
Abstract
The RNA-guided CRISPR–Cas9 technology has paved the way for rapid and cost-effective gene editing. However, there is still a great need for effective methods for rapid generation and validation of CRISPR/Cas9 gRNAs. Previously, we have demonstrated that highly efficient generation of multiplexed CRISPR guide RNA (gRNA) expression array can be achieved with Golden Gate Assembly (GGA). Here, we present an optimized and rapid method for generation and validation in less than 1 day of CRISPR gene targeting vectors. The method (LION) is based on ligation of double-stranded gRNA oligos into CRISPR vectors with GGA followed by nucleic acid purification. Using a dual-fluorescent reporter vector (C-Check), T7E1 assay, TIDE assay and a traffic light reporter assay, we proved that the LION-based generation of CRISPR vectors are functionally active, and equivalent to CRISPR plasmids generated by traditional methods. We also tested the activity of LION CRISPR vectors in different human cell types. The LION method presented here advances the rapid functional validation and application of CRISPR system for gene editing and simplified the CRISPR gene-editing procedures.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) are an adaptive immune system found in bacteria and archaea [1] that was harnessed for programmable and precise gene editing in 2012 [2, 3]. To achieve CRISPR–Cas9 gene editing, the Cas9 protein firstly binds to the conserved secondary structure (commonly known as a Cas9 interacting scaffold) of a small guide RNA (gRNA). Then, the Cas9 nuclease is guided to the corresponding target site in the genome, based on the Watson–Crick base pairing principle between the gRNA guide sequences and the target site. There is another important condition that the target site has to meet, that is the presence of a highly conserved DNA motif, known as the protospacer adjacent motif (PAM, and a downstream 5′-NGG for SpCas9). When all these targeting parameters are met, Cas9 DNA endonuclease activity is activated and causes a double-stranded DNA break (DSB) to the target site. Repair of the DSB via the endogenous DNA repair machinery most frequently leads to an introduction of small insertions or deletions, known as indels. Owing to the simplicity and high efficiency, CRISPR/Cas9 has become the state-of-the-art gene-editing tool in both academic research and gene-related applications [4].
Most applications of the CRISPR/Cas9 technology are to introduce out-of-frame mutations to essential exons of genes of interest (GOI) and hence disrupting gene functions. Compared to other gene-editing tools such as ZFNs and TALENs, CRISPR/Cas9 offers simplicity in multiplexed gene editing [5]. For example, it has been used to inactivate 62 copies of porcine endogenous retrovirus (PERV) in porcine cells [6], and it has further been shown that this technology can be applied in primary pig cells to generate PERV-inactivated pigs [7]. Apart from simple gene disruption, exogenous DNA can be efficiently integrated into specific genomic locus via CRISPR-triggered homology-directed repair (HDR). Typical applications of CRISPR-based HDR include gene tagging [8] and point mutation/correction [9]. Furthermore, the flexibility of the Cas9 protein makes this system extremely powerful. Fusion of dead Cas9 (dCas9) nuclease with a variety of functional enzymes significantly expands the applications of this technology, by including gene activation [10, 11], multicolor labeling [12], epigenome editing [13] and base editing [14, 15]. In addition, we have recently discovered that the CRISPR–Cas9 technology can be repurposed for making chromosomal DNAs into extrachromosomal circular DNAs, with a wide range of lengths (from several hundred base pairs to Mb-scale DNA) [16].
Despite the broad versatility and applications of CRISPR/Cas9, the highly variable activity among different gRNAs also increases the workload and complexity of conducing CRISPR gene-editing studies [17,18,19]. Although several in silico gRNA activity prediction models and web tools have been developed, the current models are still far from satisfactory. Currently, over 1000 bioinformatic features have been predicted to have an impact on gRNA activity, such as being rich in guanine and adenine depletion in the gRNA sequence [20], a preference for 5′GG [21] and a preference for guanine but not thymine in position 20 adjacent to PAM [22]. Previously, we also identified that the gRNA secondary structure plays a critical role in active gRNA–Cas9 complex assembly [23]. Internal gRNA interactions impede functional gRNA–Cas9 complex formation and lead to low cleavage activity [17, 24]. Target local chromatin accessibility factors also have to be considered, including DNA methylation level, H3K4me3, DNase I sensitivity and CTCF binding frequency [17, 25, 26]. For these reasons, experimentally validated gRNA activity still remains the gold standard for evaluating the efficacy of CRISPR gRNAs.
Currently, it is a generally advisable to validate and select the highest efficient gRNA among several candidate gRNAs for subsequent research or applications [27,28,29]. A number of methods have been developed to evaluate gRNA cleavage efficacy in vitro or in vivo, including T7E1 assay [2], tracking indels by decomposition (TIDE) [30], fluorescent reporter test [31, 32], indel detection by amplicons analysis (IDAA) [33], digital PCR [34], amplicons next-generation sequencing (NGS) [35, 36], and antibody staining-based flow cytometry analysis [37]. Despite the rapid development of gRNA validation methodologies, the current procedure of generating CRISPR–Cas9 gRNA vectors by E. coli transformation is one major speed-limiting step for most molecular laboratories. To bypass the laborious step of vector cloning, we here present a rapid and efficient transformation-free LION method for generating transfection-grade gRNA plasmids, based on the Golden Gate Cloning technology. We validated the activity of LION-based CRISPR vectors in human cells by a fluorescent reporter system (C-Check), T7E1, TIDE, and traffic light reporter (TLR) assay [38]. We proved that the LION CRISPR vectors are functionally active, equivalent to CRISPR plasmids generated by traditional methods, and directly useful for gene editing in different human cell types tested.
