I, Roussaki-Schulze A, Vlychou M, Katsiari C, Bogdanos DP, Sakkas LI (2017) IL-10 producing Bregs are impaired in psoriatic arthritis and psoriasis and inversely correlate with IL-17- and IFNgamma-producing T cells. Clin Immunol 184:33–41. https://doi.org/10.1016/j.clim.2017.04.010
Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, Ruether A, Schreiber S, Weichenthal M, Gladman D, Rahman P, Schrodi SJ, Prahalad S, Guthery SL, Fischer J, Liao W, Kwok PY, Menter A, Lathrop GM, Wise CA, Begovich AB, Voorhees JJ, Elder JT, Krueger GG, Bowcock AM, Abecasis GR, for the Collaborative Association Study of Psoriasis (2009) Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet 41:199–204. https://doi.org/10.1038/ng.311
Tejasvi T, Stuart PE, Chandran V, Voorhees JJ, Gladman DD, Rahman P, Elder JT, Nair RP (2012) TNFAIP3 gene polymorphisms are associated with response to TNF blockade in psoriasis. J Invest Dermatol 132:593–600. https://doi.org/10.1038/jid.2011.376
Acknowledgements
Work in the authors’ lab related to the topic of this paper is supported by the VIB and the Research Foundation—Flanders (FWO; G090914N and G035517N). I.S.A. is supported by a postdoctoral fellowship and research grants (1503418N and 1503815N) of the FWO. E.V.N. was supported by a predoctoral FWO fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Afonina, I.S., Van Nuffel, E. & Beyaert, R. Immune responses and therapeutic options in psoriasis. Cell. Mol. Life Sci. 78, 2709–2727 (2021). https://doi.org/10.1007/s00018-020-03726-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03726-1