
Abstract
Adipose tissue (AT) is an extramedullary reservoir of normal hematopoietic stem cells (HSCs). Adipocytes prevent the production of normal HSCs via secretion of inflammatory factors, and adipocyte-derived free fatty acids may contribute to the development and progression of leukemia via providing energy for leukemic cells. In addition, adipocytes are able to metabolize and inactivate therapeutic agents, reducing the concentrations of active drugs in adipocyte-rich microenvironments. The aim of this study was to detect the role of adipocytes in the progression and treatment of leukemia. Relevant literature was identified through a PubMed search (2000–2018) of English-language papers using the following terms: leukemia, adipocyte, leukemic stem cell, chemotherapy, and bone marrow. Findings suggest the striking interplay between leukemic cells and adipocytes to create a unique microenvironment supporting the metabolic demands and survival of leukemic cells. Based on these findings, targeting lipid metabolism of leukemic cells and adipocytes in combination with standard therapeutic agents might present novel treatment options.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bone marrow (BM) microenvironment contains several cells, including macrophages, endothelial cells, osteoclasts, osteoblasts, and adipocytes, which contribute to the regulation of hematopoiesis and preservation of hematopoietic stem cells (HSCs) through cell–cell contact, secretion of growth factors and cytokines [1, 2]. Adipocytes are among the most important components of BM microenvironment that are derived from mesenchymal stem cells (MSCs) [3]. The quantity of bone marrow adipocytes (BMAs) is increased with age. Adipocytes are indeed active cells in BM as they actively store free fatty acids (FFAs) and secrete adipokines [4, 5]. In addition, studies have shown that these cells induce the development and progression of tumors, leading to metastasis and resistance to chemotherapy through interaction with other stromal cells. For example, the development and progression of hematological malignancies including, i.e., multiple myeloma (MM) and acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia (CLL) are closely correlated with BM microenvironment in which BMAs play an important role. Generally, BMAs are an important element in BM that can affect their adjacent cells because of autoparacrine effect [6]. Remodeling of adipocytes (e.g., morphological changes) may indicate pathological conditions in the body. The size of adipocytes shrinks around AML cell line, and they affect the metastasis and growth of AML cells. The change in morphology is not only a function of fatty acids (FAs) release from adipocytes to adjacent cells but is dependent upon a series of reactions catalyzed by increasing expression of adipose triglyceride lipase (ATGL) and hormone sensitive lipase (HSL) genes [7]. These changes enable adipocytes to provide more energy to induce the proliferation of tumor cells, indicating the essential role of lipolysis in the alteration of adipocytes morphology [8, 9]. Furthermore, the inflammatory factors and growth differentiation factor 15 (GDF15) released by tumor cells are involved in morphological remodeling of BMAs, and their expression levels are closely related to the size of BMAs. For instance, GDF15, a transforming growth factor-beta (TGF-β) is highly expressed in leukemia cells and leads to the shrinkage of adipocytes [7]. Interestingly, the increase of FFA levels in culture system causes a faster proliferation rate of ALL cell lines in their co-culture with adipocytes. These results indicate that tumor cells and leukemic cells utilize lipolytic pathway to acquire FAs in both solid tumors and leukemia, which increases their proliferation [10, 11]. In addition, the size and number of small adipocytes stimulate the proliferation of AML cells, which can be associated with a poor prognosis of patients [12]. In summary, although adipocytes seem to be negative regulators of hematopoietic microenvironment, they can be affected by pathological changes in the context of diseases (including cardiac failure and osteoporosis (similar to other cells [13, 14]. Nowadays, knowledge of BMAs as well as interactions between them and tumor cells gradually deepens and greatly contributes to new therapies.
Mesenchymal stem cells to adipocyte axis
BM MSCs are multipotent cells with self-renewal capacity that can differentiate into adipocytes, chondrocytes, and osteocytes [15]. The differentiation of adipocytes from MSCs is a complex process involving morphological changes, cease of cell growth factor, expression of several lipogenic enzymes, extensive lipid accumulation, and sensitivity to the majority or all of the key hormones (growth hormone, glucocorticoids) [16]. A feature of adipocytes differentiation is the activation of CCAAT/EBP β and CCAAT/EBP γ transcription factors, which stimulate the expression of PPAR2β adipocyte differentiation gene, in turn stimulating the expression of genes involved in metabolism [fatty acid synthase (FAS), sterol regulatory element-binding protein-1 (SREBP-1), stearoyl-CoA desaturase (SCD), and leptin] [17, 18]. In this context, Vicent Lopez et al. examined MSCs of ALL patients at baseline and during treatment, and suggested that MSCs of these patients show increased adipogenic differentiation potential, including increasing expression of adipogenic genes such as CCAAT/enhancer binding protein/peroxisome proliferator-activated receptor γ (CEBP/PPAR γ) relative to MSCs of healthy people [19]. Wnt signaling pathway also regulates the differentiation of adipocytes. Leukemia inhibitory factor (LIF) that belongs to interleukin-6 (IL-6) family of cytokines induces autophosphorylation of Janus kinase (JAKs), leading to the activation of mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase, and protein kinase B (PI3K/Akt) pathways as well as suppressing the expression of Wnt signaling molecules, which is involved in early and late differentiation stages of adipocytes [20, 21]. This suggests that LIF probably exerts its effect on the differentiation of adipocytes through negative regulation of Wnt signaling, which reduces the differentiation of adipocytes [22]. Although evidence suggests that protein kinase A (PKA) signaling stimulates the differentiation of adipocytes [23, 24], there are reports indicating that PKA signaling can have an opposite effect on adipocytes differentiation [25, 26]. Connective tissue growth factor (CTGF) is a member of CCN (CYR61 (cysteine-rich angiogenic protein 61 or CCN1), CTGF (connective tissue growth factor or CCN2), and NOV (nephroblastoma overexpressed or CCN3)) family of proteins, the expression of which is regulated with transforming growth factor beta (TGF-ß) expression in fibroblasts, leading to adipogenic differentiation of BM MSCs and stimulating the affinity of leukemia cells and their growth in BM niche [27]. Given that MS-derived MSCs of AML patients show reduced adipocyte differentiation capacity, CTGF expression is likely to increase in leukemia cells and inhibit the ability of MSCs to differentiate into adipocytes [28]. In addition to TGF-ß pathway, Notch signaling and hypoxia-inducible factor 1-alpha (HIF-1α) transcription factor are involved in differentiation regulation of MSCs in AML patients. Briefly, a number of genes, including Jagged-1 and TGF-β, are involved in the commitment of adipocytes, which are affected in MSCs of AML patients [29, 30]. BMAs have dual functional roles on HSCs. In this regards, BMAs not only are considered as negative regulators of hematopoietic microenvironments but also they contribute to the maintenance and survival control of HSCs along with other cells [31,32,33], acting as a fuel source for HSCs through the release of lipids. Adipocytes can convey FFAs to HSCs, which results in increased survival and proliferation of AML blasts [34]. Other studies have shown that the adipogenic differentiation of MSCs in BM is associated with increased incidence of leukemia, so that BM-derived MSCs stimulate the survival of leukemia cells by altering the metabolism from pyruvate oxidation to fatty acid oxidation (FAO), uncoupling of mitochondrial oxidative phosphorylation, and regulating the anti-apoptotic mechanism in these cells [35, 36].
