
Abstract
Dendritic cells (DC) play a pivotal role in the tumor microenvironment (TME). As the primary antigen-presenting cells in the tumor, DCs modulate anti-tumor responses by regulating the magnitude and duration of infiltrating cytotoxic T lymphocyte responses. Unfortunately, due to the immunosuppressive nature of the TME, as well as the inherent plasticity of DCs, tumor DCs are often dysfunctional, a phenomenon that contributes to immune evasion. Recent progresses in our understanding of tumor DC biology have revealed potential molecular targets that allow us to improve tumor DC immunogenicity and cancer immunotherapy. Here, we review the molecular mechanisms that drive tumor DC dysfunction. We discuss recent advances in our understanding of tumor DC ontogeny, tumor DC subset heterogeneity, and factors in the tumor microenvironment that affect DC recruitment, differentiation, and function. Finally, we describe potential strategies to optimize tumor DC function in the context of cancer therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Although immunotherapy holds much promise for treating cancer, this therapy is not consistently effective, and many patients derive little or no benefits [1]. Understanding the nature of intra-tumor immune responses and the mechanisms that enable tumors to escape immune attack remains an urgent and daunting challenge for tumor immunologists. Current efforts to improve outcomes have mostly focused on ways to increase the number and specificity of cytotoxic T lymphocytes (CTLs) and to block molecules that are thought to impair CTL function in the tumor microenvironment. Dendritic cells (DCs) are the most potent professional antigen-presenting cells and play a pivotal role in adaptive immunity [2, 3]. In patients with cancer, defective DC function is considered a key cause of impaired immune responses to antigens expressed by tumors [4]. Recent efforts to define the DC compartment in tumors have revealed an unexpected level of diversity and complexity, which has opened new opportunities to improve immunotherapy. In this review, we discuss advances in the understanding of tumor DC ontogeny, tumor DC subset heterogeneity, factors in the tumor microenvironment (TME) that effect DC recruitment, differentiation, and function, and potential strategies to optimize their function.
DC overview
Dendritic cells comprise a heterogeneous population of cells specialized in antigen capture, processing, and presentation. They occur in trace numbers in all tissues, forming a sentinel network that links innate and adaptive immunity. DCs are subdivided currently into four major categories: the conventional or classic DC (cDC); interferon-producing plasmacytoid DC (pDC); monocyte-derived DC; and Langerhans cells. cDC is further classified based on location, surface markers, function, and, more recently, by transcription factor expression.
The hallmark function of DCs is to prime naïve T cells for adaptive immunity, which is influenced by their maturation status, subtype, and cell number [2]. Under the steady-state conditions, DCs exist in an immature state characterized by avid uptake of antigens and low expression of major histocompatibility complex (MHC) and co-stimulatory (e.g., CD80, CD86, and CD40) molecules. Antigen presentation by immature DCs promotes immune tolerance [5, 6]. DC maturation refers to a process that increases cell-surface expression of MHC/peptide complexes and co-stimulatory molecules and production of cytokines critical for T-cell activation [7]. This process can be initiated by the recognition of pathogen-associated molecular patterns and endogenous “danger” signals through pattern-recognition receptors and by inflammatory cytokines [8]. Immune defenses also depend on maintaining the optimal number of DCs in peripheral tissues: DC depletion decreases adaptive immune responses to nominal antigens and pathogens [9, 10], whereas persistently high numbers of lymphoid tissue DC elevate the risk of autoimmune disease [11, 12]. Many studies in patients with cancer and in animal tumor models have documented reduced numbers and immaturity of cDCs in tumors and blood [13–19].
With the exception of LCs, which self-renew mostly in situ, DCs originate from bone marrow progenitors. Findings from our lab and others over the past decade underlie the current view of DC and macrophage ontogeny [20–22] (Fig. 1). In this model, DCs arise from a bipotent bone marrow monocyte/macrophage and DC progenitor (MDP) that give rise to common monocyte progenitors (cMoP) and common DC precursors (CDPs). CDPs differentiate into interferon-producing plasmacytoid DC (pDC), which completes their development in BM, and pre-DCs, an immediate precursor of classical DC (cDC) that migrates rapidly via blood into peripheral lymphoid and non-lymphoid tissues [23–31]. Pre-DCs differentiate into two main cDC subpopulations: (1) lymphoid-tissue resident CD8α+ CD11b− cDC and tissue CD103+ CD11b− cDCs (cDC1), which specialize in cross presentation of exogenous antigens on MHC-I molecules to CD8+ T cells [32]; and (2) CD11b+ cDC (cDC2), which have a dominant role in presenting endogenous antigens on MHCII to CD4+ T cells [33]. Recent evidence suggests that commitment towards these subsets occurs in the bone marrow [31]. Analogous counterparts of cDC1 and cDC2 in the human are CD141+ DCs (also known as BDCA3+) and CD1c+ DCs (also known as BDCA1+), respectively [21]. Circulating monocytes participate in the generation of cDC in some tissues [34–36].

Current model of DC ontogeny. This figure shows the developmental pathways for DC and monocytes/macrophages in peripheral tissues and tumors. The cell-surface molecules that are used commonly to distinguish these cell populations are shown. HSC hematopoietic stem cell, CMP common myeloid progenitor, CLP common lymphoid progenitor, MDP monocyte dendritic cell progenitor, CDP common dendritic cell progenitor, Pre-cDC immediate precursor of classical DCs, pDC plasmacytoid DCs
During the steady-state conditions, DC homeostasis in lymphoid and non-lymphoid tissues reflects a balance between the influx of new precursors, DC emigration, and cell death. Flt3 ligand (Flt3L), which is expressed by stromal cells in BM and lymphoid tissues and by activated T cells, plays an essential role by driving proliferation and differentiation of Flt3+ bone marrow progenitors and pre-DCs [37–39]. In mice and humans, overexpression or injection of Flt3L markedly increases the number cDCs and pDCs in blood and in lymphoid and non-lymphoid tissues [39–42]. Notch 2 signaling controls differentiation of pre-DC-derived cDC2 in spleen and lymphotoxin β receptor signaling regulates cDC2 development as well as cDC proliferation [43, 44]. Retinoic acid signaling in pre-cDCs influences differentiation and homeostasis of CD11b+CD8α− cDCs in the spleen and CD11b+CD103+cDCs in the intestine [45].
cDC numbers expand during inflammatory processes through recruitment of new precursors, augmentation of cDC proliferation in situ, and differentiation of monocytes into ‘inflammatory’ DC [25, 46–49]. Increased expression of granulocyte/macrophage colony-stimulating factor (GM-CSF, also known as CSF-2) contributes to this response [50, 51]. Inflammation also causes egress of tissue DC into lymph nodes by modulating chemokine receptor expression and altering the structure of regional lymphatics and lymph nodes [52–54].
Transcription factors (TFs) guide DC development from hematopoietic progenitors [55]. Some TFs, such as Ikaros and PU.1, are critical for all DC as well as other myeloid populations. Gfi-1, a transcriptional repressor, promotes DC over macrophage differentiation at the MDP stage. E2-2 expression is absolutely essential for the development and maintenance of pDC [56, 57], whereas ID2 and the recently identified TF, Zbtb46, are expressed at high levels in all cDC [29, 30, 58]. CD8+ cDC and CD103+ CD11b− cDC express high levels of IRF8, Batf3, and NFIL3 [32, 59, 60]; CD11b+ cDC express high levels of relB and IRF4 [59, 61]. These findings in mice have been shown to be highly relevant to human DC ontogeny. For example, mutation of Gata2 and IRF8 causes DC deficiency syndromes that increase susceptibility to mycobacterial and fungal infections [62, 63].
