
Abstract
Macrophages play an important role in tissue development and homeostasis. They serve as a nexus between adaptive and innate immunity, and employ considerable plasticity. In cancer, they play a pivotal role in chronic inflammation and tumor growth either by directly stimulating the proliferation of cancer cells or by producing angiogenic and lymphangiogenic factors. Although numerous immune cells play an important role in the tumor microenvironment, tumor-associated macrophages (TAMs) are by far the most extensively studied. A better understanding of the role of TAMs in mediating chemo- and radiotherapy resistance and suppressing immunosurveillance has led to numerous strategies targeting TAMs as an anticancer therapy either by targeting them directly or by polarizing TAMs toward a tumoricidal phenotype.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Macrophages are phagocytic cells, critical for our innate and acquired immune response. They not only detect, engulf, and destroy cellular debris, foreign material, and pathogens but also cancer cells [1]. They are the first line of defense against anything that expresses signatures on their surface different from molecules on host cells, namely damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) [2]. Besides phagocytosis and its role in the innate immunity, they can also recruit the other immune cells such as lymphocytes and present antigens to T cells (adaptive immunity).
Macrophages have their origin in the bone marrow, where they pass a monoblast and promonocyte stage to the stage of becoming monocytes which enter the blood system. When inflammation occurs, monocytes undergo a series of changes to become a macrophage when they leave the bloodstream. A majority of macrophages, however, is of embryonic origin (tissue-resident macrophages) and are stationed in certain tissues (the mononuclear phagocytic system) where microbial invasion or accumulation of foreign material is common such as the liver, lymph nodes, and spleen [3, 4]. The main task of these strategically placed macrophages is ingesting foreign material and recruiting additional macrophages if needed. In contrast with circulating blood monocytes which have a half-life of about a day, the life span of tissue macrophages is several months or even years.
The main hallmark of macrophages is their versatile nature, whereby they can play multiple roles in the immune response. Besides the scavenger function, they can present antigens [Major Histocompatibility Complex (MHC) class II] along with dendritic cells and produce cytokines vital to the regulation of the immune response.
Monocytes can be classified as classical (in humans: CD14high, CD16−), intermediate (CD14high, CD16low), and non-classical (CD14low, CD16high) with all different phenotypes [5]. Classical monocytes are involved with phagocytosis and cytokine production, while non-classical can be pro-inflammatory depending on the context. Although considered as separate entities, the current evidence supports the existence of a monocyte continuum rather than incremental differences between the different subtypes [6]. Plasticity of monocytes is also proven by several clinical studies shown their diversity in pathological context. For example, we have shown that colon cancer cell-derived stimuli change their transcriptome [7], while others found a difference in the frequency of non-classical monocytes in breast cancer patients [8]. This plasticity makes them attractive cells for diagnosis and disease follow-up.
Similarly to the concept of a monocyte continuum, macrophages derived from all these monocyte subtypes are considered as well to be a wide spectrum of phenotypes. At its extremes, they can be divided into two main groups designated as M1 and M2, which can be identified by cell surface markers and their functional phenotype. “Killer” M1 macrophages are in vitro activated by interferon-gamma (IFN-γ), lipopolysaccharide, (LPS), and granulocyte–macrophage colony-stimulating factor (GM-CSF or also called colony-stimulating factor 2, CSF2) and secrete pro-inflammatory mediators such as interleukin (IL)-1, IL-12, IL-18, IL-23, and tumor necrosis factor (TNF). Phenotypically, they express high levels of MHC class II, CD68, and co-stimulatory molecules CD80 and CD86. Their main role is pathogen destruction and driving Type 1 T helper (Th1) responses [1]. “Repair” M2 macrophages, on the other hand, are activated in vitro by IL-4 or macrophage colony-stimulating factor (M-CSF or also called colony-stimulating factor 1, CSF1), and they are more involved in processes like wound healing and tissue repair and secrete an anti-inflammatory response by producing anti-inflammatory cytokines such as IL-10. Therefore, they are involved in angiogenesis, extracellular matrix remodeling, and resolution of inflammation [9, 10]. M2 macrophages are further divided into four major types based on their role (M2a, M2b, M2c, and M2d) [11, 12].
The dichotomy between M1 and M2 is believed now to be a more continuous spectrum where insufficient shifts between one of these types could be causative in the pathogenesis and complications of many diseases such as atherosclerosis, muscle regeneration, chronic infections, wound healing, and cancer.
Role of tumor-associated macrophages (TAMs) in cancer
Progression of cancer is not only based on the growth of malignant cells but also on behavior of the components of the tumor microenvironment (TME), which includes various immune cells as well as tumor-associated stromal fibroblasts [13]. However, just the presence of immune cells does not imply that they are activated to kill or stimulate cancer cell growth. The lymphoid component of the TME consists of tumor-infiltrating lymphocytes (TILs) as well as natural killer (NK) cells. Myeloid cells present in the TME include myeloid-derived suppressor cells (MDSCs), granulocytes, dendritic cells (DCs), and TAMs. TAMs include both M1-like cells harboring anti-tumor effector functions as well as M2-like macrophages which, similar to MDSCs, express immunosuppressive and tumor-promoting factors. Both M1- and M2-like TAMs show strong intrinsic plasticity and can cross-regulate each other’s functions and do not represent fixed, frozen phenotypic conditions [14, 15]. TAMs can have, therefore, different effects on the tumor depending on their activation state [16] and, therefore, orchestrate the intratumoral inflammation. In general, higher M1-like infiltrates in the tumor correlate with a better prognosis (anti-tumoral effect), while higher M2-like TAM infiltrate correlates with poor prognosis (pro-tumoral), although M1-like macrophages can also promote malignant transformation by inducing chronic inflammation [17, 18]. In tumors, TAMs mainly originate from bone marrow-derived monocytes [19]. This infiltration is mainly regulated by chemokines such as CCL2, CCL5, CXCL12, and CSF1 (or M-CSF). Once in the tumor, TAMs can undergo a phenotypic switch based on microenvironmental factors such as hypoxia in conjunction with cytokine availability [20]. Moreover, some studies suggest that TAMs can also be activated by exosomes derived from cancer cells [21,22,23]. Exosomes are important signal mediators transferring cancer-associated signaling molecules to surrounding cells such as immune cells.
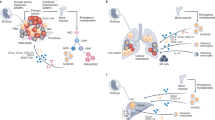
In an early phase, macrophages recognize the malignant cells and present their antigens to the effectors of the immune system. In a later phase, TAMs play a role in tumor progression by stimulating tumor growth, angiogenesis, metastasis, and immunosuppression (Fig. 1) [24]. Qian and Pollard classified TAMs into six functional subtypes: angiogenic, immunosuppressive, invasive, metastasis-associated, perivascular, and activated macrophages [25].