Overall, the mechanisms emerging for the interaction between BMAs and hematopoiesis include differentiation regulation of MSCs, supply of FFAs as a fuel source for hematopoiesis, and release of soluble adipocyte-specific intermediates that directly and indirectly affect systemic metabolism [34]. Furthermore, CD36+ leukemic stem cells are significantly protected from chemotherapy by adipose tissue niche. Fatty acid translocates or CD36 + is considered to be a marker of poor prognosis in AML patients [37]. Observations have shown that leukemic stem cells (LSCs) take refuge in adipose tissue and use it as a niche to support their metabolism. LSCs location in adipose niche creates a pro-inflammatory phenotype for CML cells, resulting in the secretion of cytokines that increase the oxidation of fatty acids in LSCs via increasing lipolysis. LSCs use these FFAs as a fuel source, leading to LSCs quiescence and resistance to chemotherapy [38].
Adipocyte as the calm before the storm in leukemia niche
Chemokines
BMAs release a series of inflammatory adipokines such as leptin, tumor necrosis factor alpha (TNF-α), and IL-6, as well as an anti-inflammatory adipokine called adiponectin, which play a role in the proliferation, migration, and metastasis of cancer cells. Therefore, BMAs are likely to provide a higher proliferation capacity for AML cells relative to FAs alone. In other words, cytokines and chemokines secreted by BMAs as well as fatty acids might induce proliferation of AML cells [39, 40]. Adiponectin, which is exclusively secreted by adipocytes, is an important regulator of energy metabolism and hematopoiesis. Adiponectin concentration increases with energy loss, suppressing the expression of adhesion molecules in vascular endothelial cells as well as production of cytokines, which inhibits inflammatory processes through two mechanisms: control of macrophage precursors and suppression of adult macrophage function. Hence, increased adiponectin levels are associated with lower levels of inflammatory markers [41, 42]. Moreover, as a hormone released by adipocytes, adiponectin induces apoptosis by activating caspases and suppressing angiogenesis [43]. In a study by Yokota et al. on adiponectin function in hematopoiesis, it was found that adiponectin inhibits the proliferation of myeloid series and induces apoptosis in myelomonocytic leukemia cells [44]. Adiponectin activates PKA, which reduces Akt activity but increases AMP-activated protein kinase (AMPK) activity. Furthermore, adiponectin-induced apoptosis inactivates PKA/AMPK pathway and increases the expression of acetyl CoA carboxylase (ACC) enzyme required for lipolysis, leading to cell cycle arrest and apoptosis of MM cells through this pathway. Inhibition of AMPK-dependent activity in these cells suppresses the ability of adipocytes to induce apoptosis [45, 46]. It can be concluded that in general the ability of adiponectin to reduce the survival of MM cells is mediated by suppression of lipolysis.
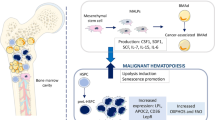
Simultaneous reduction in adiponectin and increase in leptin levels are associated with increasing risk of hematological malignancies [47, 48]. As a cytokine regulating fat metabolism, leptin is secreted in high levels during the differentiation of BMAs. Leptin receptors (OB-Rs) function through their specific receptors on cell membrane, leading to the induction of proliferation and differentiation in a variety of hematopoietic cells [33, 49]. Leptin induces and increases the survival of primary leukemia cells in AML patients [50, 51]. It also plays a role in pathophysiology of acute promyelocytic leukemia (APL), and APL cells express a high level of leptin receptor (OB-R). Leptin secretion with BMAs in the vicinity of leukemia cells can play an important role in the proliferation and survival of APL cells through a paracrine interaction in BM microenvironment. Additionally, leptin produced from adipocytes inhibits the apoptosis of APL cells through phosphorylation of signal transducer and activator of transcription 3 (STAT3), activation of MAPK pathway as well as interaction with leptin/OB-R, which requires direct cell–cell contact [52]. Leptin and its isomers are also expressed in high levels by AML cells, leading to the proliferation of AML cell lines (e.g., HEL, MO7E, and TF-1) and blasts from primary AML patients [53, 54]. Furthermore, leptin increases the production of cytokines in AML blasts, and overexpression of leptin in AML and ALL suggests an increase in adipose tissue content in leukemic BM. In other words, leptin stimulates the proliferation of leukemia cells by provoking angiogenesis, and treatment with anti OB-R in these patients reduces the rate of angiogenesis [49]. Adipocytes generally facilitate leukemia BM engraftment by stimulating the secretion of leptin and stromal cell-derived factor-1 alpha (SDF-1α). It seems that leptin and SDF-1α play an essential role in leukemogenesis and actively stimulate the development of leukemia in adipocyte-enriched BM niche [27]. SDF-1α is a chemoattractant for cells of lymphoid origin (including hematopoietic cells and leukemia cells) that is expressed in adipocytes residing in adipose tissue. Leukemia cells migrate to adipose tissue under the regulation of SDF-1α secreted by adipocytes, which binds the C-X-C chemokine receptor type 4 (CXCR4) receptor and causes intracellular changes, including cytoskeletal reorganization as well as activity of integrin and adhesion molecules [55, 56]. Research has indicated that the expression of OB-Rs in chronic myeloid leukemia (CML) patients is downregulated and that the gene of this receptor is upregulated in the chronic phase of the disease [57]. At the onset of disease, BMAs partially inhibit the expression of malignant leukemia clones. Cytokines produced by leukemia cells induce lipolysis of BM adipocytes. Then, the polyunsaturated fatty acid (PUFA) n-3 released by adipocytes disrupts the proliferation and survival of CML cells by inhibiting the PI3k pathway. However, this effect is rapidly inhibited by the leptin secreted by adipocytes, which increases lipolysis of adipocytes and protects CML cells from apoptosis by activating the PI3k pathway [58]. Various studies also show the involvement of leptin and its receptors in MM patients. The level of leptin is increased in MM patients, although it is not related to the progression of disease. However, a number of studies have shown that the expression of OB-R in MM cells can indicate the likelihood of patients’ response to treatment (Fig. 1) [40, 59].