DC and anti-tumor immune responses
Inflammation is a well-recognized component of many cancers [64]. Chemokines, cytokines, and growth factors in tumors promote the influx of infiltrating cells, including DCs, and the generation of stromal elements. Cancers express a wide range of tumor-associated and tumor-specific antigens [65, 66]. It is generally thought that DCs in tumors capture tumor antigens and migrate to draining lymph nodes, where they prime and activate tumor-specific T cells [67]. Memory and effector CTLs return to the tumor to perform immunosurveillance activities. Evidence suggests that this process can prevent tumor development and influence the rate of tumor progression [68, 69]. For most patients, however, natural anti-tumor immune defenses fail to control the cancer.
The success of cancer immunotherapy relies on augmenting numbers of functional CTL in the tumor [70]. Through intensive efforts over the past few decades, it is now possible to generate large numbers of tumor-antigen-specific T cells [65]; however, the overall efficacy of this therapy is still poor. Tumors can evade immune attack through many mechanisms, including “immunoediting”, a process that progressively selects less immunogenic tumor cells [71, 72], immune checkpoint inhibition through CTLA4, PD-1/PD-L1 and other molecules, and active suppression by myeloid-derived suppressor cells (MDSC), tumor-associated macrophages (TAMs), fibroblast stromal cells, and T regulatory cells (Tregs) [73–76]. The relative importance of each of these mechanisms likely varies with both the tumor and the host.
In cancer therapy, it has been noted that antigen-experienced stem cell-like memory and central and effector memory T cells are superior to terminal-differentiated CTL, because of their capacity to proliferate and acquire effector functions in the tumor [65]. One aspect of tumors that has been largely overlooked until recently is how intra-tumor DCs influence CTL behavior and function. Evidence from infectious disease models indicates that antigen-experienced T cells require cognate interactions with tissue DC to expand in situ and achieve full effector functions [77–80]. Confocal microscopy studies of tumors have revealed interactions between CTL and intra-tumor APCs [81, 82]; and recent studies from our lab and others showed that intra-tumor DCs were the only cells that could stimulate CTL proliferation, at least in vitro [83, 84]. Furthermore, contrary to the prevalent view that all tumor DCs are functionally defective, it has become clear that some DC subsets are uniquely equipped to stimulate anti-tumor immunity and influence tumor biology [85]. These findings suggest that manipulating tumor DC may be a useful approach to improve responses to cancer immunotherapy.
Tumor DC heterogeneity
Multi-dimensional flow cytometry and transcriptional profiling of transplantable tumors and spontaneous tumors in mice have revealed tremendous diversity in the myeloid cell compartment [85]. All cDCs in the mouse and human express the cell-surface integrin CD11c; however, other cells, including macrophages, monocytes, and activated lymphocytes, express this molecule, albeit at lower levels than cDCs, necessitating the use of multiple parameters to define each cell population. Similar to other tissues, the TME in mice contains three main subsets of CD11c+ MHCII+ cDCs: (1) CD11b− CD103+ DCs (BATF3/IR8-dependent cDC1); (2) CD11b+ CD103− CD64− F4/80− DC (IRF4-dependent cDC2); and (3) CD11b+ CD64+ F4/80+ DC, which maybe more closely aligned with monocytes and macrophages than DC. Collectively, they constitute a relatively minor population in most tumors, accounting for 5–10 % of all myeloid cells (macrophages and neutrophils predominate in most tumors). Numerous studies have documented the presence of cDCs in human cancers (reviewed in [86]); however, the cell-surface markers available offer limited interrogation of subsets, maturity, and function. pDCs are rare in mouse tumors, but are found in variety of human tumors.
Origin of tumor DC
Pre-cDCs exist in a variety of transplantable tumor models, including B16 melanoma, CT26 colon carcinoma, Lewis lung carcinoma, and EMT6 breast carcinoma [87]. Tumor pre-cDCs are morphologically, phenotypically, and functionally indistinguishable from those isolated from BM and spleen. Adoptive transfer studies of bone marrow pre-cDCs revealed that tumors recruit pre-cDCs through a CCL3-dependent mechanism, where they differentiate into proliferating cDCs [87]. Flt3L therapy promotes intra-tumor expansion of CD103+ DC progenitors (CD11c+MHCII+CD103− CD11b−) and immature CD103+ DCs [88]. Monocytes and more primitive bone marrow progenitors have also been detected in tumors, particularly in the setting of inflammation induced by anthracycline chemotherapeutic agents, and differentiate into inflammatory DCs [89, 90].
Tumor DC plasticity
The number, phenotype, and function of cDCs can change, as the tumor progresses [91, 92]. In a model of spontaneous ovarian cancer, Scarlett et al. detected increased densities of tumor infiltrating DCs, macrophages, MDSCs, and T cells, as well as a functional switch in DC from an immunostimulatory to an immunosuppressive phenotype as the tumors grew [91]. In this model, depletion of DCs at early time points accelerated tumor growth, whereas depletion at later time points led to tumor regression. Krempski et al. also showed in a transplantable ID8 mouse model of peritoneal ovarian cancer that the number of tumor-infiltrating cDCs correlated with tumor burden [92]. Furthermore, tumor cDCs progressively expressed PD-1, as well as PD-L1, which was associated with T-cell suppression and loss of tumor-infiltrating T cells [92]. Our research has shown that the TME drives immunostimulatory Gr-1− cDCs to generate a functionally defective Gr-1+ cDC subpopulation that induces T-cell tolerance [83]. Using a transgenic mouse model that allowed in vivo tracking of DCs in tumors, we also found that immunostimulatory cDCs derived from pre-DCs can lose their DC identity and evolve into CD11c−MHCII− regulatory macrophages [93] (Fig. 2). DC-derived-macrophages (DC-d-M) potently suppressed T-cell responses through the production of immunosuppressive molecules, including nitric oxide, arginase, and IL-10. A relative deficiency of GM-CSF appeared to provide a permissive signal for DC de-differentiation, as augmenting GM-CSF expression levels in the tumor blocked this process. Collectively, these findings highlight the plasticity of DC and suggest that maintenance of DC identity and function depends, at least partly, on cues received from the TME.

Model of tumor DC plasticity. Circulating pre-cDC recruited into tumors differentiate into Gr-1−cDC, which possesses the capacity to stimulate proliferation and expansion of CTLs. Under the influence of the tumor microenvironment, Gr-1−cDCs generate: (1) Gr-1+cDC, a subpopulation of maturation-resistant, IL-10-producing DCs that induce T-cell anergy, and (2) DC-derived-macrophages (DC-d-M) that potently suppress CTL proliferation by releasing IL-10, arginase-1, and nitric oxide
DC defects in cancer
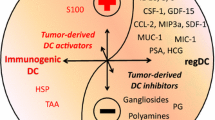
Antigen cross presentation by DCs plays a critical role in the generation of anti-tumor responses [9, 94, 95]. DCs can acquire tumor antigens from multiple sources: (1) apoptotic/necrotic tumor cells [96–98]; (2) chaperone proteins, such as heat shock proteins, that bind soluble tumor antigens [99, 100], (3) secreted vesicles (e.g., exosomes) from tumor cells [101], (4) gap junctions that transfer small antigenic protein fragments [102]; and (5) tumor plasma membrane fragments [103]. In addition, DCs can acquire preformed peptide-MHC Class I complexes through direct contact with tumor cells, a process resembling trogocytosis (also known as cross dressing) [104, 105]. Cross presentation of tumor antigens generally requires stable and high antigen expression levels and tumor cell apoptosis/necrosis to release the antigens [106]. These conditions are frequently unmet in untreated cancers, but can be induced with chemotherapy, radiation therapy, and oncolytic viral therapy.
