
Abstract
Purpose of Review
Along with a strong impact on skeletal integrity, bone marrow adipose tissue (BMAT) is an important modulator of the adult hematopoietic system. This review will summarize the current knowledge on the causal relationship between bone marrow (BM) adipogenesis and the development and progression of hematologic malignancies.
Recent Findings
BM adipocytes (BMAds) support a number of processes promoting oncogenesis, including the evolution of clonal hematopoiesis, malignant cell survival, proliferation, angiogenesis, and chemoresistance. In addition, leukemic cells manipulate surrounding BMAds by promoting lipolysis and release of free fatty acids, which are then utilized by leukemic cells via β-oxidation. Therefore, limiting BM adipogenesis, blocking BMAd-derived adipokines, or lipid metabolism obstruction have been considered as potential treatment options for hematological malignancies.
Summary
Leukemic stem cells rely heavily on BMAds within the structural BM microenvironment for necessary signals which foster disease progression. Further development of 3D constructs resembling BMAT at different skeletal regions are critical to better understand these relationships in geometric space and may provide essential insight into the development of hematologic malignancies within the BM niche. In turn, these mechanisms provide promising potential as novel approaches to targeting the microenvironment with new therapeutic strategies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Fundamental Roles of BMAT in Shaping Skeletal and Hematopoietic Integrity
Bone Marrow Adipose Tissue as Important Regulator of Bone Health and Regeneration
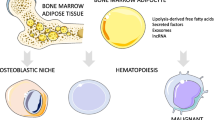
Although bone marrow adipose tissue (BMAT) can occupy the vast majority of the bone marrow (BM) in both health and disease, the bone marrow adipocytes (BMAds) have long been ignored in the physiology of aging and malignancy. However, recent evidence strongly suggests that BMAT represents unique adipose tissue, metabolically different from the other human adipose depots [1, 2]. BMAT is known to be regulated by multiple factors, including nutrient status, skeletal site integrity, and hormonal signals[3].
In turn, BMAds regulate a variety of cells and mechanisms within the microenvironment. Thus, BMAT represents an important BM denominator of both skeletal and hematopoietic systems, and several studies have revealed the specific mechanisms involved. For example, lipolysis-derived energy derived from BMAd is necessary for bone regeneration as well as myelopoiesis during stress, including caloric restriction and prolonged exposure to cold [4]. Interestingly, voluntary running exercise in rodents can reduce BMAT and degrade bone in hypocaloric states, while this bone degradation has not been observed in hypercaloric states, suggesting different energy-dependent behavior of BMAT and bone [5]. In an experimental mouse model of BMAd depletion obtained by expression of diphtheria toxin A or by deletion of peroxisome proliferator-activated receptor gamma (Pparγ), the loss of BMAds leads to augmented bone formation and the relative reduction in hematopoietic stem and progenitor cells (HSPCs). This can protect mice from bone loss induced by caloric restriction or ovariectomy, supporting bone healing process as well [6].
Notably, in order to track BM adipogenesis progression (e.g. during aging), these studies aimed to identify BMAd progenitors within BM. It has been elucidated that BM progenitors committed to BMAds inhibit hematopoiesis and bone healing by producing protease dipeptidyl peptidase-4 [7]. The overexpansion of BMAT has been observed in postmenopausal osteoporosis (PMOP), a prevalent skeletal disorder associated with menopause-related estrogen withdrawal, where BMAT plays important roles in initiation and progression of PMOP [8]. Thus, understanding the coordinated control of bone regeneration requires deciphering the cues triggered by BMAT factors.
Bone Marrow Adipogenesis and Adult Hematopoiesis
BM adipogenesis has been recognized as an emergency phenomenon that follows the production of HSPC niche factors, regulating physiological [4, 9] and stressed hematopoiesis [7]. BMAds influence survival and proliferation of hematopoietic cells at different maturation stages [9]. Particularly in humans, the impact of BMAds on steady-state hematopoiesis, hematopoietic regeneration or malignancies remains incompletely explored. Since hematopoietic stem cell transplantation (HSCT) is a potentially curative treatment for patients suffering from hematologic malignancies and other hematological disorders, host and transplanted HSPCs require support of a specialized BM microenvironment. Strategies that bring beneficial effects to the BM niche can lead to improved BM transplantation outcome [10]. Thus, estimation and profiling of BM microenvironment components, such as BMAT, may contribute to therapeutic potential of BM cell transplantation strategies I [11].
Adult hematopoiesis is orchestrated by BM niches, where disbalanced marrow osteogenesis and adipogenesis critically guide maintenance and differentiation of HSPCs [4, 7]. Although accumulation of BMAds during obesity or aging has been described as detrimental for hematopoietic regeneration as well as bone fracture healing process [7], recent studies have found that BMAT depletion reduces HSPC numbers [4]. Thus, the crosstalk of marrow adipogenesis and hematopoiesis appears to be tightly regulated, requiring detailed analyses of the role of the specific program which regulates marrow adipogenesis. As BMAT represents a unique pool of adipose tissue [1, 2], it is reasonable to re-evaluate the well-known paradigm of BM mesenchymal stroma (stem) cells (BMSCs) as the cell origin of BMAds. Therefore, deconvolution of cells committed towards osteoblastogenesis and adipogenesis within skeletal progenitors is required [12].
Recently, a novel BM cell population of marrow adipogenic lineage precursors (MALPs) has been identified in bone. MALPs have been shown to strongly express colony stimulating factor 1 (Csf1) and its expression is increased in aging. MALPs represent the major source of Csf1 in mouse BM cells when analyzed by single-cell RNA sequencing (scRNA-seq). Furthermore, MALP-derived Csf1 controls both bone remodeling and hematopoiesis and Csf1 KO mice exhibited reduced BM cellularity, a reduction in HSPCs as well as macrophage, monocyte, and erythroid progenitors [13]. Additionally, unique to BM, a so called “secondary adipogenesis pathway” can be triggered by the Adiponectin (AdipoQ)-negative stromal progenitors within BM. Activation of this pathway leads to expansion of BMAT within hematopoietic regions of BM [12]. Emerging evidence also suggests that BMAT may influence the specific mechanisms regulating erythropoiesis within the BM [14]. Additionally, increased BMAT is followed by dysregulated megakaryopoiesis and platelet production in mice [15]. These processes are of tremendous importance for defining roles of BMAds in fracture repair, where erythrocytes and platelets play crucial roles in initial hematoma formation. Besides megakaryocyte lineages, BMAd lipolysis has been shown to guide myeloid cell maintenance under caloric restriction [4], as well as short-term high fat diet (HFD) [12]. Also, BMAT expansion induced by HFD has shown to be related to decreased accumulation of B220+ B-lymphocytes in mice [16], while human BMAds impaired differentiation of umbilical cord blood HSPCs, reducing generation of committed lymphoid progenitors [17]. In addition, malignant hematopoietic cells can also affect BMAds. Interestingly, administration of PPARγ agonists induced BM adipogenesis and led to healthy hematopoiesis recovery and repression of growth of acute myeloid leukemia (AML) [18]. Taken together, these data suggest that several significant interactions occur between BMAds and HSPCs which regulate specific processes which dictate hematopoiesis. Further deciphering their mediators can reveal new biomarkers relevant for human health and diseases and to foster BM transplantation management.