Results
Establishment and optimization of the LION method
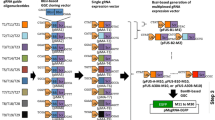
The Golden Gate Assembly (GGA) is one of the most broadly used methods for vector cloning as shown in Fig. 1, as it allows efficient enzyme digestion and ligation to occur in one reaction. Previously, we have generated several GGA-based methods for the generation of multiplexed CRISPR gRNA expression arrays [39]. As we always obtained over 95% positive rate when performing colony PCR screening of GGA-based CRISPR vectors, we propose to simplify the whole procedure of CRISPR vector generation by testing whether the GGA ligation product can be purified directly and used for gene editing (Fig. 1, methodology denoted as LION).

Schematic diagram depicting the workflow comparison between traditional and LION approaches for constructing gRNA expression vectors. a The traditional gRNA vector construction comprises Golden Gate Assembly, transformation of E. coli competent cells, bacterial colonies PCR screening, positive colony expansion by incubating at 37 °C and bacterial culture overnight, and plasmid prep before the CRISPR vector is ready for the cell transfection and activity validation. The whole traditional procedure takes approximately 2.5 days. b The LION method requires only two steps: Golden Gate Assembly and quick DNA purification to generate transfection-grade CRISPR plasmids. The whole procedure takes approximately 5 h (~ 4 h for GGA and less than 1 h for DNA purification
First, we tested the optimization of our GGA ligate reaction system by tuning the amount of backbone plasmids, as this allows us to obtain high enough amounts of DNA for transfection. We employed one of the most frequently used CRISPR gRNA expression vectors (lentiGuide-Puro, Addgene ID 52963) for this purpose. Eight different amounts (100–5000 ng, Fig. 2a) of lentiGuide-Puro were assembled with 1 μL annealed gRNA oligos (5 pmol in total) using a GGA assembly protocol (see method), followed by a standard PCR purification of the ligation product with a commercially available purification kit (see “Materials and methods”). To distinguish our constructs from plasmids generated by normal procedure (Fig. 1a), all vectors generated by the LION method (Fig. 1b) are denoted as LION vectors.

Optimization of the backbone plasmid amount used in LION and validation of LION CRISPR vector for gene editing by C-Check. a Diagram depicting the working principle of our C-check system. SSA single-strand annealing, NHEJ non-homologous end joining. The AsRed expression cassette is used as a background fluorescence for normalization of the transfection efficiency. b Left: an example of the C-Check assay in cells transfected with or without CRISPR/Cas9 plasmids. Right: FCM patterns of the C-Check efficiency test for different amounts of backbone used in LION. mock denotes cell samples transfected with the same total amount of pUC19 plasmid. c FCM quantification of C-Check cleavage efficiency between LION and pure CRISPR vectors (n = 3). w/o without. Asterisk (**) indicates a p value less than 0.01
To investigate whether the LION CRISPR vectors can be directly used for gene editing, we transfected HEK293T cells with a LION CRISPR vector, a Cas9 expression vector and the corresponding C-Check vector, a dual-fluorescent system previously developed by us to measure CRISPR activity (Fig. 2a). By using fluorescence imaging and flow cytometry (FCM), we found that all LION vectors have as active gene-editing capability as vectors generated by the usual procedures (Fig. 2b). Quantification of FCM results showed that plasmid amounts of 500, 1000, and 1500 ng gave LION vectors equally effective CRISPR gene-editing activity compared to normal plasmids (Fig. 2c). Although the 500 ng backbone yields an apparently higher efficiency, the amount of LION vector after purification is often not enough for subsequent experiments. Thus, we chose 1000 ng backbone plasmid for all following experiments.
We next optimized the amounts (5, 10, 15, 25, 40 pmol) of annealed gRNA oligos for generating LION vectors. Fluorescence imaging and FCM analysis of C-Check-based testing of LION vectors showed that all doses tested yielded a similar gene-editing activity compared to the normal CRISPR plasmid (Supplementary Figs. 1, 3a, b). Considering that there were linear DNA/oligos in the LION product after purification which might affect the gene-editing efficiency, we treated the LION vector, made from the 1000 ng backbone and 5 pmol gRNA oligo, with plasmid-safe nuclease (PSN) and the efficiency PSN LION vectors were similar to ordinary LION vectors (11.53 ± 0.35% vs. 11.67 ± 0.40%), showing that PSN treatment is not necessary for LION vector generation.