TAMs importantly contributes to all the steps of cancer progression. Pro-tumoral effects of tumor-associated macrophages (TAMs) on cancer cells and the different molecules associated with the specific roles
TAMs are abundant in all the stages of tumor progression [26]. By producing growth factors, they can stimulate carcinoma cell proliferation [27]. They can also produce proteolytic enzymes that digest the extracellular matrix to assist with tumor cell dissemination. Finally, they provide a supportive niche for metastatic tumor cells at distant sites [28].
Role of TAMs in tumor angiogenesis
Due to intensive proliferating and expanding tumor tissue, oxygen demand is surpassed by oxygen supply leading to tumor hypoxia. Cancer cells respond differently to hypoxia leading to cell death or survival depending on the time of exposure of hypoxia. Hypoxia induces the activation of a number of intracellular signaling pathways such as the major hypoxia-inducible factor (HIF) pathway, the PI3K/AKT/mTOR pathway and the NFkβ pathway [29,30,31]. In cancer, these pathways can also be stimulated in a hypoxia-independent manner by growth factors, cytokines, and chemokines or by epigenetic changes and acquired mutations of the members of these pathways, such as mutations in the receptor tyrosine kinase leading to uncontrollable cell growth. However, the generation of tumor blood supply is often a rate-limiting step during tumor progression. Hypoxia induces an imbalance between pro- and anti-angiogenic factors leading to chaotic blood vessel formation. In contrast with normal blood vessels, tumor blood vessels are often abnormal, immature, and leaky. This onset of angiogenesis is called the “angiogenic switch” and can occur at different stages during tumorigenesis [32]. The most common mechanism is sprouting angiogenesis by which new vascular branches arise from pre-existing capillaries or postcapillary venules. Other mechanisms of tumor angiogenesis are vasculogenesis, vascular mimicry, intussusception, and vascular co-option [33].
Angiogenesis is initiated by activation of the Vascular Endothelial Growth Factor Receptor 2 (VEGFR2), expressed on blood endothelial cells, by binding to the ligand VEGF, and also requires the participation of other signaling molecules such as angiopoietin-2 and delta ligand-like 4 [34]. For the last decades, there was a lot of interest in the possibility of inhibiting cancer growth by blocking angiogenesis, for example, with monoclonal antibodies directed against either the VEGF ligand or the VEGF receptors. By contrast, inflammation-associated angiogenesis was rather a neglected field until recently. There is now accumulating evidence that immune cells can be involved in the modulation of angiogenesis [35].
In 1971, Judah Folkman, already suggested that immune cells, in particular macrophages and mast cells, could be a source of pro-angiogenic factors [36]. Later on, multiple studies confirmed that several cells of the innate and adaptive immune system can play a major role in tumor angiogenesis, such as monocytes, macrophages, mast cells, neutrophils, basophils, and eosinophils [37].
Although monocytes were assumed in the past to be only a transient precursor for tissue macrophages, they have now emerged as a highly plastic and dynamic cellular system. They can promote angiogenesis by producing VEGF after the stimulation of different molecules such as cysteinyl-leukotriene D4, M-CSF, and ATP [38,39,40]. On the other hand, CD16− classical monocytes also express VEGFR1 and migrate more when activated by VEGF [41]. In 2005, De Palma et al. identified a unique subset of mouse and human monocytes expressing the tyrosine kinase receptor TIE-2 (TEMs) [42]. These monocytes are a functional distinct myeloid lineage that can induce angiogenesis and tumor growth. They have been identified in different human tumors such as kidney, colon, pancreas, liver, lung, and breast tumors, while they were excluded from surrounding healthy tissue [43,44,45]. In vitro, TIE-2 expressing monocytes migrate toward angiopoietin-2 (Ang-2) released by activated endothelial cells and angiogenic vessels [43]. Furthermore, angiopoietin-2 stimulated TIE-2 expressing monocytes also have an immunosuppressive and, therefore, pro-tumorigenic role by suppressing T-cell proliferation via IL-10 dependent mechanisms [46]. Targeting the Ang-2/TIE-2 axis might, therefore, be interesting approach to inhibit tumor growth [47].
Not only circulating monocytes but also tissue-based TAMs can play a pro-angiogenic role. Preclinical mouse models have shown the functional importance of TAMs in tumor angiogenesis. Here, tumor-infiltrating, VEGF-producing macrophages were shown to facilitate the angiogenic switch and the progression to malignancy, because inactivation of TAMs by blocking the CSF1/CSF1R pathway, or broadly depleting TAMs, or genetic VEGF deletion in macrophages delayed the angiogenic switch, whereas genetic restoration of the macrophage population rescued the angiogenic phenotype [48,49,50]. TAMs also express proteases including matrix metalloproteinases (MMPs), plasmin, urokinase plasminogen activator (uPA), and serine or cysteine proteinases which can facilitate the infiltrative growth of tumor cells in the tumor microenvironment [51].
Another key molecule in angiogenesis is placental growth factor (PLGF), member of the VEGF subfamily. PLGF, produced by both tumor and stromal cells such as macrophages, was shown not only to be a chemoattractant for TAMs but also to play a role in their abnormal polarization [52, 53].
Besides monocytes and TAMs, also other myeloid cells such as neutrophils and MDSCs can play an important role in promoting tumor angiogenesis [54]. T cells, on the other hand, can negatively or positively regulate tumor angiogenesis based on the T-cell type. CD8+ cytotoxic T lymphocytes and CD4+ Th1 cells produce IFNγ that restrains endothelial cell proliferation and induces the production of angiostatic chemokines CXCL9, 10, and 11 in TAMs [55, 56]. In contrast, regulatory T cells (Tregs) suppress INFγ-expressing CD4+ Th1 cells and secrete VEGF via hypoxia-induced CCL28, which both contribute to a pro-angiogenic tumor environment [57].
Role of TAMs in lymphangiogenesis
The lymphatic system primarily functions to regulate tissue fluid homeostasis, as well as collecting antigens and traffic immune cells from the periphery to the lymph nodes [58]. In contrast with blood vessels, they are not encircled by pericytes or smooth muscle cells, which make them highly permeable to interstitial fluids and immune cell migration. Lymphangiogenesis only takes place during embryogenesis and during pathological conditions such as tissue repair, inflammation, and tumor growth [59]. In cancer, the lymphatic system serves as a major route for tumor cell dissemination from the primary tumor site. The most important pro-lymphangiogenic factors are vascular endothelial growth factor (VEGF)-C and -D, which bind to the VEGFR3 expressed on lymphatic endothelium [60, 61]. The expression of VEGF-C in tumors correlates with poor prognosis in several tumor types, in part due to an increase in lymphangiogenesis [62]. Besides VEGFR3 also VEGFR2 and neuropilin receptor NRP-2 play a role in lymphangiogenesis [63, 64]. Several other factors with pro-lymphangiogenic activity have been identified such as hepatocyte growth factor, angiopoietin-1, fibroblast growth factor-1 and -2, platelet derived growth factor, insulin-like growth factor-1 and -2, adrenomedullin, and endothelin-1 [65].