The functional status and subset of tumor DCs affect their apparent cross-presentation capacity and immunogenicity [82, 107–109]. Stoitzner et al. reported that DCs sorted from B16 melanoma could not induce proliferation of tumor-antigen-specific CD8+ T cells or CD4+ T cells [110]. Engelhardt et al. showed by live cell imaging that tumor-antigen-bearing DCs engage tumor-antigen-specific T cells; however, these interactions were unable to fully activate T-cell effector functions, including the capacity to kill tumor cells [82]. By contrast, treatment of tumor-bearing mice with agonistic anti-CD40 monoclonal antibodies and other DC activating agents, such as TLR9 agonists, led to the influx of large numbers of T cells that were capable of eradicating established tumors [107].
Recent reports have highlighted the importance of tumor CD103+ cDCs in stimulating anti-tumor immune responses in primary cancers and metastases [84, 111, 112]. On a per cell basis, tumor CD103+cDCs stimulate naïve and primed tumor-antigen-specific T cells more effectively than tumor CD11b+cDC, which is attributed to more efficient cross-presentation machinery and higher expression levels of IL-12 [84, 113]. Tumor CD103+ also specializes in the transport of intact tumor antigens to tumor draining lymph nodes [88]. Targeted reduction/elimination of tumor CD103+DCs in BATF3 knockout mice and in Zbtb46-diphtheria toxin receptor (Zbtb46-DTR) transgenic mice attenuated responses to cancer immunotherapy. Spranger et al. reported that active oncogenic WNT/β-catenin signaling within melanoma cells in Braf V600E /Pten −/− /CAT-STA tumors inhibited T-cell priming by suppressing the CCL4-dependent recruitment of dermal CD103+ DCs. Injecting these tumors with poly:IC activated Flt3 ligand-induced bone marrow-derived DCs restored responses to anti-CTLA4 and anti-PD-L1 monoclonal antibodies [114]. Similarly, expansion and activation of CD103+ cDC in B16 melanoma with Flt3L and poly I:C treatment, respectively, enhanced responses to PD-L1 and BRAF blockade [88]. The relevance of these findings to human cancer remains to be clarified. Notably, the human equivalents of CD103+ DC and CD11b+ DC from lymphoid tissues (BDCA3+ and BDCA1+ DCs, respectively) show less striking differences in cross-presentation activity [115]. Furthermore, mouse CD11b+ cDC exhibits robust cross-presentation capacities under appropriate conditions [116], and was critical to chemotherapy-induced immune responses [89].
Immaturity and co-inhibitory molecules
Multiple inhibitory (e.g., PD-L1, TIM-3, LAG-3, CD200, CTLA4) and activating (4-1BBL, ICOS-L, CD80, and CD86) immune checkpoint molecules influence the activation, differentiation, and proliferation of T cells. T-cell interactions with immature DCs can lead to T-cell tolerance through various mechanisms, including deletion, anergy, and the generation of regulatory T cells [6, 117, 118].
PD-1 and its corresponding ligands PD-L1 and PD-L2 negatively regulate T-cell priming and effector functions [119]. PD-L1 and PD-1 expressions on tumor DCs correlated with cancer progression in an ovarian cancer model [92]. In a melanoma model, CD103+ DCs, from tumor-draining lymph nodes express high amounts of PD-L1 as compared with CD103+ DCs isolated from non-draining lymph nodes [88]. Antibody blockade of PD-L1 and PD-1 mitigates DC dysfunction as evidenced by increased NF-κB activation, increased IL-12, TNF, and IL-1β production and co-stimulatory molecule expression, and enhanced T-cell stimulatory capacity [120, 121].
Tumor DCs also suppress tumor-specific T cells through the release of biomolecules, such as nitric oxide (NO), arginase I, and indoleamine 2,3-dioxygenase (IDO) [122]. Arginase I degrades arginine, an essential amino acid for CD4+ T-cell proliferation and differentiation [119]. High expression levels of arginase I promote the accumulation of reactive oxygen intermediates, such as NO, which block CD8+ T-cell responses [123]. IDO is a tryptophan-catabolizing enzyme that plays an important role in inducing and maintaining tolerance. Tumor cells, DCs, and regulatory macrophages can express IDO. IDO activates Tregs and creates a milieu deficient in tryptophan, another amino acid used by T cells during activation [124–126]. Furthermore, IDO triggers the production of tryptophan metabolites that induce T-cell apoptosis and suppress T-cell proliferation [125].
Mechanisms of tumor DC dysfunction
Many studies document the presence of immature DCs in blood and tumors in humans and tumor-bearing mice [127, 128]. The expansion of immature DCs is often considered to be a systemic process, in which tumor-derived factors skew differentiation of bone marrow progenitors [128]. This view is based partly on in vitro studies of bone marrow cells stimulated with GM-CSF and tumor-conditioned media, and studies of mice with large tumor burdens. Recent evidence indicates that GM-CSF-stimulated bone marrow DCs are poor representatives of natural DC, and as such, may not be the best tool for evaluating the effects of cancer [129, 130]. Others and we have shown that hematopoiesis and DC development proceed normally in mice with small transplantable tumors and spontaneous tumors. In addition, studies of human breast and other cancers have shown that while intra-tumor DCs are immature, DCs at the margins of the tumor are mature [86, 131]. Collectively, these findings suggest that the local TME rather than systemic factors is more relevant. Understanding how tumors promote tumor DC dysfunction is expected to help advance cancer immunotherapy. Some of the factors and their associated mechanisms of action are reviewed below.
IL-6
Cancer cells and tumor-infiltrating DCs, monocytes, and macrophages can produce IL-6. IL-6 targets genes involved in cell-cycle progression and suppression of apoptosis [132, 133], accounting for its known role in oncogenesis [64, 134, 135]. Although IL-6 is often considered a proinflammatory cytokine, IL-6 dampens immune responses in some settings [136, 137]. High serum concentration of IL-6 correlates with poor outcome and abnormal immune responses in patients with various epithelial and lymphoid cancers and hepatocellular carcinoma [138–140]. High serum IL-6 concentrations in multiple myeloma patients were associated with lower absolute numbers of circulating precursors of myeloid DCs [141]. Peripheral blood DCs from these patients also showed significantly lower expression of HLA-DR, CD40, and CD80, and impaired stimulation of allogeneic T-cell proliferation as compared with DCs isolated from healthy controls.