Bone Marrow Adiposity in Cancer
Over the past decade, the potential pathogenic role of BMAT has been widely investigated in hematological malignancies and cancer metastasis. BMAds can promote growth of cancer cells [19] and survival during chemotherapy [20]. In bone and BM metastasis, BMAds interact with cancer cells and participate in metastatic BM niche maintenance [21]. Thus, it can be speculated that BMAds gain the features analogous to cancer-associated adipocytes (CAAs), previously described in adipocytes of solid tumors [21, 22]. CAAs are thought to be essential factors in cancer progression given their role in facilitating angiogenesis, cell growth and migration, via direct or indirect mechanisms [22, 23]. In addition to energy storage, CAAs also contribute to the secretion of hormones, cytokines, adipokines and growth factors [24]. In contrast to normal adipocytes, CAAs are typically smaller in size, possessing a lower lipid content [25]. Although adipocyte differentiation markers have been shown to be downregulated in CAAs, they secrete high levels of adipokines, and inflammatory factors such as Leptin, CCL2, CCL5, and IL-6, which would be expected only in mature adipocytes [24, 25]. Recent studies have found that BMAds can induce the formation of an invasive front within the BM of patients with cancer which has metastasized to the bone marrow [26]. Detailed immunohistochemical analysis have showed that adipocytes co-express markers found on cancer-associated fibroblast (CAF) and adipocyte markers, suggesting that adipocytes can be a good source of CAFs and identifying the induction of CAFs as a potential mechanism used by BMAds to promote cancer invasion [26]. As well, the expression of smooth muscle actin α (α-SMA) molecules, typical for CAFs, has been observed in BMAd-associated with metastatic breast cancer cells [26]. In comparison to extramedullary solid tumor sites, CAAs in BM have been shown to attract dormant cancer cells, contributing to their drug resistance and immune-evasion phenotype [26]. Similarly, BMAds associated with prostate cancer cells undergo significant metabolic reprogramming and lipolysis [21], and metastatic prostate cancer cells induce pro-inflammatory phenotype of BMAds [27], contributing to drug resistance and survival of cancer cells.
Leukemic cells are also able to perturbate the BMAds to promote their own growth via stimulation of a lipolytic state, increasing the expression of lipid transporter genes [19], or via induction of a senescence-associated secretory phenotype [28]. BM preadipocytes can be found in close proximity to leukemic cells in BM biopsies of patients with acute myeloid leukemia (AML) [29].
Bone Marrow Adipose Tissue and Clonal Hematopoiesis
While the interactions between BMAds and normal [7, 30] or leukemic hematopoietic cells [31] have been partially described in the past, little is known about the role of BMAds in the early stages of leukemia and clonal hematopoiesis (CH). Since BMAT accumulation and CH are both age-related and occur in the same location, it has been hypothesized that the accumulation of BMAds could provide selective advantages to specific DNMT3A-mutated pre-Leukemic-HSPC (preL-HSPCs) [32]. Using human and mouse preL-HSPCs and external stress it has been demonstrated that the accumulation of BMAds provides a selective advantage to the human HSPCs carrying the DMT3A R882H mutation, one of the most common mutations in CH [32]. A more pronounced engraftment has been detected when DNMT3A-mutated cells derived from 1 year old mice were injected in either normal BM, fatty BM or fatty BM with a PPARg inhibitor to impair adipogenesis. Furthermore, paracrine inflammatory signals from the BMAds can activate the IL-6 pathway in preL-HSPCs increasing the clonogenic ability of preL-HSPCs [32]. To investigate the role of BMAds and CH, several mouse lines harboring the common CH mutations Tet2, Dnmt3a, Asxl1 or Jak2 on the LepOb/Ob background have been generated. Regardless of the specific CH mutations, they showed that the obesity-induced changes may play a catalyzing role in CH-associated disease [33]. Accumulation of BMAds during aging is ubiquitous, but several factors influence this accumulation, including decline of kidney function [34], increased body mass index [35] and andropenia and menopause [36]. In male subjects BMAds increase steadily, while in females there is a drastic change after menopause, and this may be related to the hormonal changes in estrogens and testosterone [37]. Other studies have showed an increase in DNMT3A mutation, which is a CH typical mutation [38], and one can speculate that BMAd expansion during aging might correlate to expansion of DNMT3A-mutated preL-HSPC clones. This sudden increase of BMAds suggests that the dynamics of BMAds accumulation shape the CH rather than the BMAds mass. These data suggest the role of BMAds in the CH evolution.
Bone Marrow Adipose Tissue in Myelodysplastic Syndromes, Myeloproliferative Neoplasms and Acute Myeloid Leukemia
Myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) are clonal hematopoietic stem cell disorders with a poor prognosis and minimal curative approaches, especially for elderly patients due to aging comorbidities [39]. Cells in the BM microenvironment play important roles in disease development and progression of MDS and AML. One of the main components of the BM microenvironment are the BM stromal cells (BMSCs) that can give rise to osteoblast, chondrocytes, and adipocytes [40]. Balance between adipogenic and osteogenic differentiation is crucial for the regulation of hematopoiesis, and this may be affected during malignant transformation [41]. In vitro differentiation in adipo- and osteo- lineages in BMSCs (CD45−, lineage (CD235a/CD31)−, CD271+, CD73+ and CD105+) in healthy, MDS and AML-BM samples have been analyzed [46]. Although osteogenic differentiation was similar in the BMSCs in healthy, MDS- and AML-BM, MDS- and AML-BMSCs exhibited greater adipogenic differentation [42,43,44]. This data indicates an important role of the DLK1 gene in the BM niche remodeling during hematological malignancies development and disease progression [44]. Although few studies have noted the disruption of adipogenesis may also be noted in patients in AML [44, 45], mechanisms underlying the oncogenic roles of adipocytes in promoting leukemia cell growth and progression have been well established. In addition to the role of DLK1 reduction in niche remodeling, BMAds have also been found to support AML cells in vitro and in vivo via the transferring of fatty acid from adipocytes to AML blasts by using FABP4. Notably, knockdown of FABP4 in AML cells improved the survival of in vivo models [19]. Thus, the specific role that BMAds play in the development of myeloid malignancies warrants further study.