Tumor-induced lymphangiogenesis is mediated by growth factors that are produced by either the tumors themselves or by stromal cells, activated platelets or TAMs. TAMs can promote lymphangiogenesis by expressing VEGF-C and -D [66]. This process is stimulated by cancer cells which activate macrophage-derived lymphangiogenesis by producing interleukin-1α in a highly specific manner [67]. Besides VEGF-C and -D, TAMs also secrete VEGF-A, more characterized for its role in angiogenesis, though this factor plays also an important function in lymph angiogenesis. First, VEGF-A recruits TAMs mostly via the activation of VEGFR1 on macrophages [68, 69], but also it directly induces the proliferation and migration of lymphoid endothelial cells (LECs) via VEGFR2 activation [63].
Macrophages have also been shown to contribute to chemotherapy resistance, for example, by production of cathepsins, leading to increased heparanase activity which in turn induces the expression of VEGF-C, ultimately leading to lymphangiogenesis and metastasis [70, 71].
Role of TAMs in immunosuppression and regulation of adaptive immunity
One of the important roles of the immune system is to eliminate cancer cells [72]. More than 60 years ago, Prehn and Main showed, for the first time, that mice could generate immunity against carcinogen-induced tumors [73]. Since then, many developments in the field of immunology led to a better understanding of the role of immune cells in cancer. Escape of tumor cells from immunosurveillance is a critical event that regulates tumor growth and metastasis. In 2002, Dunn et al. proposed an improved version of the cancer immunosurveillance hypothesis called “tumor immunoediting” [74], resulting in one of three potential outcomes: tumor elimination, an equilibrium with the immune system, or escape from the immune system. The best understood phase of cancer immunosurveillance is the escape phase. For example, many studies have shown that patients with higher tumor-infiltrating T cells within the tumor have a better outcome [75, 76]. In addition, the more advanced tumors are, the less effector immune cells are not only present but also activated within the tumor microenvironment. For example, TAMs in hypoxic areas of the tumor respond to hypoxia with an altered gene expression profile leading to the development of a pro-tumoral phenotype that favors angiogenesis, metastasis, and suppresses anti-tumor immune response [77,78,79]. We have shown that TAMs’ localization into hypoxic tumor areas is controlled by a Sema3A/Neuropilin-1 (NRP-1) signaling axis, leading to PlexinA1/PlexinA4-dependent VEGFR1 activation [68]. Blunting the Sema3A/NRP-1 pathway restored anti-tumor immunity and abated angiogenesis by confining TAMs inside normoxic regions and, thus, inhibiting tumor growth and metastasis [68]. Also other immune cells such as CD1d-restricted invariant natural killer (iNKT) cells contribute to cancer immune surveillance. In a mouse prostate cancer model, Cortesi et al. provide evidence that iNKT cells can remodel the TME by restricting pro-angiogenic TEMs and sustaining pro-inflammatory TAMs by cooperative CD1d, CD40, and Fas engagement [80]. Therefore, low iNKT cells and high TEMs can make cancers more aggressive.
Programmed cell death protein 1 (PD-1) is an immune checkpoint receptor, upregulated on activated T cells for the induction of immune tolerance. Tumor cells frequently overexpress the ligand PD-L1, facilitating their escape from the immune system. Besides its role on inhibition of T cells, a recent publication showed that TAMs also express PD-1 with an inverse correlation between the expression of PD-1 and the phagocytic potency of the TAMs [81]. This suggests that checkpoint inhibitors used in the treatment of many cancers such as melanoma, lung, head and neck, and bladder and renal cancer may also function through a direct effect on macrophages. Moreover, recent data showed that CSF1 expression by melanoma cells may limit the immune attack by activated CD8+ T cells (adaptive resistance mechanism) and that simultaneous blocking of CSF1R with immune checkpoint targeting may be beneficial in cancers refractory to immune checkpoint blockade [82]. On the other hand, a population of PD1-negative TAMs has been involved in buffering anti-PD1 immunoglobulins by subtracting them to their target through the Fcγ receptors (FcγRs). Blockade of FcγRs before anti-PD1 administration enhances the effect of this treatment on cytotoxic T cells and induced the complete rejection of MC38 colorectal tumors in mice [83].
Although immunotherapy in cancer patients has been a clinical success, many patients experienced minimal effect or no clinical effect with the same treatment. We are far from understanding the complexity and diversity of the immune context of the TME and its influence on response to therapy [84]. Although numerous populations of immune cells have been reported to have suppressive functions in the tumor microenvironment, TAMs are the most extensively studied and well characterized.
Role of TAMs in chemoresistance and radioresistance
As TAMs can mediate chemotherapy and radiotherapy resistance by providing survival factors and activate anti-apoptotic programs, targeting TAMs for anticancer therapy has a clear rationale. For example, TAMs can limit the effect of cytoxic agents such as platinum-containing compounds, paclitaxel, gemcitabine, and doxorubicin [85,86,87,88]. De Nardo showed, in a mammary tumor-bearing mouse model, that response to chemotherapy is partly regulated by the tumor immune microenvironment and that cytotoxic drugs such as paclitaxel induce neoplastic cells to produce macrophage recruitment factors, which, in turn, enhance macrophage infiltration into mammary adenocarcinomas leading to tumor progression [85]. Another study in cervical and ovarian cancer cell lines suggests that a platinum chemotherapy-mediated increase in M2 macrophages may form an indirect mechanism for chemoresistance [86]. Finally, in a pancreatic cancer mouse model, Mitchem et al. showed that macrophages can directly induce tumor-initiating cell (TIC) properties by enhancing STAT3 activation and that STAT3+ TICs enhance TAM-mediated immunosuppression. Thus, cross-talk between TAMs and TICs through STAT3 regulates the chemotherapeutic response by repressing anti-tumor cytotoxic T-cell activity [88]. Not only chemotherapy but also radiotherapy plays a major therapeutic role in the treatment of most solid tumors. Next to inducing lethal DNA damages in the tumor, radiotherapy also impacts the tumor microenvironment with its associated immune system. For example, radiotherapy can promote tumor immune response by eliciting immunogenic cell death, and activate tumor antigen release and subsequently immune cell activation. Therefore, radiotherapy can program macrophages leading to either radiosensitization or radioresistance according to the tumor type and the radiotherapy regimen. Many studies have shown that irradiation induces macrophage infiltration in tumors that in turn limits the efficacy of radiotherapy. Moreover, macrophages are also one of the most radioresistant cells, due to a high production of anti-oxidative molecules [89]. In addition to the intrinsic radioresistance of macrophages, radiation also leads to a high recruitment of myeloid cells at the tumor site, possibly leading to tumor regrowth which was shown in an intracranial xenograft model of glioblastoma [90] as well in a pancreatic cancer mouse model [91]. Another study showed that depletion of macrophages by liposomal clodronate before radiation promoted the anti-tumor effects of radiotherapy [92]. After radiotherapy, TAMS accumulate in hypoxic regions of the tumor by induction of the expression of SDF1α, which, upon interaction with its receptor CXCR4, induce the recruitment of macrophages that restored tumor vasculature and promote tumor regrowth [93]. Likewise, the inhibition of CXCR4 was shown to significantly delayed xenograft lung tumor regrowth after radiotherapy [94]. Altogether, it is clear that strategies aiming to target TAMs or TAM functions carry the potential to synergize with standard chemo- and radiotherapeutic treatments.