We also evaluated the application of LION CRISPR vector for gene editing using T7E1 and TIDE assays, two widely used methods for evaluating CRISPR/gRNA activity [40,41,42]. Our T7E1 results showed that, consistent with the C-Check results, the gene-editing efficiency of the LION CRISPR vectors is similar to traditional CRISPR plasmids (Fig. 3c). We conducted Sanger sequencing to detect the indels spectrum induced by plasmid and LION vectors. There are obvious multi-signal peaks started around the cleavage site of gRNA in both plasmid-gRNA and LION1 vector groups (Fig. 3d), and TIDE analysis showed that the indel spectrum was similar in the two groups (Fig. S2). Taken together, our result showed that 1000 ng backbone plasmid and 5 pmol gRNA oligo presented the optimized condition for generation LION CRISPR vectors, and was therefore used for all subsequent efficiency tests.

Optimization of gRNA amounts used in LION and validation of LION CRISPR vector-based gene editing. a FCM patterns of C-Check efficiency tested for different amounts of gRNA used in LION CRISPR vector generation. b FCM quantification of C-Check cleavage efficiency in a comparison between traditional and LION CRISPR vectors (n = 3). c T7E1 assay results of LION and pure CRISPR plasmid. L indicates 100 bp DNA ladders. Lion 0, 1, 2, 5, 8 indicate 1000 ng backbone ligated with 0, 1, 2, 3, 5, 8 μL annealed gRNA (5 μM), respectively. 1PSN represents treatment of LION CRISPR made from 5 pmol annealed gRNA oligo and treated with plasmid-safe DNA nuclease. P indicates pure plasmid. CTRL indicates negative control. d Sanger sequencing to detect indel spectrums of optimized LION and pure plasmids. Red line and frame indicate the gRNA sequence. Red arrows indicate the cleavage site of CRISPR/Cas9
DNA composition of LION CRISPR vectors
Having proved that the LION CRISPR vector can be used for gene editing, we analyzed the DNA composition (linear and circular DNAs) of LION CRISPR vectors. The lentiGuide-Puro plasmid contains two BsmBI sites flanking a cloning fragment (1885 bp) for GGA-mediated insertion of gRNA guide oligos. The 1885 bp cloning fragment would be replaced with the 20 bp gRNA guide oligo upon successful GGA insertion (Fig. 4a). We predicted that three forms of DNAs are present in LION vector after purification: (1) a circular form of LION CRIPR vector; (2) a linear form of LION CRISPR vector; (3) a linear DNA of the cloning fragment. To quantify the linear and circular LION CRISPR vectors, we gel purified the band around 8300 bp, which contains the two forms of LION CRISPR DNA, and digested them with a MluI, a unique site in the lentiGuide-Puro vector (Fig. 4a, b). Our results revealed that, in all five conditions tested, the circular form of LION CRISPR vector was approximately 40% (Fig. 4c). For the control, GGA without annealing gRNA oligonucleotides yielded mainly linearized backbone vector.

DNA composition analysis of LION CRISPR vectors and ligation efficiency quantification. a lentiGuide-Puro backbone structure diagram with selected restriction enzymes cleavage sites. b Restriction enzyme digestion analysis of LION CRISPR vectors. DNA fragments of approximately 8317 bp were gel purified and further digested with MluI. c Fragment intensity analysis of LION 0, 1, 2, 3, 5, 8 and pure plasmid after MluI digestion. d Colony-forming unit analysis of LION 0, 1, 2, 3, 5, 8. e PCR screening results of the bacterial colonies made from six different conditions of LION CRISPR assembly. For negative control (Lion 0), 48 colonies were screened and for the other give conditions, 96 colonies (per condition) were PCR screened
We also quantified the ligation efficiency of LION CRISPR vectors by colony-forming unit (CFU) and colony PCR screening. Our results showed the ligation efficiency of all LION vectors tested were within a similar range of 6 × 104 colonies per mL (Fig. 4d). We further conducted colony PCR screening of 96 colonies from each group to verify correct insertion of the CRISPR gRNA guide sequences to the backbone vector. For control, we analyzed all the 48 colonies in the plates. More than 98% of the LION colonies analyzed were positive for carrying the correct CRISPR guide insertion. For the LION control, all 48 colonies as expected were negative (Fig. 4e). Results for colony PCR screening are shown in supplementary data Fig. S3.
LION CRISPR vectors are functionally similar to traditional CRISPR vectors in different target loci and cell types tested
To evaluate LION CRISPR vectors for gene editing and evaluation of CRISPR gRNA activity, we tested and compared LION CRISPR vectors to CRISPR plasmids generated by traditional approach in 20 different endogenous loci, located on different chromosomes (Supplementary Table S1). We previously validated the gene-editing activity of these 20 gRNAs [17]. First, we co-transfected HEK293T cells with CRISPR vectors and their corresponding C-Check vector and then analyzed gene-editing activity by FCM 48 h after transfection. The results showed that the efficacy of LION CRISPR vectors and normal CRISPR plasmids were quite similar in terms of gene-editing activity for all 20 sites tested (R2 = 0.958, linear regression assay) (Fig. 5a).