In vitro studies have shown that IL-6 inhibits LPS-induced DC maturation, suppresses intracellular MHC Class II expression, and lowers CCR7 expression, the chemokine receptor critical for DC migration to lymphoid tissues [136, 142–144]. IL-6 also inhibits DC differentiation from human monocytes and CD34+ myeloid progenitor cells in vitro, while promoting the development of macrophages with an alternatively activated phenotype that is associated with wound healing [145, 146]. This effect of IL-6 was linked to its ability to increase macrophage colony-stimulating factor receptor (Csf1r) expression levels [147]. Our group reported that IL-6 promotes the differentiation of immunosuppressive Gr-1+ cDCs from pre-cDCs in tumors [83]. IL-6−/− mice had a threefold reduction in the frequency of tumor Gr-1+ DCs as compared with wild-type mice, which was associated with a higher frequency and rate of proliferation of IFN-γ+ CTLs, and better responses to adoptive T cell immunotherapy [83].
IL-10
Many cells in the TME express IL-10, including Tregs, TAMs, MDSCs, DCs, and cancer cells [148]. Autocrine and paracrine IL-10 can increase the proliferation and survival of cancer cells (e.g., B16 melanoma, human stomach adenocarcinoma, and human glioblastoma multiforme) [135, 149]. IL-10 is a classic anti-inflammatory cytokine that inhibits multiple aspects of DC biology, including maturation, production of proinflammatory cytokines (e.g., IL-12 and IL-1β), and stimulation of T cells [150, 151]. Under some conditions, IL-10 stimulates the generation of ‘tolerogenic’ DCs, which express low levels of MHC and co-stimulatory molecules and produce high amounts of IL-10 [152]. Recent studies in a breast cancer model have shown that inhibition of IL-12 expression in tumor DCs is the dominant effect of macrophage-derived IL-10; blocking the IL-10 receptor (IL-10R) with neutralizing antibodies restored IL-12 expression in tumor DCs and anti-tumor T-cell responses [112]. Similarly, Vicari et al. reported that tumor DC function could be restored with a combination of anti-IL-10R antibodies and activation with CpG oligonucleotides [153].
The effect of IL-10 on cancer is complex, however, as anti-tumor effects have also been observed [154, 155]. Exogenous IL-10 inhibited growth and metastasis of mammary and ovarian carcinoma xenografts, partly through the downregulation of MHC Class I expression, which enhanced NK-cell-mediated tumor killing [154]. Mumm et al. reported that IL-10 promotes expansion of tumor-infiltrating IFN-γ+ CTLs, which enhanced tumor immunosurveillance and control of tumor growth. The reason for these discrepant findings remains unclear, but likely reflects differences in the models, TME, cellular source and bioavailability of IL-10, and the relative sensitivity of tumor infiltrating cells to IL-10, IL-12, and other molecules [112, 156].
Vascular endothelial growth factor (VEGF)
Serum VEGF levels correlate with poor prognosis in various human cancers [157, 158]. VEGF stimulates tumor endothelial cell proliferation and angiogenesis, which are essential for tumor growth and development [157]. VEGF binds to high-affinity membrane tyrosine kinase receptors—VEGFR-1, 2, 3—that are expressed mostly on endothelial cells and a few populations of hematopoietic cells, including DCs [157]. Earlier studies showed that VEGF inhibited DC differentiation and maturation in vitro through an NF-κB-dependent pathway, which could be blocked with neutralizing anti-VEGF antibodies [159, 160]. Administration of VEGF to tumor-free mice resulted in impaired DC development and accumulation of immature Gr-1+ myeloid cells (defined as MDSCs). Treatment of tumor-bearing mice with neutralizing anti-VEGF antibodies increased the number of spleen and lymph node DCs, improved DC function, and improved anti-tumor CTL responses [161–163].
STAT3
Constitutive activation of STAT3 occurs in many cancers [164]. STAT3 serves as an oncogenic driver that enhances tumor cell proliferation, survival, and invasion [165]. STAT3 activation up-regulates the expression of anti-apoptotic proteins, such as BCL-XL, MCL1, cyclin D1 and MYC, and pro-angiogenic factors, such as HIF-α, VEGF, MMP-2, and MMP-9 [166]. STAT3 activation also contributes to tumor progression by enhancing tumor inflammation and hampering anti-tumor immunity. STAT3 activation in DC has been linked to defects in differentiation, maturation, and function [167–170]. Both the intensity and duration of STAT3 signaling determine whether DC dysfunction develops [171, 172]. Pharmacologic inhibitors and genetic ablation of STAT3 signaling restore maturation responses to inflammatory stimuli in DCs incubated in tumor-conditioned medium. In a study using the Cre-loxP system to delete STAT3 in hematopoietic cells, tumor-infiltrating DC from STAT3−/− mice expressed higher levels of surface MHC Class II, CD80, and CD86, and produced more IL-12 [173], which was associated with better anti-tumor immune responses. Elevated STAT3 activity in tumor DCs is attributed mostly to paracrine stimulation by cytokines in the TME, such as IL-6, IL-10, and VEGF [169, 174]. Our studies show that tumor-derived factors stimulate autocrine IL-6 and IL-10 production in cDCs, which worked synergistically to promote DC dysfunction [175].
TIM-3
TIM-3, a receptor for galectin-9, was identified initially as a negative regulator of Th1 immunity [176], but is also expressed by myeloid cells, including DCs [177]. Chiba et al. reported that tumor-derived factors upregulate TIM-3 expression in tumor DCs [178]. TIM-3 plays dual roles in anti-tumor immunity. Under some conditions, galectin-9-TIM-3 interactions promote DC maturation and cross priming of tumor-antigen-specific T cells [178, 179]. By contrast, the interaction of TIM-3 with high-mobility group box 1 protein (HMGB1), a classic danger associated molecular pattern molecule (DAMP) in the TME, prevents tumor DCs from sensing nucleic acid danger signals released from dying tumor cells, which suppresses type 1 interferon- and IL-12-mediated anti-tumor responses [178]. Others have shown that ligating TIM-3 on bone marrow-derived and spleen DCs with cross-linking antibodies activates Bruton’s tyrosine kinase and c-SRC, which inhibits DC activation and maturation by blocking the NF-κB pathway [180].
Lipids
Intracellular fat and glycogen accumulate in DCs with exposure to maturation stimuli in vitro, and occur during normal development in lymphoid tissues [181]. Several reports suggest that accumulation of oxidized lipids, especially triacylglycerols (TAG), causes dysfunction and shortens the lifespan of DCs [19, 182–185]. DCs from mouse EL-4 lymphoma, CT-26, and B16-F10 tumors and in some cancer patients exhibit elevated TAG levels [19]. Tumor-conditioned media can stimulate the uptake of TAG in bone marrow-derived DCs and human monocyte-derived DC by regulating the expression levels of scavenger receptor A (SRA1, CD204, and MSR1), lipoprotein lipase (LPL), and fatty acid-binding protein 4 (FABP4) [19, 182, 184]. As compared with normal DCs from tumor-free mice, lipid-laden DCs stimulated T cells poorly because of defects in antigen cross presentation and increased production of IL-10 [19, 185].
Endoplasmic reticulum stress response
Tumors adapt to hypoxia, nutrient deficiency, and oxidative stress by triggering an endoplasmic reticulum (ER) stress response, also known as the unfolded protein response (UPR) [186–189]. Cubillos-Ruiz et al. recently demonstrated that the TME also induces an ER stress response in DC that is mediated by the spliced transcription factor, XBP1 [183]. Although the ER stress response contributes to normal DC development and survival [190, 191], constitutive XBP1 activation in tumor DCs induced abnormal accumulation of oxidized lipids by targeting multiple triglyceride biosynthetic genes [183]. Reactive oxygen species generated reactive lipid peroxidation byproducts (e.g., aldehyde 4-hydroxy-trans-2-nonenal (4-HNE)) that sustained XBP1 activation. Notably, lipid accumulation induced by this pathway operates independently of scavenger receptors involved in the uptake of extracellular lipids. Inactivation of XBP1 in tumor DCs improved anti-tumor T-cell immune responses and the control of tumor growth in an ovarian cancer model.