Metastasis-associated macrophages
Although most research has been focused on TAMs, less is known about the distinct role of the so-called metastasis-associated macrophages (MAMs) which include both tissue-resident macrophages as bone marrow-derived macrophages. They are located at the metastatic site promoting tumor cell extravasation, seeding, and persistent growth. Several studies demonstrated that myeloid cells and, in particular, MAMs are important for the ‘preparation’ of the metastatic niche via the release of matrix proteins at the metastatic sites, and for this reason, these cells are also entrained by the primary tumor into the premetastatic niche before the lodging of cancer cells [95,96,97]. MAMs are derived from circulating monocytes recruited by CCL2/CCR2 chemokine signaling [80]. In a metastatic breast cancer mouse model, Kitamura et al. showed that circulating monocytes differentiate into a distinct myeloid cell population characterized as CD11bhighLy6Chigh in the metastatic lung where they further differentiate into MAMs [98]. These authors also found that accumulation of the CD11bhighLy6Chigh cells was increased when micrometastases started to outgrow. MAMs are enriched for the expression of VEFGR1 and activation of this receptor has been shown to be important for metastatic growth but not for cancer cell extravasation [99, 100]. We have recently shown that VEGFR1 exposure on the cell surface can be restrained by caveolin-1 (likely through the formation of caveolae). CSF2 (or GM-CSF) keeps high the levels of caveolin-1 in the interstitial macrophages of the lungs. It follows that macrophage-associated caveolin-1 is critical for hindering metastasis and represents an intrinsic antimetastatic surveillance mechanism in the pulmonary microenvironment whereby its upregulation prevents excessive exposure of VEGFR1 and thereby limits downstream MMP9 and CSF1 expression, angiogenesis, and finally metastatic growth [99]. Since the physiology of the lung is to counter dangerous signals that are coming from the outside (i.e., airways), it is not surprising that the block of metastasis by this axis in macrophages was seen in lungs but not in livers [99]. On the contrary, MAMs can aid metastatic growth by inhibiting tissue destructive immune response, promoting angiogenesis and cellular growth. Several mechanisms through which these macrophages contribute to metastatic progression have already been explored such as stimulation of the CCL2–CCR2 and the CCL3–CCR1 axis [101,102,103,104]. Further effort will be required to understand how the pro-metastatic axis represented by the CCL2/CCR2–CCL3/CCR1 axis in MAMs is specific for breast cancer and lung metastasis or whether this pathway is also observed in the other tumors and/or metastatic sites. Further understanding of the similarities and differences in TAM and MAM function is critical for developing new therapeutic macrophage targeting agents.
Macrophages as a therapeutic target in cancer treatment
Numerous strategies are being explored to target either macrophages directly or polarizing TAMs toward a tumoricidal phenotype (Fig. 2) [105, 106]. Ways to target directly the biology of macrophages can be achieved by either blocking the myeloid growth factor receptor CSF1R or the monocyte chemoattractant protein CCL2. Zhu et al. demonstrated in a mouse model of pancreatic ductal adenocarcinoma that inhibiting CSF1R can reprogram macrophage responses enhancing antigen presentation leading to anti-tumor T-cell responses [107]. CSF1/CSF1R signaling drives the recruitment and differentiation of TAMs toward an M2 phenotype [108]. Numerous clinical studies are exploring the effect of monoclonal antibodies (ex. emactuzumab and cabiralizumab) or small molecules (ex. pexidartinib) targeting CSF1 or CSF1R, either in monotherapy as in combination with checkpoint inhibitors. The rationale for this combination treatment has been highlighted only recently. In mouse and human melanomas, CSF1 expression was found to correlate with the abundance of CD8+ cytotoxic T cells (CTLs) and CD163+ TAMs. Mechanistically, it was found that, partly because of CTL-release IFN-γ, melanoma cells increase their production of CSF1 as a kind of “defensive, immunosuppressive mechanism” to tone down the CTL response. Combination of anti-PD1 and anti-CSF1 receptor (CSF1R) antibodies induced the regression of BRAFV600E-driven, transplant mouse melanomas, implying CSF1 and TAMs as CD8+ T-cell-dependent adaptive resistance mechanism. This approach has also limitations that probably can partly explain the limited success of this approach in the clinic [109]. Quail et al. have proven that prolonged treatment with CSF1-targeted therapies causes the release of IGF-1 by the TAMs which in turn activates an IGFR-PI3 K cascade that drives resistance and tumor growth [110]. Another mechanism of resistance to anti-CSF1(R) treatment is completely engaged by the stroma. An unprecedentedly described activation of CSFR1 in cancer-associated fibroblasts (CAFs) was found recently responsible for silencing the expression of granulocytic cytokines. It follows that a CSF1 block causes the release of this cytokines with consequent recruitment of granulocytes and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC). This resistance mode is, at least in mice, overcome by the combination of CSF1R inhibitors with a CXCR2 antagonist [111]. Also targeting CCL2 led to tumor regression in a preclinical prostate cancer tumor model [112]. By blocking CCL2, not only recruitment of monocytes at the tumor site is inhibited but also the retention of metastasis-associated macrophages. Carlumab (CNT088), a human monoclonal antibody against CCL2, was evaluated in solid tumors in which it was administered either in monotherapy as well as in combination with the other chemotherapy agents [113]. The study did not show prolonged inhibition of CCL2, probably because the drug bound-free CLL2 with lesser affinity than reported from in vitro studies, making it less efficient in inhibiting CCL2 in humans [114]. Genetic and pharmacologic evidence has shown that CCL2 inhibition of MAMs in breast cancer metastasis is efficient till the treatment is endorsed [102, 115]. However, upon treatment withdrawal, a strong increase in CCL2 and other factors such as IL6 and VEGF was boosting macrophage release from the bone marrow to the metastatic site with poor disease outcome. This indicates that macrophage phenotype manipulation rather than a block in macrophage recruitment can be a safer therapeutic option [53]. Trabectedin, a marine-derived antineoplastic drug, currently used for the treatment of sarcomas, has not only direct effects against cancer cells, but also host-modulating properties such as depletion of TAMs as well as inhibition of monocyte recruitment and angiogenesis [116]. The mechanism of action is very complex, as it binds to DNA, blocks transcription, and also interferes with the DNA repair efficiency. Trabectedin is very cytotoxic for TAMs by engaging monocyte-specific TRAIL receptors 1 and 2 and mediating a caspase 8 dependent apoptosis [117].