The model of interaction between adipocytes and leukemic cells. BMAs secrete inflammatory adipokines (leptin, TNF-αand IL-6) and anti-inflammatory adipokines (adiponectin) that affect the leukemic cells residing in adipose tissue. Leukemic cells express the FFA scavenger receptor CD36+. Upregulation of CD36+ stimulates lipolysis with cytokine secretion and activates lipase that promotes the release of FFAs and increases FAO in leukemic cells. FFAs as energy source contribute to leukemic cells growth and survival and eventually lead to drug resistance. HSCs hematopoietic stem cells, IL-6 Interleukin-6, TNF-α tumor necrosis factor-alpha, ER endoplasmic reticulum, FAO free acid oxidation, CD36 cluster of differentiation 36, CCL2 C-C motif chemokine ligand 2, CXCL12 C-X-C motif chemokine 12, AKT protein kinase B, PI3K phosphoinositide-3-kinase, JAK Janus kinase, STAT3 signal transducer and activator of transcription 3, FFA free fatty acid, mTOR mammalian target of rapamycin, MIP 1 α/β macrophage inflammatory protein-1 alpha/beta, Bcl-2 B-cell lymphoma 2, PIM2 pim-2 proto-oncogene, serine/threonine kinase
Energy
Adipocytes support the survival and growth in various types of tumor cells through the induction of mitochondrial metabolism and high-energy lipid transfer [35]. Tumor cells increase glucose uptake but reduce the level of oxidized glucose in Krebs cycle, and are thus more dependent upon β-oxidation of FAs [60]. In general, the main function of adipocytes is to store energy in the form of triglycerides, which can be broken into glycerol and FFA to be released in the lipolysis process [61]. Recently, AML cells have been shown to alter the function of non-malignant adipocytes, which provide FFAs as a fuel source and lead to the stimulation of FAO in these cells as well as growth stimulation through the transfer of lipids [8]. Adipocytes also support the survival and proliferation of tumor cells, especially AML blast cells. In this way, AML cells induce the lipolysis of triglycerides stored in BMAs and alter their function [8, 38]. Afterwards, FFAs in adipocytes are transferred to AML blasts in a fatty acid-binding protein-4 (FABP4) chaperon protein dependent process. Proximity to adipocytes results in the upregulation of FABP4 within the blasts, which is used to carry the adipocyte-derived FAs into mitochondria of the cell [8, 62,63,64]. AML mitochondria use FAs as a substrate for ß-oxidation to produce the energy required for the growth and proliferation of leukemia cells [65, 66] (Fig. 1). In some cases, reduced AML survival is due to the inability of FAs to be secreted from the adipocytes that show a lower expression of FABP4. FABP4s are likely to activate peroxisome proliferator-activated receptor gamma (PPARγ) ligands within the cell in the process of apoptosis induced by FAs [67]. Increased levels of FFAs enhance the inflammatory status of leukemia cells and act as a fuel for metabolic processes of leukemia cells in adipose and hematopoietic tissues, as well as increasing the oxidation of FAs for LSCs [68]. According to studies, gonadal adipose tissue (GAT) in leukemia acts as a reservoir for LSCs and is responsible for resistance to chemotherapy, so that LSCs easily home to CD36+ GATs and in this case show a high tendency towards the microenvironment containing adipocytes to have maximal and easy access to FAs [38, 69, 70].
Lipoprotein lipase (LPL) is typically expressed in adipocytes. According to reports, LPL expression is obviously increased in CLL cells, which is associated with a poor prognosis and invasive state of the disease. LPL causes cellular uptake of lipoproteins and leads to hydrolysis of triglycerides into FFAs [71]. CLL cells store lipids in their cytoplasmic vacuoles and use FFAs to generate energy through oxidative phosphorylation [72]. FFAs bind to PPARα and activate the transcription of enzymes necessary for oxidative phosphorylation. Specifically, PPARα is overexpressed in CLL cells and its level is related to progression of the disease [73].
A study by Tung et al. showed that FFAs could lead to a higher rate of metabolism in CLL as well as causing resistance to cytotoxicity [74]. Tucci et al. also found that ALL cells stimulate lipolysis in adipocytes and use adipocyte-derived FFAs to supplement de novo lipogenesis and proliferation [10]. Since adipocytes can supply FFAs as a fuel source for cancer cells, blockage of lipolysis and FFAs oxidation, as well as control of FFAs transfer using FABP4 inhibitor between BM adipocytes and leukemic blasts [8], it can be concluded that FABP4 inhibitors may be considered as a therapeutic target reducing tumor survival and increasing the survival of leukemia patients.