Gangliosides
Gangliosides are sialic-acid-containing glycosphingolipids located in the plasma membrane of all vertebrate cells. Gangliosides which shed from various tumors have been shown to suppress anti-tumor immune responses by affecting T cells, NK cells, and DCs [192, 193]. Gangliosides derived from neuroblastoma and melanoma inhibit DC differentiation from human monocytes and mouse bone marrow cells [194, 195]. Exposure of bone marrow-derived DCs to GM1 ganglioside inhibited maturation and up-regulation of co-stimulatory molecules, reducing their capacity to prime naïve T cells. T cells primed with pre-treated ganglioside DCs also produced significantly less IFN-γ and IL-2 upon re-stimulation [192, 195].
TLR2
We recently reported that TLR2 activation is a critical proximal signal that contributes to tumor DC dysfunction [175]. Tumor-conditioned medium (TCM) generated from both murine and human cancer cell lines stimulated DC through TLR2 to produce autocrine IL-10 and IL-6. The ability of IL-6 and IL-10 to stimulate DC dysfunction, however, relied on TLR2-induced up-regulation of their cell-surface receptors, which markedly decreased the cytokine concentration threshold required to activate STAT3. This effect of TLR2 helps reconcile the relative tumor specificity of DC dysfunction, and suggests that cytokine receptor expression levels, rather than the source of the cytokines, may be the ultimate arbiter of DC fate and function in tumors. TLR2 deficiency improved the immunogenicity of intra-tumor DCs, enhanced CTL expansion, and improved anti-tumor responses to immunotherapy. Consistent with the earlier reports [134], we found that versican, an extracellular matrix glycoprotein that is overexpressed in many cancers, is a key TLR2 ligand in TCM. Interestingly, the level of versican expression in human cancers correlates inversely with prognosis and the density of CD8+ T-cell infiltration [196–198]. Additional TLR2 ligands released by cancer cells include laminin-β1, procollagen III-α1, Hsp60, and Hsp72 [134, 199, 200]. The microbiome also generates TLR2 ligands that could potentially influence the TME and tumor progression [201].
Strategies to optimize tumor DC function
Immunogenic cell death (ICD) is a recognized benefit of the conventional cancer therapies. Chemotherapy and radiation therapy induce ICD by stimulating the release of DAMPs [202–205]. Endoplasmic reticulum stress and autophagy expose calreticulin (CRT) on the outer leaflet of the plasma membrane; apoptosis releases adenosine triphosphate (ATP); and permeabilization of the nuclear membrane during necrosis releases HMGB1 [206]. DAMPs cause ICD partly through their interactions with DCs: CRT, ATP, and HMGB1 ligate, respectively, CD91, P2PX7 (a purigenic receptor), and TLR4, which facilitates DC recruitment, engulfment of tumor antigens, and antigen presentation to T cells. Vacchelli et al. reported that the efficacy of anthracycline-based chemotherapy requires DCs to express formyl peptide receptor 1, which promotes stable interactions with dying cancer cells expressing annexin-1 [207].
Studies of the conventional cancer therapy on ICD reveal the central importance of tumor DCs, and suggest that cancer immunotherapy should incorporate strategies to promote their activity. Administrating GM-CSF and Flt3 ligand systemically to increase tumor DC numbers fails to improve anti-tumor CTL responses significantly [208, 209], likely because neither growth factor supersedes the local mechanism(s) that cause tumor DC dysfunction. Successful approaches to improve DC function have generally targeted inhibitory molecules in the TME and their downstream signaling pathways or have included DC promoting agents that overwhelm negative regulators. Improving the function of tumor DCs presents several challenges, however, including the delivery of therapeutic agents into the TME at effective concentrations, potential off-target effects on other cells, and the rapid turnover of tumor DCs, which may necessitate on-going therapy for nascent DCs and their progenitors.
Concluding remarks
Anti-tumor immune responses elicited by adoptive T-cell therapy and active vaccination approaches rely on interactions between CTLs and cancer cells. The development of in vitro culture techniques to generate large quantifies of DCs in the early 1990s led to extensive exploration of tumor antigen-loaded DC as vaccination vehicles. Despite their ability to stimulate tumor-antigen-specific immune responses, DC-based vaccines have met with a little clinical success thus far [210]. These failures require reassessment of how CTL responses are regulated within TME. Recent evidence strongly suggests that secondary antigen presentation in the tumor modulates the number, duration, and quality of CTLs. As the principal antigen-presenting cells in tumors, DCs have a pivotal position in this process. The inherent plasticity of tumor DCs, however, renders them susceptible to the vagaries of the TME, which often leads to dysfunction. Progress in our understanding of tumor DC biology in both untreated tumors and in tumors treated with drugs, radiation, and checkpoint inhibitor antibodies have revealed potential targets to optimize their function. Although the identification of individual molecules that promote immune evasion will be important to improve T-cell responses, alternative approaches might focus on improving the intrinsic functional qualities of DCs. Our recent work shows that tumor DCs experience a phenotypical and functional de-differentiation process involving fundamental changes in TF expression [93], suggesting that targeting TF regulatory pathways in tumor DCs may improve immunotherapy.