Different ways to target tumor-associated macrophages (TAMs) and potential mechanisms of resistance
Another key molecule involved in innate and adaptive immunity is CD40, a member of the TNF receptor superfamily, expressed on antigen presenting cells such as monocytes, macrophages, and dendritic cells. Many studies have shown that administration of an agonistic antibody directed against CD40 produced protective T-cell immunity in murine cancer models [118, 119]. Also CD47, known as integrin association protein, belonging to the immunoglobin superfamily is an interesting therapeutic target. CD47 acts as a “don’t eat me” signal to macrophages. CD47 is found to be overexpressed on cancer cells and interaction with signal-regulatory protein alpha (SIRPα) inhibits macrophage phagocytosis, allowing cancer cells to escape immune surveillance [120, 121]. Current CD47 antagonists undergoing clinical trials include, for example, Hu5F9 (an anti-CD47 antibody that directly inhibits the CD47-SIRPα interaction) and TTI-621, (a fusion protein composed of CD47-binding domain of human SIRPα and linked to the Fc region of IgG1).
Next to strategies that deplete (anti-CSF-1 antibodies and CSF-1R inhibition) or stimulate (agonistic anti-CD40 or inhibitory anti-CD47 antibodies) TAMs also pharmacologic modulation of macrophage phenotype could produce an anti-tumour effect which has been shown in a recent publication by Guerriero et al. [122]. By utilizing a macrophage-dependent autochthonous mouse model of breast cancer, they demonstrated that treatment with a class IIa histone deacetylase (HDAC) inhibitor altered the tumor microenvironment and reduced tumor burden and metastases by modulating macrophage phenotypes. Moreover, combination with chemotherapy regimens or checkpoint inhibitors significantly enhanced the durability of tumor reduction.
Interleukin-6 (IL-6) is a cytokine relevant in many inflammatory diseases and cancer. Several agents targeting either IL-6 itself (siltuximab, olokizumab, and sirukumab) or its receptor IL6-R (tocilizumab) are currently in clinical trials for inflammatory diseases such as rheumatoid arthritis but also cancer [123]. Remarkably, tocilizumab is also used in the clinic for the treatment of severe or life-threatening chimeric antigen receptor (CAR) T-cell induced cytokine release syndrome [124].
A protein called stimulator of interferon genes (STING) is part of the innate immune system as the first line of defense against pathogens. When activated, STING enforces the production of interferons and cytokines. One of the first STING analogs DMXAA (vadimezan) showed promising results in many preclinical models, but failed in a phase III clinical trial in non-small cell lung cancer [125, 126]. The lack of efficiency might be explained by DMXAA not binding to human STING. Many companies are, therefore, currently developing STING agonists or agents targeting the STING pathway, to be tested in clinical trials either in monotherapy or in combination with checkpoint inhibitors.
Finally, as hypoxia in the tumor microenvironment is a major factor that polarizes TAMs to a pro-tumoral phenotype [68], reduction of hypoxia can be an alternative approach to induce anti-tumor TAMs. This could be achieved by inducing vascular normalization through the inhibition of VEGF and angiopoietin-2. For example, a preclinical study published by Peterson et al., showed that dual inhibition of VEGFR and Ang-2 prolonged survival in glioblastoma models by reducing tumor burden, improving vessel normalization, and altering TAMs [127]. Overall, from the original aim of depleting TAMs from the tumor, the current strategy is to re-educate macrophages towards their more ancestral function, i.e., to protect the body against harmful stimuli.
References
Mosser DM, Edwards JP (2008) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8:958–969. https://doi.org/10.1038/nri2448
Tang D, Kang R, Coyne CB et al (2012) PAMPs and DAMPs: signal 0 s that spur autophagy and immunity. Immunol Rev 249:158–175. https://doi.org/10.1111/j.1600-065X.2012.01146.x
Mass E, Ballesteros I, Farlik M et al (2016) Specification of tissue-resident macrophages during organogenesis. Science 353:aaf4238. https://doi.org/10.1126/science.aaf4238
O’Neill LAJ, Golenbock D, Bowie AG (2013) The history of Toll-like receptors—redefining innate immunity. Nat Rev Immunol 13:453–460. https://doi.org/10.1038/nri3446
Ginhoux F, Jung S (2014) Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14:392–404. https://doi.org/10.1038/nri3671
Cignarella A, Tedesco S, Cappellari R, Fadini GP (2018) The continuum of monocyte phenotypes: experimental evidence and prognostic utility in assessing cardiovascular risk. J Leukoc Biol. https://doi.org/10.1002/jlb.5ru1217-477rr
Hamm A, Prenen H, Van Delm W et al (2016) Tumour-educated circulating monocytes are powerful candidate biomarkers for diagnosis and disease follow-up of colorectal cancer. Gut 65:990–1000. https://doi.org/10.1136/gutjnl-2014-308988
Feng A-L, Zhu J-K, Sun J-T et al (2011) CD16+ monocytes in breast cancer patients: expanded by monocyte chemoattractant protein-1 and may be useful for early diagnosis. Clin Exp Immunol 164:57–65. https://doi.org/10.1111/j.1365-2249.2011.04321.x
Takeda Y, Costa S, Delamarre E et al (2011) Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 479:122–126. https://doi.org/10.1038/nature10507
Madsen DH, Leonard D, Masedunskas A et al (2013) M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol 202:951–966. https://doi.org/10.1083/jcb.201301081
Mantovani A, Sozzani S, Locati M et al (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555
Duluc D, Corvaisier M, Blanchard S et al (2009) Interferon-γ reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer 125:367–373. https://doi.org/10.1002/ijc.24401
Junttila MR, de Sauvage FJ (2013) Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501:346–354. https://doi.org/10.1038/nature12626
Sica A, Mantovani A (2012) Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 122:787–795. https://doi.org/10.1172/JCI59643
Biswas SK, Sica A, Lewis CE (2008) Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol 180:2011–2017
Zarif JC, Taichman RS, Pienta KJ (2014) TAM macrophages promote growth and metastasis within the cancer ecosystem. Oncoimmunology 3:e941734. https://doi.org/10.4161/21624011.2014.941734