Adipocytes and chemotherapy challenge
Adipose tissue is one of the major organs that metabolizes and inactivates drugs, thereby reducing the concentration of active chemotherapy agents in adipocyte-rich microenvironments, such as the bone marrow [75]. BM adipocyte niche is a dynamic tissue that is increased after various injuries such as irradiation or chemotherapy. Since adipocytes modify the pharmacokinetics of chemotherapy and provide more energy to stimulate the proliferation of tumor cells [76], it can be suggested that they contribute to the survival of tumor cells by suppressing the antitumor effect of chemotherapy agents as well as secreting adipokines. The effect of adipokines, especially inflammatory cytokines and chemokines, on tumor growth and drug resistance is a potential new target for therapeutic interventions. Furthermore, as fat deposits shelter ALL cells during chemotherapy, the microenvironment of adipocytes is considered as a protective niche for these cells [77, 78]. On the other hand, treatment of cancer causes a severe increase in total fat in the body. However, these changes reduce the cytotoxic activity of chemotherapy and lead to the emergence of drug-resistant tumor cells, which increases the risk of treatment failure [79]. These findings are of particular importance during leukemia treatment. While several studies on drug resistance have focused on gene mutations in leukemia, some studies have found that leukemia microenvironments play an important role in resistance to chemotherapy. Since adipocytes protect ALL cells from chemotherapy and absorb chemotherapy agents, these cells may migrate to adipose tissue and obtain a survival advantage. ALL cells in this environment remain in a dormant state or receive the survival signal that enables them to resist chemotherapy, which can contribute to an increase in recurrence rates [78].
Today, neoplastic diseases are treated using chemotherapy, radiation therapy, or both, which increase BM adipocytes and reduce hematopoiesis via induction of toxicity. Increasing adipogenesis in BM following chemotherapy or radiotherapy stimulates hematopoietic regeneration and can be beneficial [80]. Regular doses of chemotherapy drugs mostly eliminate HSCs, while several inflammatory factors secreted by these cells regulate adipogenic differentiation and indirectly prevent recurrence but are not potent enough to affect stromal cells [81].
Several therapeutic strategies targeting BM such as induction of systemic adiponectin levels and blockage of accelerated adipogenesis develop promising approaches to target tumor cells. Furthermore, targeting lipolysis and oxidation, which can eliminate the nutrients of tumor cells, is also effective in this process [42]. The strategies blocking the migration of ALL cells to adipose tissue may improve the patient’s outcome. In a study, it was found that CXCR4 antagonists inhibited the affinity of ALL cells to adipose tissue, which could have a beneficial effect in therapy [78].
Chemotherapy is still crucial to prevent the recurrence of AML in patients. The induction of chemotherapy improves a majority of AML patients; however, recurrence after recovery is the main factor of mortality in these patients [82]. The size of adipocytes is decreased in AML-complete remission (CR) patients, and adipocyte content is inversely related to relapse free survival (RFS). The reduced size of adipocytes is related to GDF15, which is secreted by hematopoietic cells following chemotherapy and inhibition of adipogenesis. This is probably an inflammatory response or a strategy developed by chemotherapy; therefore, in general, increasing content of adipocytes is associated with shorter RFS and an increased risk of recurrence [12, 81].
The contribution of adipocytes to resistance against chemotherapy was first described by Behan and colleagues who found that adipocytes protect ALL cells against vincristine, nilotinib, and daunorubicin, reducing the apoptosis of leukemia cells and increasing cell cycle rate. Adipocytes also increase the expression of pro-survival signals, which may alter apoptotic equilibrium towards survival [77]. The main target of this adipocyte-mediated effect is related to the upregulation of Bcl-2 (B cell lymphoma 2) and PIM2 (pim-2 proto-oncogene, serine/threonine kinase) as well as an increase in Bad phosphorylation [83, 84]. In particular, adipocytes metabolize daunorubicin into a less toxic metabolite and allow adjacent ALL cells to evade cytotoxicity from daunorubicin. It has also been reported that adipocytes protect ALL cells from oxidative stress induced by drugs or irradiation. ALL cells induce intracellular reactive oxygen species (ROS) production and lead to oxidative stress in adipocytes, and the oxidative stress response leads to secretion of factors protecting ALL cells from daunorubicin [85]. Generally, adipocytes reduce the accumulation of chemotherapy agents in ALL cells and eliminate them from leukemia environment by absorbing these agents. Adipocytes also metabolize chemotherapy agents, and their enzymes alter the structure of chemotherapy molecules, which results in lower toxicity in ALL cells [75].
Adiponectin levels are increased in CML patients after treatment with imatinib regardless of age and sex, so that its levels are tripled 3 months after treatment. The therapeutic dose of imatinib stimulates the differentiation of BM-derived adipocytes via upregulation of PPARγ and CCAAT/enhancer binding protein α [5, 86].
A high dose of synthetic glucocorticoids (GCs) such as dexamethasone is among the most effective treatments for CLL patients [87]. GCs inhibit glucose metabolism and induce oxidation of FAs in several tissues. According to studies, GCs increase the dependence of CLL cells on the oxidation of FAs by changing the expressions of PPARα and pyruvate dehydrogenase kinase isoform 4 (PDK4), and the PPARα responsible for regulating the oxidation of FAs is increased by dexamethasone [74]. GCs may also play a role in the treatment of ALL. Leptin secretion is stimulated by GCs, and hyperleptinemia has been observed in ALL cells during and after the treatment, although the expression of adiponectin is reduced in these cells [51, 88].
L-asparaginase, which breaks down asparagine (ASN) and glutamine (GLN), is the first line of treatment for ALL [89]. ALL cells are dependent upon ASN and GLN for survival and proliferation. Adipocytes protect ALL cells from L-asparaginase (ASNase) mainly via GLN secretion. Given that adipose tissue is one of the most important sources of GLN and secretes it into the interstitial fluid, and since ALL cells penetrate into adipose tissue, this tissue is a likely shelter to protect ALL cells from ASNase activity, which occurs after chemotherapy in BM [90]. Therefore, it can be generally stated that adipocytes may have an active role in the survival of leukemia cells against various chemotherapy agents.