Alterations of BMAds were also described during the onset of chronic myeloid leukemia (CML), including dysregulation in the cytokine production and promotion of a pro-inflammatory environment. The number of pre-adipocytes increases during the chronic phase of CML and decreases during the blast phase. Adipocyte number alteration is associated with the upregulation of Leptin receptor and genes involved in the fatty acid synthesis in CML cells [46].
The BM of patients with MDS, AML, myeloproliferative disorders and neoplasms all demonstrate a reduction in BMAds [14, 31]. In myelofibrosis (MF), the reduction in the number of adipocytes is also associated with a reduction in the diameter and area of BMAds, together with a reduction in the number of AdipoQ+ BMAds in MF patients [47]. Therefore, different myeloid malignancies may influence BMAd and BMSCs-derived adipocytes by unique mechanisms, and although not currently studied, may change in earlier versus later stage disease. These and other questions must be studied further to best understand the specific crosstalk between malignant cells and the niche.
BMAT in Lymphoproliferative Malignancies
Dynamic role of BMAds in Acute Lymphoblastic Leukemia (ALL)
The BMAd niche plays a dynamic role during acute lymphoblastic leukemia (ALL) spanning from a depleted state at disease onset to reconstitution during remission and response to therapy. This dynamism is due to the inhibition of protein synthesis in ALL cells mediated by BMAds. Notably, during the remission phase, the returning adipocytes are smaller when compared with healthy donors [47]. Bioactive phospholipids, including sphingosine-1-phosphate (S1P), ceramide-1-phosphate (C1P) play important roles in adipose tissue dysfunction, metabolic syndrome, obesity, and diabetes. These molecules were shown to affect HSPCs [48] as well as leukemic cells [49] (Table 1). In addition, AdipoQ, an adipocyte-derived hormone, can inhibit B lymphopoiesis through induction of cyclooxygenase-2 (COX-2) expression and subsequent prostaglandin E2 (PGE2) synthesis by in vitro differentiated adipocytic cells [50, 51]. Interestingly PGE2 increases intracellular cAMP in T-ALL cells and re-sensitizes leukemic cells to dexamethasone [52]. These findings indicate that lipids participate in regulation of HSPCs and leukemic cells and might represent further targets of revealing BMAd-leukemic cell interplay.
Chemoresistance Mechanism Mediated by BMAds in ALL
ALL cells promote the release of free fatty acids (FFAs) by BMAds, which are then utilized by ALL cells via b-oxidation. This mechanism contributes to the chemotherapy resistance against vincristine and daunorubicin and may explain the inferior prognosis of obese patients with ALL [53]. BMAds protect ALL cells from chemotherapy, supporting their survival [54]. Exposure of human B-ALL cells to secreted molecules of adipocyte causes increased response in the Wnt16-mediated pathways, B cell development, chemoresistance and increasing of b-oxidation of fatty acids (FAO) [55].
Region-Specific BMAds in the Context of T-cell ALL (T-ALL)
As the content and profile of BMAT are skeletal site-specific, it is important to reveal whether the impact of BMAT and BMAds on malignant HSPCs vary between distinct skeletal regions. Xenograft and murine models of T-cell ALL (T-ALL) have shown that the BM content plays an important role not in the engraftment but in the absolute number of leukemic cells. Thus, lower number of leukemic cells was found in the tail, filled with constitutive BMAT (cBMAT) compared to the thoracic segments filled with regulated BMAT (rBMAT). These results indicated that the T-ALL cells can infiltrate in all the BM locations, but their expansion is strongly delayed in the cBMAT locations of long bones, rich in adipocytes and poor in hematopoietic cells [56].
BMAds and Multiple Myeloma (MM)
BMAd content increases with aging and so the incidence of multiple myeloma (MM). BMAds support myeloma cells via BMAd-derived factors such as chemokine ligand 2 and C-X-C Motif chemokine Ligand 12 [57, 58]. Adipsin promotes chemoresistance of MM cells [59], while Leptin and other adipokines promote cell proliferation [59]. MM cells modulate BMAds, contributing to the myeloma-induced bone disease. Moreover, BMAds isolated from MM patients showed senescent features that may alter bone formation and present a dramatic downregulation of the osteogenic and adipogenic gene expression compared to the healthy controls [60]. Impairment of adipogenesis was reproduced in vitro in BMSCs from MM patients and murine BMSCs exposed to human MM cell line MM.1S [28, 61]. It is possible that changes triggered by MM cells in BMSCs are transient, and collectively represent metabolic adaptation to the demands of neoplastic microenvironment. A recent study revealed that an increased BMAd density in patients with monoclonal gammopathy of undetermined significance (MGUS) is associated with higher rates of progression towards MM [61, 62]. These findings provide evidence that BMAT contributes to the progression of and subsequent consequences of hematological malignancies and therefore should be investigated and considered as potentially relevant biomarker as well as therapeutic target.
Disrupting Bone Marrow Adipocyte–Leukemic Cell Interaction as Novel Therapeutic Strategy
Various BMAd-derived factors impact malignant cell survival and proliferation, angiogenesis process, as well as chemotherapy protection [63], acting directly through cell–cell contacts or indirectly through paracrine secretion [27]. Therefore, targeting BMAd and BMAT has been recognized as a promising strategy for hematological malignancies treatment. In this context, different strategies have been proposed including the blocking of adipocyte-derived adipokines, limiting adipogenesis or targeting lipid metabolism (Table 1).