Qian B-Z, Pollard JW (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141:39–51. https://doi.org/10.1016/j.cell.2010.03.014
Elinav E, Nowarski R, Thaiss CA et al (2013) Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 13:759–771. https://doi.org/10.1038/nrc3611
Franklin RA, Liao W, Sarkar A et al (2014) The cellular and molecular origin of tumor-associated macrophages. Science (80-) 344:921–925. https://doi.org/10.1126/science.1252510
Henze A-T, Mazzone M (2016) The impact of hypoxia on tumor-associated macrophages. J Clin Invest 126:3672–3679. https://doi.org/10.1172/JCI84427
Xiao M, Zhang J, Chen W, Chen W (2018) M1-like tumor-associated macrophages activated by exosome-transferred THBS1 promote malignant migration in oral squamous cell carcinoma. J Exp Clin Cancer Res 37:143. https://doi.org/10.1186/s13046-018-0815-2
Chen X, Ying X, Wang X et al (2017) Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol Rep 38:522–528. https://doi.org/10.3892/or.2017.5697
Hsieh C-H, Tai S-K, Yang M-H (2018) Snail-overexpressing cancer cells promote M2-like polarization of tumor-associated macrophages by delivering MiR-21-abundant exosomes. Neoplasia 20:775–788. https://doi.org/10.1016/j.neo.2018.06.004
Mittal D, Gubin MM, Schreiber RD, Smyth MJ (2014) New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol 27:16–25. https://doi.org/10.1016/j.coi.2014.01.004
Qian B-Z, Pollard JW (2012) New tricks for metastasis-associated macrophages. Breast Cancer Res 14:316. https://doi.org/10.1186/bcr3143
Ruffell B, Coussens LM (2015) Macrophages and therapeutic resistance in cancer. Cancer Cell 27:462–472. https://doi.org/10.1016/j.ccell.2015.02.015
Mantovani A, Allavena P, Sica A, Balkwill F (2008) Cancer-related inflammation. Nature 454:436–444. https://doi.org/10.1038/nature07205
Mantovani A, Allavena P (2015) The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med 212:435–445. https://doi.org/10.1084/jem.20150295
Koong AC, Chen EY, Mivechi NF et al (1994) Hypoxic activation of nuclear factor-kappa B is mediated by a Ras and Raf signaling pathway and does not involve MAP kinase (ERK1 or ERK2). Cancer Res 54:5273–5279
Minet E, Arnould T, Michel G et al (2000) ERK activation upon hypoxia: involvement in HIF-1 activation. FEBS Lett 468:53–58
Keith B, Johnson RS, Simon MC (2011) HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer 12:9–22. https://doi.org/10.1038/nrc3183
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3:401–410. https://doi.org/10.1038/nrc1093
Leite de Oliveira R, Hamm A, Mazzone M (2011) Growing tumor vessels: more than one way to skin a cat—implications for angiogenesis targeted cancer therapies. Mol Aspects Med 32:71–87. https://doi.org/10.1016/j.mam.2011.04.001
Potente M, Gerhardt H, Carmeliet P (2011) Basic and therapeutic aspects of angiogenesis. Cell 146:873–887. https://doi.org/10.1016/j.cell.2011.08.039
Varricchi G, Loffredo S, Galdiero MR et al (2018) Innate effector cells in angiogenesis and lymphangiogenesis. Curr Opin Immunol 53:152–160. https://doi.org/10.1016/j.coi.2018.05.002
Sherwood LM, Parris EE, Folkman J (1971) Tumor Angiogenesis: therapeutic Implications. N Engl J Med 285:1182–1186. https://doi.org/10.1056/NEJM197111182852108
De Palma M, Biziato D, Petrova TV (2017) Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer 17:457–474. https://doi.org/10.1038/nrc.2017.51
Eubank TD, Galloway M, Montague CM et al (2003) M-CSF induces vascular endothelial growth factor production and angiogenic activity from human monocytes. J Immunol 171:2637–2643
Hill LM, Gavala ML, Lenertz LY, Bertics PJ (2010) Extracellular ATP may contribute to tissue repair by rapidly stimulating purinergic receptor X7-dependent vascular endothelial growth factor release from primary human monocytes. J Immunol 185:3028–3034. https://doi.org/10.4049/jimmunol.1001298
Poulin S, Thompson C, Thivierge M et al (2011) Cysteinyl-leukotrienes induce vascular endothelial growth factor production in human monocytes and bronchial smooth muscle cells. Clin Exp Allergy 41:204–217. https://doi.org/10.1111/j.1365-2222.2010.03653.x
Czepluch FS, Olieslagers S, van Hulten R et al (2011) VEGF-A-induced chemotaxis of CD16+ monocytes is decreased secondary to lower VEGFR-1 expression. Atherosclerosis 215:331–338. https://doi.org/10.1016/j.atherosclerosis.2011.01.004
De Palma M, Venneri MA, Galli R et al (2005) Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell 8:211–226. https://doi.org/10.1016/j.ccr.2005.08.002
Venneri MA, Palma MD, Ponzoni M et al (2007) Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 109:5276–5285. https://doi.org/10.1182/blood-2006-10-053504
Matsubara T, Kanto T, Kuroda S et al (2013) TIE2-expressing monocytes as a diagnostic marker for hepatocellular carcinoma correlates with angiogenesis. Hepatology 57:1416–1425. https://doi.org/10.1002/hep.25965
Turrini R, Pabois A, Xenarios I et al (2017) TIE-2 expressing monocytes in human cancers. Oncoimmunology 6:e1303585. https://doi.org/10.1080/2162402X.2017.1303585
Coffelt SB, Chen Y-Y, Muthana M et al (2011) Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol 186:4183–4190. https://doi.org/10.4049/jimmunol.1002802
Mazzieri R, Pucci F, Moi D et al (2011) Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19:512–526. https://doi.org/10.1016/j.ccr.2011.02.005
Lin EY, Li J-F, Gnatovskiy L et al (2006) Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66:11238–11246. https://doi.org/10.1158/0008-5472.CAN-06-1278
Stockmann C, Doedens A, Weidemann A et al (2008) Deletion of vascular endothelial growth factor in myeloid cells accelerates tumorigenesis. Nature 456:814–818. https://doi.org/10.1038/nature07445
Priceman SJ, Sung JL, Shaposhnik Z et al (2010) Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood 115:1461–1471. https://doi.org/10.1182/blood-2009-08-237412
Kessenbrock K, Plaks V, Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141:52–67. https://doi.org/10.1016/j.cell.2010.03.015
Fischer C, Jonckx B, Mazzone M et al (2007) Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 131:463–475. https://doi.org/10.1016/j.cell.2007.08.038
Rolny C, Mazzone M, Tugues S et al (2011) HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 19:31–44. https://doi.org/10.1016/j.ccr.2010.11.009