CRISPR gRNA activity comparison of LION and pure plasmids in different target loci and cell types. a FCM quantification of LION and traditional CRISPR plasmid-mediated gene editing by C-Check tests in 20 different target sites (n = 3). b Comparison of LION and traditional CRISPR plasmid-mediated gene-editing efficiency in HEK293T cells by T7E1 assay analysis. c Dual-cutting efficiency comparison of LION and traditional CRISPR plasmids in HEK293T cells (n = 3). N (the second lane in LION5 + LION6 group) indicates this lane was NOT analyzed. Plasmid-E denotes lentiGuide-Puro vector without gRNA. d TIDE analysis of LION and pure plasmids in two different loci within three different human cell lines (n = 3). Asterisk (*) indicates a p value less than 0.05 and (**) less than 0.01
We also evaluated the gene-editing activity of LION CRISPR vectors in HEK293T cells by the T7E1 assay. Genome regions which harbor the target sites were amplified and subjected to a T7E1 assay 48 h after transfection. Consistent with the C-Check result, T7E1 analysis showed that the LION CRISPR vectors and normal CRISPR plasmids are similar in their gene-editing activity as tested in the HEK293T cells (Fig. 5b). The lengths of the T7E1 digestion products are provided in Table S2.
Dual gRNAs-directed DNA cleavage is a commonly used strategy for rapid endogenous gene knockout and long non-coding RNA inactivation [43,44,45,46,47]. Recently, we discovered that extrachromosomal circular DNA/genes can also be generated by dual gRNAs [16]. In addition, the efficiency of homology-directed repair (HDR) can be increased when employing paired gRNAs to introduce DSBs [48, 49]. Thus, we also tested and compared dual-cutting efficiency of LION CRISPR vectors with normal CRISPR plasmids in two genomic loci. HEK293T cells were transfected with either pairs of LION CRISPR vectors or traditional CRISPR plasmids, and genotyping PCR was conducted 48 h after transfection. Compared to LION control (LION0, GGA without annealed gRNA oligos) and empty plasmid control (plasmid-E), our results showed that both the LION and traditional CRISPR plasmids introduce significant dual cutting with similar efficiency (Fig. 5c).
Finally, we evaluated the applicability of LION CRISPR vectors in three different human cell lines: an immortalized human mammary fibroblast line (MJ), an ovarian cancer cell line (Skov-3), and a cervical cancer cell line (Hela). Two different genomic target sites were evaluated (gRNA 13 and 14). TIDE assay showed that both LION and traditional CRISPR plasmids yield similar gene-editing efficiency for the two target sites in the three cell lines (Fig. 5d). We also observed that the gene-editing activity of gRNA 14 in MJ and Hela cells varied significantly between LION vectors and traditional CRISPR plasmids, which might be cell type specific. Taken together, our results further support that the CRISPR plasmids generated by the LION approach have similar gene-editing activity to normal CRISPR plasmids, and are an applicable for different genomic loci and different cell types.
LION CRISPR vectors favor DSB repair by HDR
Repair of the DNA double-stranded breaks (DSBs) in mammalian cells is predominantly mediated by non-homologous end joining (NHEJ). The low efficiency of DSB repair by the homology-directed repair (HDR) is still one of the unmet technology challenges in gene editing. Previously, a study from Jacob Corn’s group showed that inclusion of linear non-homologous DNA can increase CRISPR gene-editing efficiency [50]. We used the traffic light reporter (TLR) system, which was developed for quantification of both NHEJ and HDR efficiency in one assay [38, 51,52,53,54,55,56] (Fig. 6a). We first generated a LION TLR gRNA vector using our optimized LION protocol. Next, we transfected HEK293 TLR cells with either the LION TLR gRNA or a normal TLR gRNA plasmid, a TLR donor plasmid, and a Cas9 expression plasmid. For the Cas9 expression plasmid, two versions of Cas9 were evaluated: the original wild-type SpCas9 and the recombination SpCas9–RecA that we recently generated [53]. Three days after transfection, the HEK293 TLR cells were analyzed by FCM (Fig. 6b–d). We quantified the percentage of Venus-positive cells (HDR repair, Fig. 6b), RFP-positive cells (NHEJ repair, Fig. 6c), and the relation of Venus to RFP-positive cells (HDR vs. NHEJ efficiency, Fig. 6d). Consistent with our previous findings, SpCas9–RecA favors DSB repair by NHEJ compared to SpCas9. Interestingly, we also found that the LION TLR gRNA favors DSBs repairs by HDR compared to traditional TLR gRNA (Fig. 6b–d) for both SpCas9 and SpCas9–RecA (increased by over 50%). Our results suggest that the LION approach can not only be used for rapid generation and validation of CRISPR vector, but also for enhancing DSB repair by HDR.