Obesity is amongst the main risk factors contributing to poor survival and recurrence rates of ALL and AML [91, 92]. Although localized marrow adipocytes are considered as a supportive factor of leukemic cell proliferation and chemo-resistance, it is suggested that reduced marrow adipocyte content is considered as a good prognostic factor in AML patients during remission. Increased expression and secretion of GDF15 by marrow hematopoietic cells after chemotherapy inhibited MSCs adipogenesis [92]. It can be concluded that obesity and excess adipose tissue can be a preservative pool for LSCs´ proliferation and survival, which make LSCs refractory to chemotherapy. Therefore, reduced marrow adipocyte volume would be a potential therapeutic strategy against leukemia [81, 92].
Discussion
Nowadays, the focus of studies on neoplastic niche indicates the key role of intracellular signals in cell-to-cell communication of BM microenvironment and eventual response of leukemia cells to drugs. BM microenvironment is involved in leukemogenesis by increasing adhesion of leukemia cells as well as providing growth and suppression factors of leukemic niche immune system [35]. Therefore, BMAs, which are considered as a protective niche for leukemia cells that play an active role in supporting neoplastic cells, can be considered as an appropriate therapeutic option for limiting the growth of leukemia cells. Although some studies have suggested BMAs merely as negative regulators of hematopoiesis [32], it has recently been shown that they can exert a dual role on the function of HSCs [33]. In confirmation of this recent finding, it has been indicated that BMAs can apply a wide range of positive and negative effects on HSCs and hematopoietic progenitors by secreting various factors such as cytokines and adipokines [93,94,95]. For example, in addition to inhibitory effect on hematopoiesis, BMAs can help preserve and maintain HSCs along with other cells in the niche [31, 33]. BMAs release factors such as TNF-α and adiponectin that impair the proliferation of normal hematopoietic cells but contribute to the growth of malignant cells (e.g., MM cells) by inhibiting apoptosis, proliferation, and migration [42, 77]. AML blasts show high growth and proliferation rates in adipocyte-rich environments, and evidence has shown that AML cells rely on adipocytes for survival and proliferation within BM [8]. Studies have also shown that several factors contribute to obesity and recurrence in ALL, including adipocyte secretion, oxidative stress response, and pharmacokinetic changes in chemotherapeutic agents that induce drug resistance [61, 85]. This matter has been of interest for researchers given the role of adipose tissue as a main reservoir of normal HSCs as well as being a poor prognosis factor in obese patients [96]. Such evidence supports the hypothesis that adipose tissue contributes to the protection of cancer cells and the relapse of disease. Pharmacokinetic changes of chemotherapy agents following excessive adiposity, accumulation of lipophilic chemotherapies in adipose tissue that increases the distribution volume and decreases the exposure of cancer cells to chemotherapy factors, increasing accumulation of fat in BM, and excessive increase in body fat during leukemia treatment are in favor of the above hypothesis [75]. It should be noted that a number of researches have shown that leukemic cells increase TGF-β expression and inhibit adipogenic differentiation of mesenchymal BM cells, preventing the formation of new BM adipose tissue by disrupting BM adipocyte niche [29, 97]. Perhaps this effect is only exerted on nascent adipogenesis that may have little significance in the development of leukemia, and the older adipose tissue is the most crucial factor behind the support of neoplastic cells.
In the end, adipocytes change the apoptotic balance of leukemia cells towards survival, increasing the expression of pro-survival signals that reduce cytotoxic activity of chemotherapy agents, lead to emergence of resistant tumor cells, and increase the risk of treatment failure [75, 96].
References
Askmyr M, Quach J, Purton LE (2011) Effects of the bone marrow microenvironment on hematopoietic malignancy. Bone 48(1):115–120
Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H et al (2014) Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab 20(2):368–375
Wöhrer S, Rabitsch W, Shehata M, Kondo R, Esterbauer H, Streubel B et al (2007) Mesenchymal stem cells in patients with chronic myelogenous leukaemia or bi-phenotypic Ph+ acute leukaemia are not related to the leukaemic clone. Anticancer Res 27(6):3837–3841
Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I (2014) Bone marrow fat: linking adipocyte-induced inflammation with skeletal metastases. Cancer Metastasis Rev 33(2–3):527–543
Fitter S, Vandyke K, Schultz CG, White D, Hughes TP, Zannettino AC (2010) Plasma adiponectin levels are markedly elevated in imatinib-treated chronic myeloid leukemia (CML) patients: a mechanism for improved insulin sensitivity in type 2 diabetic CML patients? J Clin Endocrinol Metab 95(8):3763–3767
Johrer K, Ploner C, Thangavadivel S, Wuggenig P, Greil R (2015) Adipocyte-derived players in hematologic tumors: useful novel targets? Expert Opin Biol Ther 15(1):61–77
Lu W, Wan Y, Li Z, Zhu B, Yin C, Liu H et al (2018) Growth differentiation factor 15 contributes to marrow adipocyte remodeling in response to the growth of leukemic cells. J Exp Clin Cancer Res 66(1):37