Abbreviations
- CDPs:
-
Common DC precursors
- cMoP:
-
Common monocyte progenitors
- CTLs:
-
Cytotoxic T lymphocytes
- DAMP:
-
Danger-associated molecular patterns
- DCs:
-
Dendritic cells
- ER:
-
Endoplasmic reticulum
- FABP4:
-
Fatty acid-binding protein 4
- Flt3L:
-
FMS-like tyrosine kinase 3 ligand
- GM-CSF:
-
Granulocyte/macrophage colony-stimulating factor
- HMGB1:
-
High mobility group box 1
- ICD:
-
Immunogenic cell death
- IDO:
-
Indoleamine 2,3-dioxygenase
- IL-10R:
-
Interleukin-10 receptor
- LC:
-
Langerhans cells
- LPL:
-
Lipoprotein lipase
- MDP:
-
Monocyte/macrophage and DC progenitor
- MDSC:
-
Myeloid-derived suppressor cells
- MHC:
-
Major histocompatibility complex
- NO:
-
Nitric oxide
- pDC:
-
Plasmacytoid dendritic cells
- STAT3:
-
Signal transducer and activator of transcription 3
- TAG:
-
Triacylglycerols
- TAMs:
-
Tumor-associated macrophages
- TF:
-
Transcription factor
- TIM-3:
-
T-cell immunoglobulin and mucin-domain containing-3
- TLR:
-
Toll-like receptor
- TME:
-
Tumor microenvironment
- Tregs:
-
Regulatory T cells
- UPR:
-
Unfolded protein response
- VEGF:
-
Vascular endothelial growth factor
References
Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME (2008) Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 8:299–308
Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392:245–252
Steinman RM, Banchereau J (2007) Taking dendritic cells into medicine. Nature 449:419–426
Rabinovich GA, Gabrilovich D, Sotomayor EM (2007) Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol 25:267–296
Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC (2001) Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med 194:769–779
Steinman RM, Hawiger D, Nussenzweig MC (2003) Tolerogenic dendritic cells. Annu Rev Immunol 21:685–711
Reis ESC (2006) Dendritic cells in a mature age. Nat Rev Immunol 6:476–483
Akira S, Uematsu S, Takeuchi O (2006) Pathogen recognition and innate immunity. Cell 124:783–801
Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA (2002) In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity 17:211–220
Zammit DJ, Cauley LS, Pham QM, Lefrancois L (2005) Dendritic cells maximize the memory CD8 T cell response to infection. Immunity 22:561–570
Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, Wang J (2006) Dendritic cell apoptosis in the maintenance of immune tolerance. Science 311:1160–1164
Chen M, Wang J (2010) Programmed cell death of dendritic cells in immune regulation. Immunol Rev 236:11–27
Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI (2001) Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol 166:678–689
Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, Carbone DP, Gabrilovich DI (2000) Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res 6:1755–1766
Vicari AP, Treilleux I, Lebecque S (2004) Regulation of the trafficking of tumour-infiltrating dendritic cells by chemokines. Semin Cancer Biol 14:161–169
Hoffmann TK, Muller-Berghaus J, Ferris RL, Johnson JT, Storkus WJ, Whiteside TL (2002) Alterations in the frequency of dendritic cell subsets in the peripheral circulation of patients with squamous cell carcinomas of the head and neck. Clin Cancer Res 8:1787–1793
Della Bella S, Gennaro M, Vaccari M, Ferraris C, Nicola S, Riva A, Clerici M, Greco M, Villa ML (2003) Altered maturation of peripheral blood dendritic cells in patients with breast cancer. Br J Cancer 89:1463–1472
Vicari AP, Caux C, Trinchieri G (2002) Tumour escape from immune surveillance through dendritic cell inactivation. Semin Cancer Biol 12:33–42
Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B, Knight SC, Padhya T, McCaffrey TV, McCaffrey JC, Antonia S, Fishman M, Ferris RL, Kagan VE, Gabrilovich DI (2010) Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med 16:880–886
Merad M, Sathe P, Helft J, Miller J, Mortha A (2013) The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol 31:563–604
Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, Segura E, Tussiwand R, Yona S (2014) Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol 14:571–578
Ginhoux F, Jung S (2014) Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14:392–404
Diao J, Winter E, Chen W, Cantin C, Cattral MS (2004) Characterization of distinct conventional and plasmacytoid dendritic cell-committed precursors in murine bone marrow. J Immunol 173:1826–1833
Diao J, Winter E, Cantin C, Chen W, Xu L, Kelvin D, Phillips J, Cattral MS (2006) In situ replication of immediate dendritic cell (DC) precursors contributes to conventional dc homeostasis in lymphoid tissue. J Immunol 176:7196–7206
Naik SH, Metcalf D, van Nieuwenhuijze A, Wicks I, Wu L, O’Keeffe M, Shortman K (2006) Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol 7:663–671
Naik SH, Sathe P, Park H-Y, Metcalf D, Proietto AI, Dakic A, Carotta S, O’Keeffe M, Bahlo M, Papenfuss A, Kwak J-Y, Wu L, Shortman K (2007) Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol 8:1217–1226
Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG (2007) Identification of clonogenic common Flt3+M-CSFR+ plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol 8:1207–1216
Liu K, Victora GD, Schwickert TA, Guermonprez P, Meredith MM, Yao K, Chu FF, Randolph GJ, Rudensky AY, Nussenzweig M (2009) In vivo analysis of dendritic cell development and homeostasis. Science 324:392–397
Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM (2012) Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med 209:1135–1152
Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, Nussenzweig MC (2012) Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med 209:1153–1165
Schlitzer A, Sivakamasundari V, Chen J, Sumatoh HR, Schreuder J, Lum J, Malleret B, Zhang S, Larbi A, Zolezzi F, Renia L, Poidinger M, Naik S, Newell EW, Robson P, Ginhoux F (2015) Identification of cDC1- and cDC2-committed DC progenitors reveals early lineage priming at the common DC progenitor stage in the bone marrow. Nat Immunol 16:718–728
Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM (2008) Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322:1097–1100
Suzuki S, Honma K, Matsuyama T, Suzuki K, Toriyama K, Akitoyo I, Yamamoto K, Suematsu T, Nakamura M, Yui K, Kumatori A (2004) Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha− dendritic cell development. Proc Natl Acad Sci USA 101:8981–8986
Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, Price J, Yin N, Bromberg J, Lira SA, Stanley ER, Nussenzweig M, Merad M (2009) The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med 206:3115–3130
Varol C, Landsman L, Fogg DK, Greenshtein L, Gildor B, Margalit R, Kalchenko V, Geissmann F, Jung S (2007) Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J Exp Med 204:171–180
Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, Liu K, Jakubzick C, Ingersoll MA, Leboeuf M, Stanley ER, Nussenzweig M, Lira SA, Randolph GJ, Merad M (2009) Origin of the lamina propria dendritic cell network. Immunity 31:513–525
Karsunky H, Merad M, Cozzio A, Weissman IL, Manz MG (2003) Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med 198:305–313
Liu K, Nussenzweig MC (2010) Origin and development of dendritic cells. Immunol Rev 234:45–54
Waskow C, Liu K, Darrasse-Jeze G, Guermonprez P, Ginhoux F, Merad M, Shengelia T, Yao K, Nussenzweig M (2008) The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol 9:676–683
Maraskovsky E, Daro E, Roux E, Teepe M, Maliszewski CR, Hoek J, Caron D, Lebsack ME, McKenna HJ (2000) In vivo generation of human dendritic cell subsets by Flt3 ligand. Blood 96:878–884
Manfra DJ, Chen SC, Jensen KK, Fine JS, Wiekowski MT, Lira SA (2003) Conditional expression of murine Flt3 ligand leads to expansion of multiple dendritic cell subsets in peripheral blood and tissues of transgenic mice. J Immunol 170:2843–2852
Kingston D, Schmid MA, Onai N, Obata-Onai A, Baumjohann D, Manz MG (2009) The concerted action of GM-CSF and Flt3-ligand on in vivo dendritic cell homeostasis. Blood 114:835–843