HDR and NHEJ efficiency evaluation by traffic light reporter (TLR) assay. a Diagram depicting the work principle of TLR system. b FCM detection of HDR and NHEJ events after either LION- or plasmid-TLR gRNA transfection in HEK293 TLR cells. A TLR HDR donor vector was co-transfected in each group. Two versions of Cas9 were evaluated (the original wild-type SpCas9 and the recombinant SpCas9–RecA) in both LION and plasmid groups. SpCas9 without gRNA was used as negative control group. c FCM quantification of GFP-positive cells (n = 3) and d RFP-positive cells as percentage in LION and plasmid groups (n = 3). e GFP- and RFP-positive cells ratio in the two groups (n = 3). Asterisk (*) indicates a p value less than 0.05 and (**) less than 0.01
Discussion
As there is a large variation in activity among different gRNAs, it is generally required to test the activity of several gRNAs to achieve the highest efficient gRNA before subsequent CRISPR gene-editing applications [27,28,29]. Generation of CRISPR vectors by traditional molecular cloning requires several laborious steps including transformation, colony screening and miniprep. In this study, we established a cost-effective and simple approach (LION) to generate transfection-grade CRISPR vectors and proved that the gene-editing efficacy of LION vectors is comparable with that of normal CRISPR plasmids. As our LION method simply relies on ligation and purification, it allows rapid and high-throughput generation and validation of CRISPR vectors and gRNA activity. Based on our results, we proposed a streamlined protocol for LION-based CRISPR gene-editing projects: (1) design CRISPR gRNAs using in silico web tool, normally an average of 3 gRNAs per gene depending on the application; (2) synthesize gRNA guide oligos and generate CRISPR vectors by LION; (3) evaluate gRNA gene-editing activity by, e.g., C-Check, TIDE or other similar methods that can quantify CRISPR activity; (4) select the gRNA with highest activity and conduct downstream CRISPR editing.
Although we have shown that the LION CRISPR vectors are compatible with gene knockout and knockin, the LION CRISPR vector also has its limitations. First, only a relatively low amount of LION CRISPR vector can be generated (approximately 1 µg with our optimized protocols). Although several assemblies can be performed to increase the yield, the LION plasmids are still not compatible with all downstream applications. If for instance the CRISPR vectors will be used for viral packaging, we suggest combining LION with a traditional plasmid generation approach. Thus, the LION approach can be used to quickly select the best performing gRNA, and the plasmid prep can be made from the best one for downstream applications. Another characteristic of the LION CRISPR vector is the mixture of both linear and circular CRISPR and DNA in the purified LION DNA. The presence of linear DNA will increase the chance of inserting DNA at the DSB sites [50]. Although it is unclear how the LION CRISPR vector can tune DSB repair more toward HDR, the linear non-homologous DNAs in the LION vector might trigger the cellular DSB repair machineries and enhance DSB repair by HDR.
There is an increasing demand for CRISPR gene editing for various kinds of applications. Since the CRISPR–Cas9 system was harnessed for gene editing in 2012 [3], applications of CRISPR in gene editing have significantly increased. Modifications of both the Cas9 protein and the gRNA have dramatically expanded the scope of genetic manipulation applications. The LION method developed in this study will contribute to the available tool box of CRISPR to facilitate and simplify CRISPR gene-editing applications.
Materials and methods
Oligonucleotides
All oligonucleotides were ordered from Merck KGaA, Darmstadt, Germany. The sequences of all oligonucleotides used can be found in Tables S1 and S2.
Plasmids
The following plasmids were used in this study: lentiGuide-Puro (Addgene plasmid # 52963), pSpCas9(BB)-2A-Puro (PX459) (Addgene plasmid # 62988), pUC19 (Addgene plasmid # 50005), pTLR repair vector (Addgene plasmid # 64322), C-Check vector (Addgene plasmid # 66817), and SpCas9–RecA (Addgene plasmid #87263). pMAX-GFP (VDF-1012) was supplied by the Amaxa Nucleofector kit.
CRISPR/Cas9 gRNA design
All gRNAs used in this study were designed using the online CRISPR design tool “CRISPOR” (http://crispor.tefor.net/crispor.py). Selection of candidate gRNAs is in accordance with the following set of rules: (1) minimized off-target potential. There were at least three mismatches in the candidate gRNAs compared to elsewhere in the whole human genome reference (GRCh37/hg19); (2) predicted efficiency score (> 30) provided by CRISPOR; (3) no BsmBI restriction site (CGTCTC, an important feature for LION and GGA) and absence of poly-thymine (max 3) in the sense strand of guide sequences; (4) strong secondary structure avoided in the guide sequences (m-fold). All the chosen gRNAs guide sequences were started with a “G” to ensure the efficient transcription by U6 promotor. If not, an additional “G” was added to the 5′end of guide sequences. Golden Gate Assembly linkers were added to the 5′end of gRNA oligos (5′-CACC-3′ for sense strand and 5′-AAAC-3′ for antisense strand) before oligonucleotide synthesis.
LION ligation and gRNA expression plasmid preparation
To anneal gRNA oligos, two complementary gRNA oligos (100 μM) were mixed in 10 × NEB Buffer 2 (1 μL gRNA sense strand (SS, 100 µM), 1 μL antisense strand (AS, 100 µM), 2 μL 10 × NEB Buffer 2, and ddH2O to a total volume of 20 μL). The oligos were denatured at 95 °C for 5 min and ramped down to 25 °C at a ramping rate of − 5 °C/min. We used Golden Gate Assembly strategy to construct the LION products and gRNA expression plasmids. Briefly, the assembly reaction mixture includes × μL (see experimental condition) annealed gRNA, × ng (see experimental group and condition) lentiGuide-Puro vector, 1 μL T4 DNA ligase (ThermoFisher Scientific), 1 μL FastDigest Esp3I (BsmBI, ThermoFisher Scientific), and 2 μL 10 × T4 ligase buffer in a total volume of 20 μL. Golden Gate Assembly was performed in a thermal cycler using the following program: 10 cycles of 37 °C for 5 min and 22 °C for 10 min; 37 °C for 30 min; 75 °C for 15 min. The ligation product was stored at 4 °C or used immediately.