Shafat MS, Oellerich T, Mohr S, Robinson SD, Edwards DR, Marlein CR et al (2017) Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 129(10):1320–1332
Feldman BJ, Streeper RS, Farese RV, Yamamoto KR (2006) Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects. Proc Natl Acad Sci USA 103(42):15675–15680
Tucci J, Sheng X, Mittelman SD (2014) Acute lymphoblastic leukemia cells stimulate adipocyte lipolysis and utilize adipocyte-derived free-fatty acids for proliferation. AACR. https://doi.org/10.1158/1538-7445
Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K (2013) Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res 52(4):585–589
Lu W, Weng W, Zhu Q, Zhai Y, Wan Y, Liu H et al (2018) Small bone marrow adipocytes predict poor prognosis in acute myeloid leukemia. Haematologica. 103(1):e21–e24
Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank A-M, Bocian C et al (2017) Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell 20(6):771–846
Scheller EL, Rosen CJ (2014) What’s the matter with MAT? Marrow adipose tissue, metabolism, and skeletal health. Ann N Y Acad Sci 1311:14–30
Ohishi M, Schipani E (2010) Bone marrow mesenchymal stem cells. J Cell Biochem 109(2):277–282
Torii I, Morikawa S, Nakano A, Morikawa K (2003) Establishment of a human preadipose cell line, HPB-AML-I: Refractory to PPARγ-mediated adipogenic stimulation. J Cell Physiol 197(1):42–52
Falconi D, Oizumi K, Aubin JE (2007) Leukemia inhibitory factor influences the fate choice of mesenchymal progenitor cells. Stem Cells 25(2):305–312
Reiter SS, Halsey CH, Stronach BM, Bartosh JL, Owsley WF, Bergen WG (2007) Lipid metabolism related gene-expression profiling in liver, skeletal muscle and adipose tissue in crossbred Duroc and Pietrain pigs. Comp Biochem Physiol Part D 2(3):200–206
Vicente Lopez A, Vazquez Garcia MN, Melen GJ, Entrena Martinez A, Cubillo Moreno I, Garcia-Castro J et al (2014) Mesenchymal stromal cells derived from the bone marrow of acute lymphoblastic leukemia patients show altered BMP4 production: correlations with the course of disease. PLoS One 9(1):e84496
Ikeda S, Itoh S, Yamamoto Y, Yamauchi Y, Matsushita K, Naruse H et al (2016) Developmental stage-dependent effects of leukemia inhibitory factor on adipocyte differentiation of murine bone marrow stromal cells. Cell Biochem Biophys 74(1):11–17
Hogan JC, Stephens JM (2005) Effects of leukemia inhibitory factor on 3T3-L1 adipocytes. J Endocrinol 185(3):485–496
Chen X, Hausman BS, Luo G, Zhou G, Murakami S, Rubin J et al (2013) Protein kinase inhibitor gamma reciprocally regulates osteoblast and adipocyte differentiation by downregulating leukemia inhibitory factor. Stem Cells 31(12):2789–2799
Fox KE, Fankell DM, Erickson PF, Majka SM, Crossno JT Jr, Klemm DJ (2006) Depletion of cAMP-response element-binding protein/ATF1 inhibits adipogenic conversion of 3T3-L1 cells ectopically expressing CCAAT/enhancer-binding protein (C/EBP) alpha, C/EBP beta, or PPAR gamma 2. J Biol Chem 281(52):40341–40353
Petersen RK, Madsen L, Pedersen LM, Hallenborg P, Hagland H, Viste K et al (2008) Cyclic AMP (cAMP)-mediated stimulation of adipocyte differentiation requires the synergistic action of Epac- and cAMP-dependent protein kinase-dependent processes. Mol Cell Biol 28(11):3804–3816
Li F, Wang D, Zhou Y, Zhou B, Yang Y, Chen H et al (2008) Protein kinase A suppresses the differentiation of 3T3-L1 preadipocytes. Cell Res 18(2):311–323
Li H, Fong C, Chen Y, Cai G, Yang M (2010) Beta-adrenergic signals regulate adipogenesis of mouse mesenchymal stem cells via cAMP/PKA pathway. Mol Cell Endocrinol 323(2):201–207
Battula VL, Chen Y, Cabreira Mda G, Ruvolo V, Wang Z, Ma W et al (2013) Connective tissue growth factor regulates adipocyte differentiation of mesenchymal stromal cells and facilitates leukemia bone marrow engraftment. Blood 122(3):357–366
Battula VL, Le PM, Sun JC, Nguyen K, Yuan B, Zhou X et al (2017) AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2:13
Boyd AL, Reid JC, Salci KR, Aslostovar L, Benoit YD, Shapovalova Z et al (2017) Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol 19(11):1336
Takam Kamga P, Bassi G, Cassaro A, Midolo M, Di Trapani M, Gatti A et al (2016) Notch signalling drives bone marrow stromal cell-mediated chemoresistance in acute myeloid leukemia. Oncotarget 7(16):21713–21727
Mattiucci D, Maurizi G, Izzi V, Cenci L, Ciarlantini M, Mancini S et al (2018) Bone marrow adipocytes support hematopoietic stem cell survival. J Cell Physiol 233(2):1500–1511
Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ (2009) Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460(7252):259–263
Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O (2017) Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol 19(8):891–903
Veldhuis-Vlug AG, Rosen CJ (2018) Clinical implications of bone marrow adiposity. J Intern Med 283(2):121–139
Tabe Y, Yamamoto S, Saitoh K, Sekihara K, Monma N, Ikeo K et al (2017) Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res 77(6):1453–1464
Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B et al (2010) Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 120(1):142–156
Perea G, Domingo A, Villamor N, Palacios C, Junca J, Torres P et al (2005) Adverse prognostic impact of CD36 and CD2 expression in adult de novo acute myeloid leukemia patients. Leukemia Res 29(10):1109–1116
Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M et al (2016) Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 19(1):23–37