Targeting Adipogenic Cells and Adipokines in Bone Marrow
Results of preclinical studies in T-ALL and B-ALL mice models supports the therapeutic potential of BMAT targeting. These investigations have showed that fasting, known to increase BMAT, results in decreased Leptin production by adipocytes, which correlates with inhibited engraftment and progression of T-ALL and B-ALL blasts in leukemia mice models [27]. In addition, increased Leptin receptor (LepR) expression in leukemia cells and its downstream signaling were identified as main mechanisms of inhibition of leukemia initiation and reversion of the leukemic progression emphasizing Leptin/LepR axis as potential leukemia target [64]. Another approach utilized AdipoQ potential to suppress adipogenic differentiation and established its pharmacological enhancement by apolipoprotein mimetic L-4F which exerted antitumor effects in myeloma mouse models [65] (Table 1). Moreover, recent scRNA-seq studies of murine BM stroma identified pro-hematopoietic factors (SDF-1, SCF, IL-7, IL-15) in adipogenic-primed clusters of LepR + cells with specific relevance for leukemia treatment [65]. Comparative gene expression analyses of human extramedullary adipose tissue and BMAT samples confirmed an enrichment of inflammatory genes in BMAds [66] that have been previously tested for targeting hematological malignancies [67].
BMAds are capable of metabolizing and inactivating drugs, and therefore can reduce chemotherapeutic concentration in BM, thus supressing their antitumor effects [67]. In this respect, the dynamic nature of BMAT that can increase in response to irradiation or chemotherapy may contribute to subsequent treatment failure [68]. Accordingly, reduced BMAd content is considered as a favorable prognostic factor in leukemia patients during remission [69]. Improved chemotherapy efficacy and a reduction of relapse in AML patients was correlated with inhibition of BMSC adipogenesis via Growth/differentiation factor 15 (GDF-15) secretion from BM hematopoietic cells [69]. Moreover, transient receptor potential vanilloid 4 (TRPV4) was identified as a negative regulator of GDF15-induced BMAT remodeling. Importantly, targeting this protein by using a TRPV4 agonist 4aPDD increased the survival of AML-bearing mice, suggesting its’ therapeutic potential [70]. Additionally, transforming growth factor-β type II receptor (TGFβRII) has been reported as the main receptor for GDF15 on BMAds [69]. Chemotherapy also induces an increase in total fat depots where leukemic cells can be recruited as shown in murine models [70, 71]. As well, by combining immunohistochemistry of BM and 3T3-L1 preadipocytes the BMAT protects ALL cells from treatment with L-asparaginase mainly via glutamine secretion important for ALL survival [71, 72] (Table 1). Taken together, BMAds strongly affect leukemic cell survival and therapy efficiency which indicate that further investigations are necessary to define targets of BMAd-leukemic cell interactions.
Targeting BMAd Metabolism
Leukemia cells highly depend on FAO metabolism to fulfill demands for cell proliferation. Therefore, blocking free FAs transfer from BMAds to leukemic blasts [19] represents another proposed approach of targeting BMAd-leukemic cell interactions. FAO inhibition was found to induce metabolic imbalance and AML apoptosis by induction of integrated stress response mediator Activating transcription factor 4 (ATF4) [73]. Also, inhibition of CPT1 (carnitine O-palmitoyltransferase 1), the rate-limiting FAO enzyme which conjugates FA with carnitine for translocation to the mitochondrial matrix, showed anti-AML effects [74, 75]. Namely, the pharmacological CPT1A inhibitor etomoxir sensitized leukemic cells to ABT-737-induced apoptosis, as well as to the chemotherapeutic cytarabine (AraC). Another CPT1A inhibitor, ST1326, has been found to provoke dose- and time-dependent growth arrest, mitochondrial damage, and apoptosis in primary ALL, CLL and AML cells [75], while avocado-derived FAO inhibitor, avocatin B, decreases NADPH inducing ROS-dependent AML cell death [76]. The fatty acid synthase inhibitor, orlistat, was also demonstrated to inhibit CLL cell growth via apoptosis induction, while its simultaneous application with fludarabine further strengthened its antileukemic activity [77].
Yet, FAO inhibitors have limited efficacy as triggering adaptive and alternative metabolic pathways that support AML survival. This was confirmed in vitro in co-cultures of human adipogenic-differentiated BMSCs and AML cells where decreased anti-leukemia effects of avocatin B was evidenced along with increased compensatory glycolysis which provided continued supply of ATP [76]. However, combinatorial regimes of conventional anti-neoplastic therapies with FAO inhibitors that were tested in preclinical studies resulted in highly synergistic effects. While AraC-resistant AML cells displayed increased FAO and oxidative phosphorylation (OXPHOS), addition of etomoxir induced energy shift to low OXPHOS increasing sensitivity of leukemic cells to AraC [78]. Similarly, avocatin B exerted synergistic effects with AraC by stimulating apoptosis in AML cells co-cultured with BMSC-derived adipocytes through ATF4 activation [76]. Moreover, inhibition of FAO by etomoxir significantly increased sensitivity of childhood ALL cells to L-asparaginase [79]. Furthermore, FAO inhibitor AIC-47 can reverse imatinib-induced FAO upregulation through CPT1C modulation in Ph-positive ALL and CML cells [80, 81], and PPARα and FAO enzyme inhibitors were found to increase cytotoxicity of dexamethasone in CLL cells in vitro and in vivo [82]. Collectively, it appears that modulation of lipid metabolism in leukemic cells represents a promising strategy to target hematological malignancies and their crosstalk with BMAds.
Potential novel strategies of targeting BM adipocytic niche for hematologic malignancy treatment are focused on FA uptake and shuttling. Namely the pharmacologic (BMS3094013 and SBFI- 26) or genetic (CRISPR/Cas9) inhibition of fatty acid binding protein 4 (FABP4) in various multiple myeloma cell lines induced their apoptosis and metabolic changes by reducing MYC signaling [83]. However, FABP inhibition in vivo showed variable effects in MM suggesting the necessity of its optimization. In addition, previous study provided preclinical evidence that targeting lipoprotein lipase (LPL) activator, apolipoprotein C2 (APOC2) and FA transporter/translocase CD36 delayed AML progression in mice model indicating this signalling axis as potential therapeutic target [84]. Moreover, reduction of FA uptake by CD36 inhibition was found to reduce IL-6-mediated chemoresistance [85]. However, further research is needed for defining precise BMAd-related targets of hematologic malignancies and effective clinical translation of these targeting approaches (Fig. 1).