For generation of traditional gRNA expression plasmids, 1 µL ligation product was used to transform competent E. coli cells and PCR screenings were performed to find positive gRNA-insertion colonies. Then positive colonies were expanded for plasmid preparation. To optimize the amount of backbone plasmid for LION reaction, we tested the ligation of 100 ng, 200 ng, 500 ng, 1000 ng, 1500 ng, 2000 ng, 3000 ng, and 5000 ng backbones (lentiGuide-Puro) with 1 µL annealed gRNA oligo in the Golden Gate Assembly system. Subsequently, we tested different amounts of annealed gRNA oligos (1 µL, 2 µL, 3 µL, 5 µL, 8 µL) with 1000 ng lentiGuide-Puro backbone in the Golden Gate Assembly system. The ligated products were purified using the NucleoSpin Gel and PCR Clean-up Kit (Macherey–Nagel), eluted in 30 µL ddH2O and stored at 4 °C or used directly. Normally (for 1000 ng backbone ligated with 1 µL gRNA), the final concentration of purified LION product yield is approximately 30 ng/µL.
C-Check reporter vector construction
The original C-Check vector was digested with BsaI and the 6689 bp fragment was gel purified to be used as backbone. Fragments containing two gRNAs target sites were amplified by PCR and cloned into the C-Check backbone. All the primers for cloning of target sites are listed in Supplementary Table S2.
Cell culture and transfection
Human embryonic kidney 239T (HEK293T) cells, MJ cells, Skov-3 cells, Hela cells (ATCC), and HEK293 Traffic light reporter cells (TLR cells, it is a gift from Ralf Kuehn’s Lab) were used in this study. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (LONZA) supplemented with 10% fetal bovine serum (FBS) (Gibco), 1% GlutaMAX (Gibco), and penicillin/streptomycin (100 units penicillin and 0.1 mg streptomycin/mL) in a 37 °C incubator with 5% CO2 atmosphere and maximum humidity. All the cells were passaged every 2–3 days when reaching approximately 80–90% confluence. This was done by gently washing cells twice (equal volume as growth medium) with phosphate buffered saline (PBS, 1 ×) without calcium and magnesium, followed by cell detachment by 0.05% trypsin–EDTA treatment for 5 min at 37 °C.
All transfections were conducted with the X-tremeGENE 9 transfection reagent (Roche) in 24-well plates following the manufacturer’s instruction. Concisely, 60,000 cells, counted with nucleocounter NC-100 (ChemoMetec), were seeded into a 24-well plate 1 day before transfection. For each transfection, a total of 250 ng plasmid DNA (or LION products) and 0.75 µL X-tremeGENE 9 transfection reagent were mixed in 25 µL OptiMEM (Gibco) and incubated at room temperature for 15 min. The transfection mixture was distributed dropwise to the cells. For C-Check test, 62.5 ng plasmid/LION, 62.5 ng Cas9 vector (PX459) and 125 ng C-Check vector were mixed together and co-transfected into HEK293T cells; For T7EI and TIDE test, 125 ng plasmid/LION and 125 ng PX459 were co-transfected; For TLR test, 62.5 ng plasmid/LION, 125 ng pTLR repair vector and 62.5 ng of the different Cas9 expression vectors (SpCas9, SpCas9–RecA) were co-transfected into the TLR cell lines, respectively; For dual-cutting test, equal amounts of paired-plasmid/LION (62.5 ng + 62.5 ng) and 125 ng PX459 were co-transfected into HEK293T cells. As transfection control, pUC19 or GFP plasmid was used. The culture medium was changed 24 h after transfection and the cells were harvested 48 h later for further analysis.
Flow cytometry (FCM)
Cells in 24-well plates, dissociated with 100 µL 0.5% trypsin–EDTA, were suspended in 100 µL 5% FBS-PBS and transferred to a 96 deep-well plate on ice. Cells were spun down at 2000 rpm for 5 min and the supernatant was removed by gently inverting the plate. Then the cell pellets were re-suspended in 600 µl PBS and subjected to FCM analysis immediately. FCM was performed using a BD LSRFortessa (supported by the FACS CORE facility, Department of Biomedicine, Aarhus University) with at least 30,000 events collected for each sample. Data were analyzed using Flowjo software.
Cell lysis and polymerase chain reaction (PCR)
For cell lysis, the cells were disassociated with 100 µL 0.5% trypsin–EDTA and suspended in 200 µL culture medium. Then cells were spun down at 2000 rpm for 5 min and the supernatant was discarded. The cell pellet was lysed with 300 µL cell lysis buffer (50 mM KCl, 1.5 mM MgCl2, 10 mM Tris–Cl, pH 8.5, 0.5% Nonidet P40, 0.5% Tween and 400 µg/mL proteinase K). The cells were lysed at 65 °C for 30 min, followed by inactivation of proteinase K at 95 °C for 10 min in a thermal cycler. 1 µL of the cell lysate was used for the subsequent PCR test.