Kabashima K, Banks TA, Ansel KM, Lu TT, Ware CF, Cyster JG (2005) Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity 22:439–450
Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, Klinakis A, Charo IF, Jung S, Gommerman JL, Ivanov II, Liu K, Merad M, Reizis B (2011) Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity 35:780–791
Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, Ji Y, Sukumar M, Muranski P, Scott CD, Hall JA, Ferreyra GA, Leonardi AJ, Borman ZA, Wang J, Palmer DC, Wilhelm C, Cai R, Sun J, Napoli JL, Danner RL, Gattinoni L, Belkaid Y, Restifo NP (2013) Retinoic acid controls the homeostasis of pre-cDC-derived splenic and intestinal dendritic cells. J Exp Med 210:1961–1976
Diao J, Winter E, Chen W, Xu F, Cattral MS (2007) Antigen transmission by replicating antigen-bearing dendritic cells. J Immunol 179:2713–2721
Sponaas AM, Cadman ET, Voisine C, Harrison V, Boonstra A, O’Garra A, Langhorne J (2006) Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J Exp Med 203:1427–1433
Nakano H, Lin KL, Yanagita M, Charbonneau C, Cook DN, Kakiuchi T, Gunn MD (2009) Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nat Immunol 10:394–402
Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, Jeffrey KL, Anthony RM, Kluger C, Nchinda G, Koh H, Rodriguez A, Idoyaga J, Pack M, Velinzon K, Park CG, Steinman RM (2010) Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell 143:416–429
Metcalf D, Nicola NA, Mifsud S, Di Rago L (1999) Receptor clearance obscures the magnitude of granulocyte-macrophage colony-stimulating factor responses in mice to endotoxin or local infections. Blood 93:1579–1585
Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, Bogunovic M, Gautier EL, Miller J, Leboeuf M, Lu G, Aloman C, Brown BD, Pollard JW, Xiong H, Randolph GJ, Chipuk JE, Frenette PS, Merad M (2012) GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 36:1031–1046
MacPherson GG, Jenkins CD, Stein MJ, Edwards C (1995) Endotoxin-mediated dendritic cell release from the intestine. Characterization of released dendritic cells and TNF dependence. J Immunol 154:1317–1322
Jakubzick C, Bogunovic M, Bonito AJ, Kuan EL, Merad M, Randolph GJ (2008) Lymph-migrating, tissue-derived dendritic cells are minor constituents within steady-state lymph nodes. J Exp Med 205:2839–2850
Angeli V, Ginhoux F, Llodra J, Quemeneur L, Frenette PS, Skobe M, Jessberger R, Merad M, Randolph GJ (2006) B cell-driven lymphangiogenesis in inflamed lymph nodes enhances dendritic cell mobilization. Immunity 24:203–215
Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, Hashimoto D, Chow A, Price J, Greter M, Bogunovic M, Bellemare-Pelletier A, Frenette PS, Randolph GJ, Turley SJ, Merad M (2012) Deciphering the transcriptional network of the dendritic cell lineage. Nat Immunol 13:888–899
Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, Holter W, Rauch A, Zhuang Y, Reizis B (2008) Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 135:37–48
Ghosh HS, Cisse B, Bunin A, Lewis KL, Reizis B (2010) Continuous expression of the transcription factor e2-2 maintains the cell fate of mature plasmacytoid dendritic cells. Immunity 33:905–916
Hacker C, Kirsch RD, Ju XS, Hieronymus T, Gust TC, Kuhl C, Jorgas T, Kurz SM, Rose-John S, Yokota Y, Zenke M (2003) Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol 4:380–386
Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O’Shea JJ, Singh H, Ozato K (2005) IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol 174:2573–2581
Kashiwada M, Pham NL, Pewe LL, Harty JT, Rothman PB (2011) NFIL3/E4BP4 is a key transcription factor for CD8alpha(+) dendritic cell development. Blood 117:6193–6197
Wu L, D’Amico A, Winkel KD, Suter M, Lo D, Shortman K (1998) RelB is essential for the development of myeloid-related CD8alpha− dendritic cells but not of lymphoid-related CD8alpha+ dendritic cells. Immunity 9:839–847
Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, Haniffa M, Ceron-Gutierrez L, Bacon CM, Menon G, Trouillet C, McDonald D, Carey P, Ginhoux F, Alsina L, Zumwalt TJ, Kong XF, Kumararatne D, Butler K, Hubeau M, Feinberg J, Al-Muhsen S, Cant A, Abel L, Chaussabel D, Doffinger R, Talesnik E, Grumach A, Duarte A, Abarca K, Moraes-Vasconcelos D, Burk D, Berghuis A, Geissmann F, Collin M, Casanova JL, Gros P (2011) IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med 365:127–138
Dickinson RE, Griffin H, Bigley V, Reynard LN, Hussain R, Haniffa M, Lakey JH, Rahman T, Wang XN, McGovern N, Pagan S, Cookson S, McDonald D, Chua I, Wallis J, Cant A, Wright M, Keavney B, Chinnery PF, Loughlin J, Hambleton S, Santibanez-Koref M, Collin M (2011) Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood 118:2656–2658
Grivennikov SI, Greten FR, Karin M (2010) Immunity, inflammation, and cancer. Cell 140:883–899
Restifo NP, Dudley ME, Rosenberg SA (2012) Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12:269–281
Vormehr M, Diken M, Boegel S, Kreiter S, Tureci O, Sahin U (2016) Mutanome directed cancer immunotherapy. Curr Opin Immunol 39:14–22
Finn OJ (2008) Cancer immunology. N Engl J Med 358:2704–2715
MacKie RM, Reid R, Junor B (2003) Fatal melanoma transferred in a donated kidney 16 years after melanoma surgery. N Engl J Med 348:567–568
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964
Budhu S, Loike JD, Pandolfi A, Han S, Catalano G, Constantinescu A, Clynes R, Silverstein SC (2010) CD8 + T cell concentration determines their efficiency in killing cognate antigen-expressing syngeneic mammalian cells in vitro and in mouse tissues. J Exp Med 207:223–235
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22:329–360
DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T (2012) Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482:405–409
Sica A, Bronte V (2007) Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest 117:1155–1166
Beyer M, Schultze JL (2006) Regulatory T cells in cancer. Blood 108:804–811
Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, Fearon DT (2010) Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 330:827–830
Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, Schrump DS, Steinberg SM, Rosenberg SA, Robbins PF (2012) Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood 119:5688–5696
Bennett CL, Chakraverty R (2012) Dendritic cells in tissues: in situ stimulation of immunity and immunopathology. Trends Immunol 33:8–13
Wakim LM, Waithman J, van Rooijen N, Heath WR, Carbone FR (2008) Dendritic cell-induced memory T cell activation in nonlymphoid tissues. Science 319:198–202
McGill J, Van Rooijen N, Legge KL (2008) Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J Exp Med 205:1635–1646
McGill J, Van Rooijen N, Legge KL (2010) IL-15 trans-presentation by pulmonary dendritic cells promotes effector CD8 T cell survival during influenza virus infection. J Exp Med 207:521–534
Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, Cavanagh LL, von Andrian UH, Ertl HC, Haydon PG, Weninger W (2006) Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J Exp Med 203:2749–2761
Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, Krummel MF (2012) Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell 21:402–417
Diao J, Zhao J, Winter E, Cattral MS (2011) Tumors suppress in situ proliferation of cytotoxic T cells by promoting differentiation of Gr-1(+) conventional dendritic cells through IL-6. J Immunol 186:5058–5067
Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, Van’t Veer LJ, Sperling AI, Wolf DM, Krummel MF (2014) Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 26:638–652
Broz ML, Krummel MF (2015) The emerging understanding of myeloid cells as partners and targets in tumor rejection. Cancer Immunol Res 3:313–319
Karthaus N, Torensma R, Tel J (2012) Deciphering the message broadcast by tumor-infiltrating dendritic cells. Am J Pathol 181:733–742