Interactions of bone marrow adipose tissue and malignant (leukemic) hematopoietic cells. BMAds produce factors which support malignant (leukemic) hematopoietic cells survival, proliferation and chemoresistance. Malignant cells increase lipolysis and senescence in BMAd and their precursors, leading to formation of cancer-associated BMAd phenotype. Abbreviations: FAO-fatty acid oxidation; LPL-lipoprotein lipase; OXPHOS-oxidative phosphorylation, HSPC-hematopoietic stem and progenitor cell; BMAd-bone marrow adipocytes; MALPs-Marrow adipogenic lineage precursors. Grey box: BM adipogenesis, blue box: malignant hematopoiesis
Modelling of Bone Marrow Adipose Tissue in Hematologic Malignancies
During the states of malignant marrow disease, neoplastic cells invade BM, interacting with BM niches inhabited by osteoblast, osteoclasts, BMAds, BMSCs, erythrocytes, megakaryocytes, and leukocytes [86]. Although accepted to be insufficiently translatable to humans, animal models remain the model system to study interactions of BMAds and hematopoietic cells in vivo in both non-malignant and malignant contexts [87, 88]. Yet, much of the currently available evidence investigating these interactions are based on in vitro observed interactions of BMAds and cancer cells, mostly by the co-culturing of BMSC-derived adipocytes and cancer cell lines or patient-derived cells along with animal models [19, 53, 86].
In fact, development of an optimal BMAT in vitro model dependent on efficiently recapitulated model of BM microenvironment [87]. Progress in this field has been achieved with generation of BMSC-derived humanized ossicles which allow engraftment of primary patient-derived leukemic cells in vivo [89]. However, differences in tumor microenvironment and peri-tumoral niches, limit representativity of xenotransplants [90]. To implement the principle to replace, reduce and refine (3Rs) animal experiments, there is an urgent need to develop and fabricate 3D BM models which can secure BM functionality in health and disorders [87, 89,90,91]. The majority of approaches are based on niche extracellular matrix proteins (collagen IV, fibronectin, or glycosaminoglycans) in combination with BMSCs and CD34+ cells. These systems are additionally supplemented with fibroblast growth factor, WNT ligands, stem cell factor (SCF) or CXCL12 which are regularly produced by BMSCs as well as bone morphogenic proteins (BMPs) or angiopoietin that support maintenance of both HSPCs and leukemic cells [91]. In addition, tissue-engineered bone models, composed of BMSCs, osteoblast, osteocytes, mineralized bony matrix and myeloma cells, allows non-destructive imaging over long periods to study bone cells behaviour during myeloma cell colonization [92]. A recent study demonstrated organoids generated from induced pluripotent stem cells (iPSCs) committed to mesenchymal, endothelial, and hematopoietic lineages. These organoids possessed key features of human BM— stroma, lumen-forming sinusoids, and myeloid cells including proplatelet-forming megakaryocytes, further supporting the engraftment and survival of cells from patients with hematologic malignancies Collectively, these organoids recapitulated cellular and molecular crosstalk between hematopoietic, endothelial, and stromal cells within the BM microenvironment [93]. Yet, these organoids did not reproduce BMAd-cancer cell interactions, indicating the need for further refinement of this approach.
Recapitulation of BMAd requires implementation of basic knowledge on adipocyte physiology and mechanobiology. For adipose tissue research, complex 3D platforms bridge the gaps between 2D cultures and in vivo models, bringing about more reliable data. Applying 3D adipocyte spheroids, biomaterial-based 3D culture, 3D bioprinting, and microphysiological systems may offer new opportunities to discover drugs targeting BMAds [94]. Successfully established 3D spheroids and organoids of white adipose tissue containing immune cells [95, 96] and beige adipose vascularized organoids [96] have been reported.
The first 3D BMAT model was derived from human or mouse bone BMSCs. These models were stable for 3 months in vitro, while importantly, myeloma cell lines (5TGM1, OPM-2 and MM1.S) can be cultured in this system for at least 2 weeks. Proteomic analyses coupled with the KEGG pathway revealed that 3D BMAT was less inflammatory than 2D culture [87]. Another study reported an engineered BMAT analogue made of a GelMA (gelatin methacryloyl) hydrogel/medical-grade polycaprolactone (mPCL) scaffold composite (to structurally and biologically mimic key aspects of the BM microenvironment), and bioreactor-derived mechanic loading which supported adipogenesis of BMSCs [97, 98]. Further development of more complex 3D structures resembling BM and simultaneously BMAT will significantly contribute to obtaining more translatable and predictive data of BMAT behavior, influence on homeostatic and pathological states, and anti-cancer drug efficiency.
Conclusion
In terms of skeletal region selection, material isolation, and applying suitable technique, the modeling of BMAT still represents a challenging topic. However, recent findings bring significant new data on BMAT roles in health and diseases, and particularly in hematologic malignancies. It can be concluded that, limiting BM adipogenesis, blocking BMAd-derived adipokines, or lipid metabolism manipulation represent promising opportunities for more precise treatment options for hematological malignancies. Further development of advanced ex vivo and in vivo models of BMAT integrity should boost the development and testing of innovative anti-cancer therapies.
Data Availability
No datasets were generated or analysed during the current study.
References
Suchacki KJ et al. Bone marrow adipose tissue is a unique adipose subtype with distinct roles in glucose homeostasis. Nat Commun. 2020;11(1):3097. https://doi.org/10.1038/s41467-020-16878-2.
Attané C, et al. Human Bone Marrow Is Comprised of Adipocytes with Specific Lipid Metabolism. Cell Rep. 2020;30(4):949-958.e6. https://doi.org/10.1016/j.celrep.2019.12.089.
Rosen CJ and Horowitz MC. Nutrient regulation of bone marrow adipose tissue: skeletal implications of weight loss. Nat Rev Endocrinol. 2023;19(11):626–638. https://doi.org/10.1038/s41574-023-00879-4.
Li Z, et al. Lipolysis of bone marrow adipocytes is required to fuel bone and the marrow niche during energy deficits. Elife. 2022;11:e78496. https://doi.org/10.7554/eLife.78496.
Little-Letsinger SE, et al. Exercise and Diet: Uncovering Prospective Mediators of Skeletal Fragility in Bone and Marrow Adipose Tissue. Curr Osteoporos Rep. 2020;18(6):774–789. https://doi.org/10.1007/s11914-020-00634-y.
Li Z, et al. Constitutive bone marrow adipocytes suppress local bone formation. JCI Insight. 2022;7(21):e160915. https://doi.org/10.1172/jci.insight.160915.
Ambrosi TH, et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell. 2017;20(6):771-784.e6. https://doi.org/10.1016/j.stem.2017.02.009.