PCR reactions for CRISPR gRNA vector assembly screen were performed with the DreamTaq polymerase (Thermo Fisher Scientific). PCR reactions for C-Check vector construction, T7E1 assay and Sanger sequencing were performed with the high-fidelity platinum Pfx polymerase in the presence of 2 × enhancer solution (#11708013, Thermo Fisher Scientific).
T7 endonuclease I assay
Before T7EI assay, the DNA region harboring CRISPR gRNA target sites were amplified and gel tested to make sure that there were unique bands. All primers used for PCR amplification are listed in Supplementary Table S2. Then, 18 µL PCR products mixed with 2 µL NEB buffer 2 were denatured at 95 °C for 5 min and ramped down to 25 °C at − 5 °C/min. 0.2 µL T7 endonuclease I was added into the mixture and incubated at 37 °C for 45 min. Then the entire digestion product (20.2 µL) was tested in 2% agarose gel. Cells transfected with an EGFP plasmid were used as controls.
Sanger sequencing and TIDE analysis
All Sanger sequencing in this study was conducted using the Mix2Seq Kit in Eurofins Genomics (Munich, Germany). Sanger sequencing results were analyzed with Genious. TIDE (tracking indels by decomposition) analysis was conducted using the online tool “TIDE” (https://tide.nki.nl).
Statistics
All data are presented as mean ± standard deviation. Student’s T test and one-way analysis of variance (ANOVA) with Pearson correction were used for statistical analyses. All statistical analyses were conducted utilizing GraphPad Prism 7. p values less than 0.05 were considered statistically significant.
Abbreviations
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas9:
-
CRISPR-associated protein 9
- gRNA:
-
Guide RNA
- DSB:
-
DNA double-stranded break
- TIDE:
-
Tracking indels by decomposition
- C-Check:
-
A dual-fluorescent surrogate reporter system for checking CRISPR cleavage activity in vivo
- GGA:
-
Golden Gate Assembly
- LION:
-
LIgation of double-stranded gRNA oligos into CRISPR vector by GGA followed by nucleic acid purificatiON
- SpCas9-RecA:
-
Streptococcus pyogenes Cas9 fused to E. coli recombinant RecA protein
- TLR:
-
Traffic light reporter
- FCM:
-
Flow cytometry
References
Jansen R et al (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43(6):1565–1575
Cong L et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Jinek M et al (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821
Wang H, La Russa M, Qi LS (2016) CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem 85:227–264
Mali P et al (2013) RNA-guided human genome engineering via Cas9. Science 339(6121):823–826
Yang L et al (2015) Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science 350(6264):1101–1104
Niu D et al (2017) Inactivation of porcine endogenous retrovirus in pigs using CRISPR–Cas9. Science 357(6357):1303–1307
Lackner DH et al (2015) A generic strategy for CRISPR–Cas9-mediated gene tagging. Nat Commun 6:10237
Guo D et al (2017) Creating a patient carried Men1 gene point mutation on wild type iPSCs locus mediated by CRISPR/Cas9 and ssODN. Stem Cell Res 18:67–69
Cheng AW et al (2013) Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23(10):1163–1171
Liao HK et al (2017) In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 171(7):1495 e15–1507 e15
Ma H et al (2015) Multicolor CRISPR labeling of chromosomal loci in human cells. Proc Natl Acad Sci USA 112(10):3002–3007
Lin L et al (2018) Genome-wide determination of on-target and off-target characteristics for RNA-guided DNA methylation by dCas9 methyltransferases. Gigascience 7(3):1–19
Gaudelli NM et al (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551(7681):464–471
Komor AC et al (2017) Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T: A base editors with higher efficiency and product purity. Sci Adv 3(8):eaao4774
Henrik Devitt Møller LL, Xi X, Trine SP, Jinrong H, Luhan Y, Eigil K, Uffe BJ, Xiuqing Z, Xin L, Xun X, Jian W, Huanming Y, George MC, Lars B, Birgitte R, Yonglun L (2018) CRISPR-C: circularization of genes and chromosome by CRISPR in human cells. Nucleic Acids Res 2018:gky767
Jensen KT et al (2017) Chromatin accessibility and guide sequence secondary structure affect CRISPR–Cas9 gene editing efficiency. FEBS Lett 591(13):1892–1901
Bassett AR et al (2014) Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep 6(6):1178–1179
Wang T et al (2014) Genetic screens in human cells using the CRISPR–Cas9 system. Science 343(6166):80–84
Moreno-Mateos MA et al (2015) CRISPRscan: designing highly efficient sgRNAs for CRISPR–Cas9 targeting in vivo. Nat Methods 12(10):982–988
Gagnon JA et al (2014) Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One 9(5):e98186
Chari R et al (2015) Unraveling CRISPR–Cas9 genome engineering parameters via a library-on-library approach. Nat Methods 12(9):823–826
Lim Y et al (2016) Structural roles of guide RNAs in the nuclease activity of Cas9 endonuclease. Nat Commun 7:13350
Thyme SB et al (2016) Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nat Commun 7:11750