Frączak E, Olbromski M, Piotrowska A, Glatzel-Plucińska N, Dzięgiel P, Dybko J et al (2018) Bone marrow adipocytes in haematological malignancies. Acta Histochem 120(1):22–27
Caers J, Deleu S, Belaid Z, De Raeve H, Van Valckenborgh E, De Bruyne E et al (2007) Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia 21(7):1580
Avcu F, Ural AU, Yilmaz MI, Bingol N, Nevruz O, Caglarc K (2006) Association of plasma adiponectin concentrations with chronic lymphocytic leukemia and myeloproliferative diseases. Int J Hematol 83(3):254–258
Jöhrer K, Ploner C, Thangavadivel S, Wuggenig P, Greil R (2015) Adipocyte-derived players in hematologic tumors: useful novel targets? Expert Opin Biol Therss 15(1):61–77
Petridou E, Mantzoros C, Dessypris N, Dikalioti S, Trichopoulos D (2006) Adiponectin in relation to childhood myeloblastic leukaemia. Br J Cancer 94(1):156
Yokota T, Oritani K, Takahashi I, Ishikawa J, Matsuyama A, Ouchi N et al (2000) Adiponectin, a new member of the family of soluble defense collagens, negatively regulates the growth of myelomonocytic progenitors and the functions of macrophages. Blood 96(5):1723–1732
Yamauchi T, Kamon J, Ya Minokoshi, Ito Y, Waki H, Uchida S et al (2002) Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 8(11):1288
Medina E, Oberheu K, Polusani S, Ortega V, Velagaleti G, Oyajobi B (2014) PKA/AMPK signaling in relation to adiponectin’s antiproliferative effect on multiple myeloma cells. Leukemia 28(10):2080–2089
Dalamaga M, Karmaniolas K, Panagiotou A, Hsi A, Chamberland J, Dimas C et al (2009) Low circulating adiponectin and resistin, but not leptin, levels are associated with multiple myeloma risk: a case-control study. Cancer Causes Control 20(2):9–193
Kelesidis I, Kelesidis T, Mantzoros CS (2006) Adiponectin and cancer: a systematic review. Br J Cancer 94(9):1221–1225
Han TJ, Wang X (2015) Leptin and its receptor in hematologic malignancies. Int J Clin Exp Med 8(11):19840–19849
Hino M, Nakao T, Yamane T, Ohta K, Takubo T, Tatsumi N (2000) Leptin receptor and leukemia. Leuk Lymphoma 36(5–6):457–461
Kohler J, Moon R, Wright S, Willows E, Davies J (2011) Increased adiposity and altered adipocyte function in female survivors of childhood acute lymphoblastic leukaemia treated without cranial radiation. Horm Res Paediatr 75(6):433–440
Tabe Y, Konopleva M, Munsell MF, Marini FC, Zompetta C, McQueen T et al (2004) PML-RARα is associated with leptin-receptor induction: the role of mesenchymal stem cell–derived adipocytes in APL cell survival. Blood 103(5):1815–1822
Foss B, Mentzoni L, Bruserud O (2001) Effects of vascular endothelial growth factor on acute myelogenous leukemia blasts. J Hematother Stem Cell Res℃ 10(1):81–93
Gorska E, Popko K, Wasik M (2013) Leptin receptor in childhood acute leukemias. Adv Exp Med Biol 756:155–161
Juarez J, Bradstock KF, Gottlieb DJ, Bendall LJ (2003) Effects of inhibitors of the chemokine receptor CXCR4 on acute lymphoblastic leukemia cells in vitro. Leukemia 17(7):1294–1300
Hosokawa Y, Hosokawa I, Ozaki K, Nakae H, Murakami K, Miyake Y et al (2005) CXCL12 and CXCR4 expression by human gingival fibroblasts in periodontal disease. Clin Exp Immunol 141(3):467–474
Diaz-Blanco E, Bruns I, Neumann F, Fischer JC, Graef T, Rosskopf M et al (2007) Molecular signature of CD34(+) hematopoietic stem and progenitor cells of patients with CML in chronic phase. Leukemia 21(3):494–504
Beaulieu A, Poncin G, Belaid-Choucair Z, Humblet C, Bogdanovic G, Lognay G et al (2011) Leptin reverts pro-apoptotic and antiproliferative effects of α-linolenic acids in BCR-ABL positive leukemic cells: involvement of PI3K pathway. PLoS One 6(10):e25651
Mouzaki A, Panagoulias I, Dervilli Z, Zolota V, Spadidea P, Rodi M et al (2009) Expression patterns of leptin receptor (OB-R) isoforms and direct in vitro effects of recombinant leptin on OB-R, leptin expression and cytokine secretion by human hematopoietic malignant cells. Cytokine 48(3):203–211
Jones RG, Thompson CB (2009) Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 23(5):537–548
Sheng X, Mittelman SD (2014) The role of adipose tissue and obesity in causing treatment resistance of acute lymphoblastic leukemia. Front Pediatr 2:53
Schlottmann I, Ehrhart-Bornstein M, Wabitsch M, Bornstein SR, Lamounier-Zepter V (2014) Calcium-dependent release of adipocyte fatty acid binding protein from human adipocytes. Int J Obes (Lond) 38(9):1221–1227
Cao H, Sekiya M, Ertunc ME, Burak MF, Mayers JR, White A et al (2013) Adipocyte lipid chaperone AP2 is a secreted adipokine regulating hepatic glucose production. Cell Metab 17(5):768–778
Hotamisligil GS, Bernlohr DA (2015) Metabolic functions of FABPs–mechanisms and therapeutic implications. Nat Rev Endocrinol 11(10):592–605
Maher M, Diesch J, Casquero R, Buschbeck M (2018) Epigenetic-transcriptional regulation of fatty acid metabolism and its alterations in leukaemia. Front Genet 9:405
Yan F, Shen N, Pang JX, Zhang YW, Rao EY, Bode AM et al (2017) Fatty acid-binding protein FABP4 mechanistically links obesity with aggressive AML by enhancing aberrant DNA methylation in AML cells. Leukemia 31(6):42–1434
Abdelwahab SA, Owada Y, Kitanaka N, Adida A, Sakagami H, Ono M et al (2007) Enhanced expression of adipocyte-type fatty acid binding protein in murine lymphocytes in response to dexamethasone treatment. Mol Cell Biochem 299(1–2):99–107
Kiraly O, Gong G, Olipitz W, Muthupalani S, Engelward BP (2015) Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLoS Genet 11(2):e1004901