Li J, et al. The relationship between bone marrow adipose tissue and bone metabolism in postmenopausal osteoporosis. Cytokine Growth Factor Rev. 2020;52:88–98. https://doi.org/10.1016/j.cytogfr.2020.02.003.
Zhou BO et al. Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol. 2017;19:891-903. https://doi.org/10.1038/ncb3570.
Labella R, et al. Bone Marrow Adipose Tissue: Regulation of Osteoblastic Niche, Hematopoiesis and Hematological Malignancies. Stem Cell Rev Rep. 2023;19(5):1135–51. https://doi.org/10.1007/s12015-023-10531-3.
Peci F, et al. The cellular composition and function of the bone marrow niche after allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2022;57(9):1357–1364. https://doi.org/10.1038/s41409-022-01728-0.
Zhang X, et al. A bone-specific adipogenesis pathway in fat-free mice defines key origins and adaptations of bone marrow adipocytes with age and disease. Elife. 2021;10:e66275. https://doi.org/10.7554/eLife.66275.
Zhong L, et al. Csf1 from marrow adipogenic precursors is required for osteoclast formation and hematopoiesis in bone. Elife. 2023;12:e82112. https://doi.org/10.7554/eLife.82112.
Liu T, et al. Bone marrow adiposity modulation after long duration spaceflight in astronauts. Nat Commun. 2023 Aug 9;14:4799https://doi.org/10.1038/s41467-023-40572-8.
Valet C, et al. Adipocyte Fatty Acid Transfer Supports Megakaryocyte Maturation. Cell Rep. 2020;32:107875. https://doi.org/10.1016/j.celrep.2020.107875.
Adler BJ, et al. High fat diet rapidly suppresses B lymphopoiesis by disrupting the supportive capacity of the bone marrow niche. PLoS One. 2014;9:e90639. https://doi.org/10.1371/journal.pone.0090639.
Bilwani FA, et al. Adipocyte-derived soluble factor(s) inhibits early stages of B lymphopoiesis. J Immunol. 2012;189:4379–86. https://doi.org/10.4049/jimmunol.1201176.
Boyd AL, et al. Acute myeloid leukaemia disrupts endogenous myelo-erythropoiesis by compromising the adipocyte bone marrow niche. Nat Cell Biol. 2017;19(11):1336–47. https://doi.org/10.1038/ncb3625.
Shafat MS, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. 2017;129:1320–32.
Tabe Y, Konopleva M. Leukemia Stem Cells Microenvironment. Adv Exp Med Biol. 2017;1041:19–32.
Diedrich JD, et al. Bone marrow adipocytes promote the Warburg phenotype in metastatic prostate tumors via HIF-1α activation. Oncotarget. 2016;7:64854-64877. https://doi.org/10.18632/oncotarget.11712.
Wu Q et al. Cancer-associated adipocytes: key players in breast cancer progression. J Hematol Oncol J Hematol Oncol 12, 95 (2019).
Wu Q, Li B, Sun S, Sun S. Unraveling Adipocytes and Cancer Links: Is There a Role for Senescence? Front Cell Dev Biol. 2020;8:282.
Lapeire L, et al. Cancer-Associated Adipose Tissue Promotes Breast Cancer Progression by Paracrine Oncostatin M and Jak/STAT3 Signaling. Cancer Res. 2014;74:6806–19.
Dirat B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71:2455–65.
Sato S, et al. Bone marrow adipocytes induce cancer-associated fibroblasts and immune evasion, enhancing invasion and drug resistance. Cancer Sci. 2023;114:2674–88.
Herroon MK, et al. Prostate Tumor Cell-Derived IL1β Induces an Inflammatory Phenotype in Bone Marrow Adipocytes and Reduces Sensitivity to Docetaxel via Lipolysis-Dependent Mechanisms. Mol Cancer Res. 2019;17(12):2508-2521. https://doi.org/10.1158/1541-7786.MCR-19-0540.
Fairfield H, et al. Myeloma-Modified Adipocytes Exhibit Metabolic Dysfunction and a Senescence-Associated Secretory Phenotype. Cancer Res. 2021;81:634–47.
Bianco P, et al. Alkaline phosphatase positive precursors of adipocytes in the human bone marrow. Br J Haematol. 1988;68(4):401–3. https://doi.org/10.1111/j.1365-2141.1988.tb04225.x.
Naveiras O, et al. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460(7252):259-63https://doi.org/10.1038/nature08099.
Zinngrebe J, et al. Adipocytes in hematopoiesis and acute leukemia: friends, enemies, or innocent bystanders? Leukemia. 2020;34(9):2305–2316. https://doi.org/10.1038/s41375-020-0886-x.
Zioni N, et al. Inflammatory signals from fatty bone marrow support DNMT3A driven clonal hematopoiesis. Nat Commun. 2023;14:2070.
Pasupuleti SK, et al. Obesity-induced inflammation exacerbates clonal hematopoiesis. J Clin Invest. 2023;133(11):e163968 https://doi.org/10.1172/JCI163968.
Woods GN, et al. Chronic Kidney Disease Is Associated With Greater Bone Marrow Adiposity. J Bone Miner Res. 2018;33(12):2158–2164. https://doi.org/10.1002/jbmr.3562.
Bredella MA, et al. Vertebral bone marrow fat is positively associated with visceral fat and inversely associated with IGF-1 in obese women. Obesity (Silver Spring). 2011;19(1):49–53. https://doi.org/10.1038/oby.2010.106.
Mistry SD, et al. Sex hormones are negatively associated with vertebral bone marrow fat. Bone. 2018;108:20–24. https://doi.org/10.1016/j.bone.2017.12.009.
Griffith JF, et al. Bone marrow fat content in the elderly: a reversal of sex difference seen in younger subjects. J Magn Reson Imaging. 2012;36(1):225–30. https://doi.org/10.1002/jmri.23619.
De-Morgan A, et al. Male predominance in AML is associated with specific preleukemic mutations. Leukemia. 2021;35(3):867–70. https://doi.org/10.1038/s41375-020-0935-5.
Klepin HD. Myelodysplastic Syndromes and Acute Myeloid Leukemia in the Elderly. Clin Geriatr Med. 2016;32(1):155-73. https://doi.org/10.1016/j.cger.2015.08.010.
Gao Q, et al. Bone Marrow Mesenchymal Stromal Cells: Identification, Classification, and Differentiation. Front Cell Dev Biol. 2022;9:787118. https://doi.org/10.3389/fcell.2021.787118.