Nozawa H, Chiu C, Hanahan D (2006) Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci USA 103:12493–12498. https://doi.org/10.1073/pnas.0601807103
Mantovani A (2010) Molecular pathways linking inflammation and cancer. Curr Mol Med 10:369–373
DeNardo DG, Barreto JB, Andreu P et al (2009) CD4+ T Cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16:91–102. https://doi.org/10.1016/j.ccr.2009.06.018
Facciabene A, Peng X, Hagemann IS et al (2011) Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 475:226–230. https://doi.org/10.1038/nature10169
Tammela T, Alitalo K (2010) Lymphangiogenesis: molecular mechanisms and future promise. Cell 140:460–476. https://doi.org/10.1016/j.cell.2010.01.045
Wang Y, Oliver G (2010) Current views on the function of the lymphatic vasculature in health and disease. Genes Dev 24:2115–2126. https://doi.org/10.1101/gad.1955910
Joukov V, Pajusola K, Kaipainen A et al (1996) A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J 15:290–298
Achen MG, Jeltsch M, Kukk E et al (1998) Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci U S A 95:548–553
Alitalo A, Detmar M (2012) Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 31:4499–4508. https://doi.org/10.1038/onc.2011.602
Hong Y-K (2004) VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the 1 1 and 2 1 integrins. FASEB J 18:1111–1113. https://doi.org/10.1096/fj.03-1179fje
Yuan L, Moyon D, Pardanaud L et al (2002) Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development 129:4797–4806
Christiansen A, Detmar M (2011) Lymphangiogenesis and cancer. Genes Cancer 2:1146–1158. https://doi.org/10.1177/1947601911423028
Tammela T, Petrova TV, Alitalo K (2005) Molecular lymphangiogenesis: new players. Trends Cell Biol 15:434–441. https://doi.org/10.1016/j.tcb.2005.06.004
Watari K, Shibata T, Kawahara A et al (2014) Tumor-derived interleukin-1 promotes lymphangiogenesis and lymph node metastasis through M2-type macrophages. PLoS One 9:e99568. https://doi.org/10.1371/journal.pone.0099568
Casazza A, Laoui D, Wenes M et al (2013) Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 24:695–709. https://doi.org/10.1016/j.ccr.2013.11.007
Cursiefen C, Chen L, Borges LP et al (2004) VEGF-A stimulates lymphangiogenesis and hemangiogenesis in inflammatory neovascularization via macrophage recruitment. J Clin Invest 113:1040–1050. https://doi.org/10.1172/JCI20465
Shree T, Olson OC, Elie BT et al (2011) Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev 25:2465–2479. https://doi.org/10.1101/gad.180331.111
Alishekevitz D, Gingis-Velitski S, Kaidar-Person O et al (2016) Macrophage-induced lymphangiogenesis and metastasis following paclitaxel chemotherapy is regulated by VEGFR3. Cell Rep 17:1344–1356. https://doi.org/10.1016/j.celrep.2016.09.083
Thomas L (1982) On immunosurveillance in human cancer. Yale J Biol Med 55:329–333
Prehn RT, Main JM (1957) Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst 18:769–778
Dunn GP, Bruce AT, Ikeda H et al (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3:991–998. https://doi.org/10.1038/ni1102-991
Pagès F, Mlecnik B, Marliot F et al (2018) International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 391:2128–2139. https://doi.org/10.1016/S0140-6736(18)30789-X
Galon J, Costes A, Sanchez-Cabo F et al (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (80-) 313:1960–1964. https://doi.org/10.1126/science.1129139
Burke B, Giannoudis A, Corke KP et al (2003) Hypoxia-induced gene expression in human macrophages. Am J Pathol 163:1233–1243. https://doi.org/10.1016/S0002-9440(10)63483-9
Doedens AL, Stockmann C, Rubinstein MP et al (2010) macrophage expression of hypoxia-inducible factor-1 suppresses T-cell function and promotes tumor progression. Cancer Res 70:7465–7475. https://doi.org/10.1158/0008-5472.CAN-10-1439
Movahedi K, Laoui D, Gysemans C et al (2010) Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 70:5728–5739. https://doi.org/10.1158/0008-5472.CAN-09-4672
Cortesi F, Delfanti G, Grilli A et al (2018) Bimodal CD40/Fas-dependent crosstalk between iNKT cells and tumor-associated macrophages impairs prostate cancer progression. Cell Rep 22:3006–3020. https://doi.org/10.1016/j.celrep.2018.02.058
Gordon SR, Maute RL, Dulken BW et al (2017) PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545:495–499. https://doi.org/10.1038/nature22396
Neubert NJ, Schmittnaegel M, Bordry N et al (2018) T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med 10:eaan3311. https://doi.org/10.1126/scitranslmed.aan3311
Arlauckas SP, Garris CS, Kohler RH et al (2017) In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med 9:eaal3604. https://doi.org/10.1126/scitranslmed.aal3604
Binnewies M, Roberts EW, Kersten K et al (2018) Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 24:541–550. https://doi.org/10.1038/s41591-018-0014-x
DeNardo DG, Brennan DJ, Rexhepaj E et al (2011) Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov 1:54–67. https://doi.org/10.1158/2159-8274.CD-10-0028
Dijkgraaf EM, Heusinkveld M, Tummers B et al (2013) Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res 73:2480–2492. https://doi.org/10.1158/0008-5472.CAN-12-3542
Jinushi M, Chiba S, Yoshiyama H et al (2011) Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci USA 108:12425–12430. https://doi.org/10.1073/pnas.1106645108
Mitchem JB, Brennan DJ, Knolhoff BL et al (2013) Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res 73:1128–1141. https://doi.org/10.1158/0008-5472.CAN-12-2731
Tsai C-S, Chen F-H, Wang C-C et al (2007) Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int J Radiat Oncol Biol Phys 68:499–507. https://doi.org/10.1016/j.ijrobp.2007.01.041
Stafford JH, Hirai T, Deng L et al (2016) Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol 18:797–806. https://doi.org/10.1093/neuonc/nov272
Kalbasi A, Komar C, Tooker GM et al (2017) Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res 23:137–148. https://doi.org/10.1158/1078-0432.CCR-16-0870
Meng Y, Beckett MA, Liang H et al (2010) Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res 70:1534–1543. https://doi.org/10.1158/0008-5472.CAN-09-2995
Kioi M, Vogel H, Schultz G et al (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest 120:694–705. https://doi.org/10.1172/JCI40283