Thomas D, Majeti R (2016) Burning fat fuels leukemic stem cell heterogeneity. Cell Stem Cell 19(1):1–2
Khandekar MJ, Cohen P, Spiegelman BM (2011) Molecular mechanisms of cancer development in obesity. Nat Rev Cancer 11(12):886–895
Rozovski U, Hazan-Halevy I, Barzilai M, Keating MJ, Estrov Z (2016) Metabolism pathways in chronic lymphocytic leukemia. Leuk Lymphoma 57(4):758–765
Rozovski U, Grgurevic S, Bueso-Ramos C, Harris DM, Li P, Liu Z et al (2015) Aberrant LPL expression, driven by STAT3, mediates free fatty acid metabolism in CLL cells. Mol Cancer Res 13(5):944–953
Ruby MA, Goldenson B, Orasanu G, Johnston TP, Plutzky J, Krauss RM (2010) VLDL hydrolysis by LPL activates PPAR-alpha through generation of unbound fatty acids. J Lipid Res 51(8):2275–2281
Tung S, Shi Y, Wong K, Zhu F, Gorczynski R, Laister RC et al (2013) PPARalpha and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia. Blood 122(6):969–980
Sheng X, Parmentier JH, Tucci J, Pei H, Cortez-Toledo O, Dieli-Conwright CM et al (2017) Adipocytes sequester and metabolize the chemotherapeutic daunorubicin. Mol Cancer Res 15(12):1704–1713
Cahu X, Calvo J, Poglio S, Prade N, Colsch B, Arcangeli M-L et al (2017) Bone marrow sites differently imprint dormancy and chemoresistance to T-cell acute lymphoblastic leukemia. Blood Adv 1(20):1760–1772
Behan JW, Yun JP, Proektor MP, Ehsanipour EA, Arutyunyan A, Moses AS et al (2009) Adipocytes impair leukemia treatment in mice. Cancer Res 69(19):7867–7874
Pramanik R, Sheng X, Ichihara B, Heisterkamp N, Mittelman SD (2013) Adipose tissue attracts and protects acute lymphoblastic leukemia cells from chemotherapy. Leuk Res 37(5):503–509
Carneiro IP, Mazurak VC, Prado CM (2016) Clinical implications of sarcopenic obesity in cancer. Curr Oncol Rep 18(10):62
Bolan PJ, Arentsen L, Sueblinvong T, Zhang Y, Moeller S, Carter JS et al (2013) Water-fat MRI for assessing changes in bone marrow composition due to radiation and chemotherapy in gynecologic cancer patients. J Magn Reson Imaging 38(6):1578–1584
Liu H, Zhai Y, Zhao W, Wan Y, Lu W, Yang S et al (2018) Consolidation chemotherapy prevents relapse by indirectly regulating bone marrow adipogenesis in patients with acute myeloid leukemia. Cell Physiol Biochem 45(6):2389–2400
Dombret H, Gardin C (2016) An update of current treatments for adult acute myeloid leukemia. Blood 127(1):53–61
Miyashita T, Reed JC (1993) Bcl-2 oncoprotein blocks chemotherapy-induced apoptosis in a human leukemia cell line. Blood 81(1):151–157
Zhang Y, Wang Z, Li X, Magnuson N (2008) Pim kinase-dependent inhibition of c-Myc degradation. Oncogene 27(35):4809
Sheng X, Tucci J, Parmentier JH, Ji L, Behan JW, Heisterkamp N et al (2016) Adipocytes cause leukemia cell resistance to daunorubicin via oxidative stress response. Oncotarget 7(45):73147–73159
Fitter S, Dewar AL, Kostakis P, To LB, Hughes TP, Roberts MM et al (2008) Long-term imatinib therapy promotes bone formation in CML patients. Blood 111(5):2538–2547
Spaner DE (2012) Oral high-dose glucocorticoids and ofatumumab in fludarabine-resistant chronic lymphocytic leukemia. Leukemia 26(5):1144–1145
Wallace AM, Tucker P, Williams DM, Hughes IA, Ahmed SF (2003) Short-term effects of prednisolone and dexamethasone on circulating concentrations of leptin and sex hormone-binding globulin in children being treated for acute lymphoblastic leukaemia. Clin Endocrinol (Oxf) 58(6):770–776
Avramis VI, Tiwari PN (2006) Asparaginase (native ASNase or pegylated ASNase) in the treatment of acute lymphoblastic leukemia. Int J Nanomed 1(3):241–254
Ehsanipour EA, Sheng X, Behan JW, Wang X, Butturini A, Avramis VI et al (2013) Adipocytes cause leukemia cell resistance to L-asparaginase via release of glutamine. Cancer Res 30–2998(10):7398
Mittelman SD, Orgel E (2018) Adipocyte metabolism of the chemotherapy daunorubicin. Oncoscience 5(5–6):146
Ye H, Adane B, Khan N, Ashton JM, Balys M, Stevens BM et al (2015) Adipose tissue functions as a reservoir for leukemia stem cells and confers chemo-resistance. Am Soc Hematol 6:845
Spindler TJ, Tseng AW, Zhou X, Adams GB (2013) Adipocytic cells augment the support of primitive hematopoietic cells in vitro but have no effect in the bone marrow niche under homeostatic conditions. Stem Cells Dev 23(4):434–441
Corre J, Planat-Benard V, Corberand JX, Pénicaud L, Casteilla L, Laharrague P (2004) Human bone marrow adipocytes support complete myeloid and lymphoid differentiation from human CD34+ cells. Br J Haematol 127(3):344–347
Glettig DL, Kaplan DL (2013) Extending human hematopoietic stem cell survival in vitro with adipocytes. Biores Open Access 2(3):179–185
Shafat MS, Gnaneswaran B, Bowles KM, Rushworth SA (2017) The bone marrow microenvironment–home of the leukemic blasts. Blood Rev 31(5):277–286
van Zoelen EJ, Duarte I, Hendriks JM, van der Woning SP (2016) TGFβ-induced switch from adipogenic to osteogenic differentiation of human mesenchymal stem cells: identification of drug targets for prevention of fat cell differentiation. Stem Cell Res Ther 7(1):123
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
For this type of study, informed consent is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Samimi, A., Ghanavat, M., Shahrabi, S. et al. Role of bone marrow adipocytes in leukemia and chemotherapy challenges. Cell. Mol. Life Sci. 76, 2489–2497 (2019). https://doi.org/10.1007/s00018-019-03031-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-019-03031-6