Wu J, et al. The Differentiation Balance of Bone Marrow Mesenchymal Stem Cells Is Crucial to Hematopoiesis. Stem Cells Int. 2018;2018:1540148. https://doi.org/10.1155/2018/1540148.
Le Y, et al. Adipogenic Mesenchymal Stromal Cells from Bone Marrow and Their Hematopoietic Supportive Role: Towards Understanding the Permissive Marrow Microenvironment in Acute Myeloid Leukemia. Stem Cell Rev Rep. 2016;12:235–44.
Wu Y, et al. Impaired Expression of Focal Adhesion Kinase in Mesenchymal Stromal Cells from Low-Risk Myelodysplastic Syndrome Patients. Front Oncol. 2017;7:164.
Weickert M-T, et al. Bone marrow stromal cells from MDS and AML patients show increased adipogenic potential with reduced Delta-like-1 expression. Sci Rep. 2021;11:5944.
Battula, V. L. et al. AML-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight 2, e90036, 90036 (2017).
Beaulieu A, et al. Leptin reverts pro-apoptotic and antiproliferative effects of α-linolenic acids in BCR-ABL positive leukemic cells: involvement of PI3K pathway. PLoS ONE. 2011;6:e25651.
Dello Spedale Venti, M. et al. Morphological and Immunophenotypical Changes of Human Bone Marrow Adipocytes in Marrow Metastasis and Myelofibrosis. Front. Endocrinol. 13, (2022).
Kim CH, et al. Conditioning for hematopoietic transplantation activates the complement cascade and induces a proteolytic environment in bone marrow: a novel role for bioactive lipids and soluble C5b–C9 as homing factors. Leukemia. 2012;26(1):106–16. https://doi.org/10.1038/leu.2011.185.
Abdelbaset-Ismail A, et al. Bioactive Phospholipids Enhance Migration and Adhesion of Human Leukemic Cells by Inhibiting Heme Oxygenase 1 (HO-1) and Inducible Nitric Oxygenase Synthase (iNOS) in a p38 MAPK-Dependent Manner. Stem Cell Rev Rep. 2019;15(1):139–54. https://doi.org/10.1007/s12015-018-9853-6.
Yokota T, et al. Paracrine regulation of fat cell formation in bone marrow cultures via adiponectin and prostaglandins. J Clin Invest. 2002;109(10):1303–10. https://doi.org/10.1172/JCI14506.
Yokota T, et al. Adiponectin, a fat cell product, influences the earliest lymphocyte precursors in bone marrow cultures by activation of the cyclooxygenase-prostaglandin pathway in stromal cells. J Immunol. 2003;171(10):5091–9. https://doi.org/10.4049/jimmunol.171.10.5091.
Roderick JE, et al. Prostaglandin E2 stimulates cAMP signaling and resensitizes human leukemia cells to glucocorticoid-induced cell death. Blood. 2021;137(4):500–12. https://doi.org/10.1182/blood.2020005712.
Heydt Q, et al. Adipocytes disrupt the translational programme of acute lymphoblastic leukaemia to favour tumour survival and persistence. Nat Commun. 2021;12:5507.
Tucci J, et al. Adipocytes Provide Fatty Acids to Acute Lymphoblastic Leukemia Cells. Front Oncol. 2021;11:665763.
Geitgey DK, et al. The ‘omics of obesity in B-cell acute lymphoblastic leukemia. JNCI Monogr. 2023;2023:12–29.
Cahu X, et al. Bone marrow sites differently imprint dormancy and chemoresistance to T-cell acute lymphoblastic leukemia. Blood Adv. 2017;1:1760–72.
Trotter TN, et al. Adipocyte-Lineage Cells Support Growth and Dissemination of Multiple Myeloma in Bone. Am J Pathol. 2016;186:3054–63.
Bullwinkle EM, et al. Adipocytes contribute to the growth and progression of multiple myeloma: Unraveling obesity related differences in adipocyte signaling. Cancer Lett. 2016;380:114–21.
Liu Z, et al. Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget. 2015;6:34329–41.
Caers J, et al. Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia. 2007;21:1580–4.
Fairfield H, et al. Multiple Myeloma Cells Alter Adipogenesis, Increase Senescence-Related and Inflammatory Gene Transcript Expression, and Alter Metabolism in Preadipocytes. Front Oncol. 2020;10:584683.
El-Masri BM, et al. Bone marrow adipocytes provide early sign for progression from MGUS to multiple myeloma. Oncotarget. 2024;15:20-26https://doi.org/10.18632/oncotarget.28548.
Li J, et al. Bone marrow adipocytes and lung cancer bone metastasis: unraveling the role of adipokines in the tumor microenvironment. Front Oncol. 2024;14:1360471https://doi.org/10.3389/fonc.2024.1360471.
Lu Z, et al. Fasting selectively blocks development of acute lymphoblastic leukemia via leptin-receptor upregulation. Nat Med. 2017;23:79–90. https://doi.org/10.1038/nm.4252.
Fowler JA, et al. Host-derived adiponectin is tumor suppressive and a novel therapeutic target for multiple myeloma and the associated bone disease. Blood. 2011;118:5872–82.
Miggitsch C, et al. Human bone marrow adipocytes display distinct immune regulatory properties. EBioMedicine. 2019;46:387–98.
Jöhrer K, et al. Adipocyte-derived players in hematologic tumors: useful novel targets? Expert Opin Biol Ther. 2014;15(1):61–77. https://doi.org/10.1517/14712598.2015.970632.
Li ZJ et al. Development, regulation, metabolism and function of bone marrow adipose tissues. Bone. 2018;110:134–140. https://doi.org/10.1016/j.bone.2018.01.008.
Liu H, et al. Consolidation chemotherapy prevents relapse by indirectly regulating bone marrow adipogenesis in patients with acute myeloid leukemia. Cell Physiol Biochem. 2018;45:2389–400.
Yang S, et al. Leukemia cells remodel marrow adipocytes via TRPV4-dependent lipolysis. Haematologica. 2020;105(11):2572–83. https://doi.org/10.3324/haematol.2019.225763.
Behan JW, et al. Diet-induced obesity alters vincristine pharmacokinetics in blood and tissues of mice. Pharm Res. 2010;61:385–90.
Ehsanipour EA, et al. Adipocytes cause leukemia cell resistance to l-asparaginase via release of glutamine. Cancer Res. 2013;73:2998–3006.