Kozin SV, Kamoun WS, Huang Y et al (2010) Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res 70:5679–5685. https://doi.org/10.1158/0008-5472.CAN-09-4446
Kaplan RN, Riba RD, Zacharoulis S et al (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438:820–827. https://doi.org/10.1038/nature04186
Erler JT, Bennewith KL, Cox TR et al (2009) Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15:35–44. https://doi.org/10.1016/j.ccr.2008.11.012
Hiratsuka S, Duda DG, Huang Y et al (2011) C-X-C receptor type 4 promotes metastasis by activating p38 mitogen-activated protein kinase in myeloid differentiation antigen (Gr-1)-positive cells. Proc Natl Acad Sci 108:302–307. https://doi.org/10.1073/pnas.1016917108
Kitamura T, Doughty-Shenton D, Cassetta L et al (2017) Monocytes differentiate to immune suppressive precursors of metastasis-associated macrophages in mouse models of metastatic breast cancer. Front Immunol 8:2004. https://doi.org/10.3389/fimmu.2017.02004
Celus W, Di Conza G, Oliveira AI et al (2017) Loss of caveolin-1 in metastasis-associated macrophages drives lung metastatic growth through increased angiogenesis. Cell Rep 21:2842–2854. https://doi.org/10.1016/j.celrep.2017.11.034
Qian B-Z, Zhang H, Li J et al (2015) FLT1 signaling in metastasis-associated macrophages activates an inflammatory signature that promotes breast cancer metastasis. J Exp Med 212:1433–1448. https://doi.org/10.1084/jem.20141555
Qian B, Deng Y, Im JH et al (2009) A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One 4:e6562. https://doi.org/10.1371/journal.pone.0006562
Qian B-Z, Li J, Zhang H et al (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475:222–225. https://doi.org/10.1038/nature10138
Kitamura T, Qian B-Z, Soong D et al (2015) CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med 212:1043–1059. https://doi.org/10.1084/jem.20141836
Headley MB, Bins A, Nip A et al (2016) Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531:513–517. https://doi.org/10.1038/nature16985
Quail DF, Joyce JA (2017) Molecular pathways: deciphering mechanisms of resistance to macrophage-targeted therapies. Clin Cancer Res 23:876–884. https://doi.org/10.1158/1078-0432.CCR-16-0133
Noy R, Pollard JW (2014) Tumor-associated macrophages: from mechanisms to therapy. Immunity 41:49–61. https://doi.org/10.1016/j.immuni.2014.06.010
Zhu Y, Knolhoff BL, Meyer MA et al (2014) CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res 74:5057–5069. https://doi.org/10.1158/0008-5472.CAN-13-3723
Pyonteck SM, Akkari L, Schuhmacher AJ et al (2013) CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 19:1264–1272. https://doi.org/10.1038/nm.3337
Peyraud F, Cousin S, Italiano A (2017) CSF-1R inhibitor development: current clinical status. Curr Oncol Rep 19:70. https://doi.org/10.1007/s11912-017-0634-1
Quail DF, Bowman RL, Akkari L et al (2016) The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science (80-) 352:aad3018. https://doi.org/10.1126/science.aad3018
Kumar V, Donthireddy L, Marvel D et al (2017) Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC Infiltration of tumors. Cancer Cell 32:654.e5–668.e5. https://doi.org/10.1016/j.ccell.2017.10.005
Loberg RD, Ying C, Craig M et al (2007) Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res 67:9417–9424. https://doi.org/10.1158/0008-5472.CAN-07-1286
Brana I, Calles A, LoRusso PM et al (2015) Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol 10:111–123. https://doi.org/10.1007/s11523-014-0320-2
Sandhu SK, Papadopoulos K, Fong PC et al (2013) A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol 71:1041–1050. https://doi.org/10.1007/s00280-013-2099-8
Bonapace L, Coissieux M-M, Wyckoff J et al (2014) Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 515:130–133. https://doi.org/10.1038/nature13862
D’Incalci M, Badri N, Galmarini CM, Allavena P (2014) Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br J Cancer 111:646–650. https://doi.org/10.1038/bjc.2014.149
Guerriero JL (2018) Macrophages: the road less traveled, changing anticancer therapy. Trends Mol Med 24:472–489. https://doi.org/10.1016/j.molmed.2018.03.006
Beatty GL, Chiorean EG, Fishman MP et al (2011) CD40 Agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science (80-) 331:1612–1616. https://doi.org/10.1126/science.1198443
Beatty GL, Li Y, Long KB (2017) Cancer immunotherapy: activating innate and adaptive immunity through CD40 agonists. Expert Rev Anticancer Ther 17:175–186. https://doi.org/10.1080/14737140.2017.1270208
Folkes AS, Feng M, Zain JM et al (2018) Targeting CD47 as a cancer therapeutic strategy. Curr Opin Oncol. https://doi.org/10.1097/cco.0000000000000468
Willingham SB, Volkmer J-P, Gentles AJ et al (2012) The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci 109:6662–6667. https://doi.org/10.1073/pnas.1121623109
Guerriero JL, Sotayo A, Ponichtera HE et al (2017) Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543:428–432. https://doi.org/10.1038/nature21409
Poh AR, Ernst M (2018) Targeting macrophages in cancer: from bench to bedside. Front Oncol 8:49. https://doi.org/10.3389/fonc.2018.00049
Le RQ, Li L, Yuan W et al (2018) FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T Cell-induced severe or life-threatening cytokine release syndrome. Oncologist 23:943–947. https://doi.org/10.1634/theoncologist.2018-0028
Daei Farshchi Adli A, Jahanban-Esfahlan R, Seidi K et al (2018) An overview on Vadimezan (DMXAA): the vascular disrupting agent. Chem Biol Drug Des 91:996–1006. https://doi.org/10.1111/cbdd.13166
Cheng B, Yuan W-E, Su J et al (2018) Recent advances in small molecule based cancer immunotherapy. Eur J Med Chem 157:582–598. https://doi.org/10.1016/j.ejmech.2018.08.028
Peterson TE, Kirkpatrick ND, Huang Y et al (2016) Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci USA 113:4470–4475. https://doi.org/10.1073/pnas.1525349113
Acknowledgements
Hans Prenen is a Senior Clinical investigator of the Belgian Foundation against Cancer. This study was funded by H2020 European Research Council (Grant nos. OxyMO, 308459).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Prenen, H., Mazzone, M. Tumor-associated macrophages: a short compendium. Cell. Mol. Life Sci. 76, 1447–1458 (2019). https://doi.org/10.1007/s00018-018-2997-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2997-3