Chen Y et al (2017) Using local chromatin structure to improve CRISPR/Cas9 efficiency in zebrafish. PLoS One 12(8):e0182528
Smith JD et al (2016) Quantitative CRISPR interference screens in yeast identify chemical–genetic interactions and new rules for guide RNA design. Genome Biol 17:45
Liang P et al (2015) CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6(5):363–372
Ruan GX et al (2017) CRISPR/Cas9-mediated genome editing as a therapeutic approach for Leber congenital amaurosis 10. Mol Ther 25(2):331–341
Liang G et al (2016) Selection of highly efficient sgRNAs for CRISPR/Cas9-based plant genome editing. Sci Rep 6:21451
Brinkman EK et al (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42(22):e168
Zhou Y et al (2016) Enhanced genome editing in mammalian cells with a modified dual-fluorescent surrogate system. Cell Mol Life Sci 73(13):2543–2563
Zhang JH et al (2014) Improving the specificity and efficacy of CRISPR/CAS9 and gRNA through target specific DNA reporter. J Biotechnol 189:1–8
Yang Z et al (2015) Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res 43(9):e59
Rose JC et al (2017) Rapidly inducible Cas9 and DSB-ddPCR to probe editing kinetics. Nat Methods 14(9):891–896
Jewkes R et al (2015) Relationship between single and multiple perpetrator rape perpetration in South Africa: a comparison of risk factors in a population-based sample. BMC Public Health 15:616
Ran FA et al (2013) Genome engineering using the CRISPR–Cas9 system. Nat Protoc 8(11):2281–2308
Doench JG et al (2014) Rational design of highly active sgRNAs for CRISPR–Cas9-mediated gene inactivation. Nat Biotechnol 32(12):1262–1267
Chu VT et al (2015) Increasing the efficiency of homology-directed repair for CRISPR–Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33(5):543–548
Vad-Nielsen J et al (2016) Golden Gate Assembly of CRISPR gRNA expression array for simultaneously targeting multiple genes. Cell Mol Life Sci 73(22):4315–4325
Sentmanat MF et al (2018) A survey of validation strategies for CRISPR–Cas9 editing. Sci Rep 8(1):888
Vouillot L, Thelie A, Pollet N (2015) Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda) 5(3):407–415
Germini D et al (2017) A one-step PCR-based assay to evaluate the efficiency and precision of genomic DNA-editing tools. Mol Ther Methods Clin Dev 5:43–50
Chen X et al (2014) Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci Rep 4:7581
Paix A et al (2014) Scalable and versatile genome editing using linear DNAs with microhomology to Cas9 Sites in Caenorhabditis elegans. Genetics 198(4):1347–1356
Adikusuma F, Pfitzner C, Thomas PQ (2017) Versatile single-step-assembly CRISPR/Cas9 vectors for dual gRNA expression. PLoS One 12(12):e0187236
Zhen S et al (2017) Inhibition of long non-coding RNA UCA1 by CRISPR/Cas9 attenuated malignant phenotypes of bladder cancer. Oncotarget 8(6):9634–9646
Han J et al (2014) Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol 11(7):829–835
Zhang JP et al (2017) Efficient precise knockin with a double cut HDR donor after CRISPR/Cas9-mediated double-stranded DNA cleavage. Genome Biol 18(1):35
Zheng Q et al (2014) Precise gene deletion and replacement using the CRISPR/Cas9 system in human cells. Biotechniques 57(3):115–124
Richardson CD et al (2016) Non-homologous DNA increases gene disruption efficiency by altering DNA repair outcomes. Nat Commun 7:12463
Certo MT et al (2011) Tracking genome engineering outcome at individual DNA breakpoints. Nat Methods 8(8):671–676
Yanik M et al (2018) Development of a reporter system to explore MMEJ in the context of replacing large genomic fragments. Mol Ther Nucleic Acids 11:407–415
Lin L et al (2017) Fusion of SpCas9 to E. coli Rec A protein enhances CRISPR–Cas9 mediated gene knockout in mammalian cells. J Biotechnol 247:42–49
Kuhar R et al (2014) Novel fluorescent genome editing reporters for monitoring DNA repair pathway utilization at endonuclease-induced breaks. Nucleic Acids Res 42(1):e4
Gomez-Cabello D et al (2013) New tools to study DNA double-strand break repair pathway choice. PLoS One 8(10):e77206
Davis L, Maizels N (2014) Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc Natl Acad Sci USA 111(10):E924–E932
Acknowledgements
We thank Fred Dubee for his generous help with language editing. This project was supported by the Sanming Project of Medicine in Shenzhen (SZSM201612074), BGI-Qingdao, and BGI-Shenzhen. Y.L was partially supported by the Danish Research Council for Independent Research (DFF–1337-00128), the Sapere Aude Young Research Talent Prize (DFF-1335-00763A), and Aarhus University Strategic Grant (AU-iCRISPR). L.L. was supported by the Lundbeck Foundation (R219-2016-1375) and the DFF Sapere Aude Starting Grant (8048-00072A).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiang, X., Luo, L., Nodzyński, M. et al. LION: a simple and rapid method to achieve CRISPR gene editing. Cell. Mol. Life Sci. 76, 2633–2645 (2019). https://doi.org/10.1007/s00018-019-03064-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03064-x