Tabe Y, et al. Inhibition of FAO in AML co-cultured with BM adipocytes: mechanisms of survival and chemosensitization to cytarabine. Sci Rep. 2018;8:16837. https://doi.org/10.1038/s41598-018-35198-6.
Samudio I, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120:142–56. https://doi.org/10.1172/JCI38942.
Ricciardi MR, et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126:1925–9. https://doi.org/10.1182/blood-2014-12-617498.
Tcheng M, et al. The mitochondria target drug avocatin B synergizes with induction chemotherapeutics to induce leukemia cell death. Leuk Lymphoma. 2017;58:986–8. https://doi.org/10.1080/10428194.2016.1218005.
Pallasch CP, et al. Targeting Lipid Metabolism by the Lipoprotein Lipase Inhibitor Orlistat Results in Apoptosis of B-Cell Chronic Lymphocytic Leukemia Cells. Leukemia. 2008;22(3):585–92. https://doi.org/10.1038/sj.leu.2405058.
Farge T, et al. Chemotherapy resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7:716–35. https://doi.org/10.1158/2159-8290.CD-16-0441.
Hermanova I, et al. Pharmacological inhibition of fatty-acid oxidation synergistically enhances the effect of l-asparaginase in childhood ALL cells. Leukemia. 2016;30:209–18. https://doi.org/10.1038/leu.2015.213.
Shinohara H, et al. Perturbation of energy metabolism by fatty-acid derivative AIC-47 and imatinib in BCR-ABL-harboring leukemic cells. Cancer Lett. 2016;371:1–11. https://doi.org/10.1016/j.canlet.2015.11.020.
Shinohara H, et al. Potent antiproliferative effect of fatty-acid derivative AIC-47 on leukemic mice harboring BCR-ABL mutation. Cancer Sci. 2019;110:751–60. https://doi.org/10.1111/cas.13913.
Tung S, Shi Y, et al. PPARα and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia. Blood. 2013;122:969–80. https://doi.org/10.1182/blood-2013-03-489468.
Farrell M, et al. Targeting the fatty acid binding proteins disrupts multiple myeloma cell cycle progression and MYC signaling. Elife. 2023;12:e81184. https://doi.org/10.7554/eLife.81184.
Zhang T, et al. Apolipoprotein C2 - CD36 Promotes Leukemia Growth and Presents a Targetable Axis in Acute Myeloid Leukemia. Blood Cancer Discov. 2020;1(2):198–213. https://doi.org/10.1158/2643-3230.BCD-19-0077.
Zhang Y, et al. IL-6 promotes chemoresistance via upregulating CD36 mediated fatty acids uptake in acute myeloid leukemia. Exp Cell Res. 2022;415:113112. https://doi.org/10.1016/j.yexcr.2022.113112.
Amorim T, et al. Young minds, deeper insights: a recap of the BMAS Summer School 2023, ranging from basic research to clinical implications of bone marrow adipose tissue. Biol Open. 2024;13(2):bio060263. https://doi.org/10.1242/bio.060263.
Fairfield H, et al. Development of a 3D bone marrow adipose tissue model. Bone. 2019;118:77–88. https://doi.org/10.1016/j.bone.2018.01.023.
Pereira AR, et al. Approaches to mimic the complexity of the skeletal mesenchymal stem/stromal cell niche in vitro. Eur Cell Mater. 2019;37:88–112. https://doi.org/10.22203/eCM.v037a07.
Reinisch A, et al. Generation and use of a humanized bone-marrow-ossicle niche for hematopoietic xenotransplantation into mice. Nat Protoc. 2017 Oct;12(10):2169–2188. https://doi.org/10.1038/nprot.2017.088.
Raic A, et al. 3D models of the bone marrow in health and disease: yesterday, today and tomorrow. MRS Commun. 2019;9(1):37–52. https://doi.org/10.1557/mrc.2018.203.
Yoshihara H, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1(6):685–97. https://doi.org/10.1016/j.stem.2007.10.020.
Reagan MR, et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood. 2014;124(22):3250–9. https://doi.org/10.1182/blood-2014-02-558007.
Khan AO, et al. Human Bone Marrow Organoids for Disease Modeling, Discovery, and Validation of Therapeutic Targets in Hematologic Malignancies. Cancer Discov. 2023;13(2):364–85. https://doi.org/10.1158/2159-8290.CD-22-0199.
Heejeong Yoon and Tae-Eun Park. Engineered adipose tissue platforms: recent breakthroughs and future perspectives. Organoid. 2023;3:e1 Published online January 25, 2023. https://doi.org/10.51335/organoid.2023.3.e1
Taylor J, et al. Generation of immune cell containing adipose organoids for in vitro analysis of immune metabolism. Sci Rep. 2020;10(1):21104. https://doi.org/10.1038/s41598-020-78015-9.
Robledo F, et al. Spheroids derived from the stromal vascular fraction of adipose tissue self-organize in complex adipose organoids and secrete leptin. Stem Cell Res Ther. 2023;14(1):70. https://doi.org/10.1186/s13287-023-03262-2.
Escudero M, et al. Scalable Generation of Pre-Vascularized and Functional Human Beige Adipose Organoids. Adv Sci (Weinh). 2023;10(31):e2301499. https://doi.org/10.1002/advs.202301499.
Ravichandran A, et al. Engineering a 3D bone marrow adipose composite tissue loading model suitable for studying mechanobiological questions. Mater Sci Eng C Mater Biol Appl. 2021;128:112313. https://doi.org/10.1016/j.msec.2021.112313.
Acknowledgements
Illustration has been created using smart.servier.com.
Funding
The work of DT, IODJ, and AJ is supported by the Ministry of Science Technological Development and Innovation of the Republic of Serbia [Contract Number 451–03-66/2024–03/200015 with Institute for Medical Research University of Belgrade, National Institute of Republic of Serbia].
Author information
Authors and Affiliations
Contributions
DT, MV and RL conceived the topic and subtopics of the review. DT wrote the first draft of the paper. BC performed the language correction, editing, and pointing of contextual clinical relevance. All co-authors reviewed associated literature, reviewed the manuscript, edited the manuscript, and approved the final content of the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Trivanović, D., Vujačić, M., Labella, R. et al. Molecular Deconvolution of Bone Marrow Adipose Tissue Interactions with Malignant Hematopoiesis: Potential for New Therapy Development. Curr Osteoporos Rep 22, 367–377 (2024). https://doi.org/10.1007/s11914-024-00879-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-024-00879-x