Diao J, Zhao J, Winter E, Cattral MS (2010) Recruitment and differentiation of conventional dendritic cell precursors in tumors. J Immunol 184:1261–1267
Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, Chakarov S, Rivera C, Hogstad B, Bosenberg M, Hashimoto D, Gnjatic S, Bhardwaj N, Palucka AK, Brown BD, Brody J, Ginhoux F, Merad M (2016) Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 44:924–938
Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, Portela Catani JP, Hannani D, Duret H, Steegh K, Martins I, Schlemmer F, Michaud M, Kepp O, Sukkurwala AQ, Menger L, Vacchelli E, Droin N, Galluzzi L, Krzysiek R, Gordon S, Taylor PR, Van Endert P, Solary E, Smyth MJ, Zitvogel L, Kroemer G (2013) Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity 38:729–741
Ma Y, Mattarollo SR, Adjemian S, Yang H, Aymeric L, Hannani D, Portela Catani JP, Duret H, Teng MW, Kepp O, Wang Y, Sistigu A, Schultze JL, Stoll G, Galluzzi L, Zitvogel L, Smyth MJ, Kroemer G (2014) CCL2/CCR2-dependent recruitment of functional antigen-presenting cells into tumors upon chemotherapy. Cancer Res 74:436–445
Scarlett UK, Rutkowski MR, Rauwerdink AM, Fields J, Escovar-Fadul X, Baird J, Cubillos-Ruiz JR, Jacobs AC, Gonzalez JL, Weaver J, Fiering S, Conejo-Garcia JR (2012) Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J Exp Med 209:495–506
Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, Goode EL, Kalli KR, Knutson KL (2011) Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol 186:6905–6913
Diao J, Mikhailova A, Tang M, Gu H, Zhao J, Cattral MS (2012) Immunostimulatory conventional dendritic cells evolve into regulatory macrophage-like cells. Blood 119:4919–4927
Lin ML, Zhan Y, Villadangos JA, Lew AM (2008) The cell biology of cross-presentation and the role of dendritic cell subsets. Immunol Cell Biol 86:353–362
van der Bruggen P, Van den Eynde BJ (2006) Processing and presentation of tumor antigens and vaccination strategies. Curr Opin Immunol 18:98–104
Berg M, Wingender G, Djandji D, Hegenbarth S, Momburg F, Hammerling G, Limmer A, Knolle P (2006) Cross-presentation of antigens from apoptotic tumor cells by liver sinusoidal endothelial cells leads to tumor-specific CD8+ T cell tolerance. Eur J Immunol 36:2960–2970
Blachere NE, Darnell RB, Albert ML (2005) Apoptotic cells deliver processed antigen to dendritic cells for cross-presentation. PLoS Biol 3:e185
Fonseca C, Dranoff G (2008) Capitalizing on the immunogenicity of dying tumor cells. Clin Cancer Res 14:1603–1608
Binder RJ, Kelly JB 3rd, Vatner RE, Srivastava PK (2007) Specific immunogenicity of heat shock protein gp96 derives from chaperoned antigenic peptides and not from contaminating proteins. J Immunol 179:7254–7261
Giodini A, Cresswell P (2008) Hsp90-mediated cytosolic refolding of exogenous proteins internalized by dendritic cells. EMBO J 27:201–211
Zeelenberg IS, Ostrowski M, Krumeich S, Bobrie A, Jancic C, Boissonnas A, Delcayre A, Le Pecq JB, Combadiere B, Amigorena S, Thery C (2008) Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res 68:1228–1235
Neijssen J, Herberts C, Drijfhout JW, Reits E, Janssen L, Neefjes J (2005) Cross-presentation by intercellular peptide transfer through gap junctions. Nature 434:83–88
Harshyne LA, Watkins SC, Gambotto A, Barratt-Boyes SM (2001) Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J Immunol 166:3717–3723
Dolan BP, Gibbs KD Jr, Ostrand-Rosenberg S (2006) Dendritic cells cross-dressed with peptide MHC class I complexes prime CD8+ T cells. J Immunol 177:6018–6024
Dolan BP, Gibbs KD Jr, Ostrand-Rosenberg S (2006) Tumor-specific CD4+ T cells are activated by “cross-dressed” dendritic cells presenting peptide-MHC class II complexes acquired from cell-based cancer vaccines. J Immunol 176:1447–1455
Melief CJ (2008) Cancer immunotherapy by dendritic cells. Immunity 29:372–383
van Mierlo GJ, Boonman ZF, Dumortier HM, den Boer AT, Fransen MF, Nouta J, van der Voort EI, Offringa R, Toes RE, Melief CJ (2004) Activation of dendritic cells that cross-present tumor-derived antigen licenses CD8+ CTL to cause tumor eradication. J Immunol 173:6753–6759
Gerner MY, Casey KA, Mescher MF (2008) Defective MHC class II presentation by dendritic cells limits CD4 T cell help for antitumor CD8 T cell responses. J Immunol 181:155–164
McDonnell AM, Prosser AC, van Bruggen I, Robinson BW, Currie AJ (2010) CD8alpha+ DC are not the sole subset cross-presenting cell-associated tumor antigens from a solid tumor. Eur J Immunol 40:1617–1627
Stoitzner P, Green LK, Jung JY, Price KM, Atarea H, Kivell B, Ronchese F (2008) Inefficient presentation of tumor-derived antigen by tumor-infiltrating dendritic cells. Cancer Immunol Immunother 57:1665–1673
Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gerard A, Krummel MF (2016) Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature 531:513–517
Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, Coussens LM (2014) Macrophage IL-10 blocks CD8(+) T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26:623–637
Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126:205–218
Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523:231–235
Segura E, Durand M, Amigorena S (2013) Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med 210:1035–1047
Desch AN, Gibbings SL, Clambey ET, Janssen WJ, Slansky JE, Kedl RM, Henson PM, Jakubzick C (2014) Dendritic cell subsets require cis-activation for cytotoxic CD8 T-cell induction. Nat Commun 5:4674
Melief CJ (2003) Mini-review: regulation of cytotoxic T lymphocyte responses by dendritic cells: peaceful coexistence of cross-priming and direct priming? Eur J Immunol 33:2645–2654
Steinman RM, Nussenzweig MC (2002) Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc Natl Acad Sci USA 99:351–358
Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL (2015) Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol 194:2985–2991
Karyampudi L, Lamichhane P, Scheid AD, Kalli KR, Shreeder B, Krempski JW, Behrens MD, Knutson KL (2014) Accumulation of memory precursor CD8 T cells in regressing tumors following combination therapy with vaccine and anti-PD-1 antibody. Cancer Res 74:2974–2985
Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM, Hartmann LC, Janco JM, Dong H, Hedin KE, Dietz AB, Goode EL, Knutson KL (2016) PD-1 blunts the function of ovarian tumor-infiltrating dendritic cells by inactivating NF-kappaB. Cancer Res 76:239–250
Munn DH, Bronte V (2016) Immune suppressive mechanisms in the tumor microenvironment. Curr Opin Immunol 39:1–6
Norian LA, Rodriguez PC, O’Mara LA, Zabaleta J, Ochoa AC, Cella M, Allen PM (2009) Tumor-infiltrating regulatory dendritic cells inhibit CD8 + T cell function via l-arginine metabolism. Cancer Res 69:3086–3094
Baban B, Chandler PR, Sharma MD, Pihkala J, Koni PA, Munn DH, Mellor AL (2009) IDO activates regulatory T cells and blocks their conversion into Th17-like T cells. J Immunol 183:2475–2483
Popov A, Schultze JL (2008) IDO-expressing regulatory dendritic cells in cancer and chronic infection. J Mol Med (Berl) 86:145–160
Munn DH, Mellor AL (2007) Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest 117:1147–1154
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268
Gabrilovich D (2004) Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol 4:941–952
Guilliams M, Malissen B (2015) A death notice for in-vitro-generated GM-CSF dendritic cells? Immunity 42:988–990
Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, Goubau D, Reis e Sousa C (2015) GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity 42:1197–1211
Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, Valladeau J, Davoust J, Palucka KA, Banchereau J (1999) In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med 190:1417–1426
Ishihara K, Hirano T (2002) IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev 13:357–368
Haura EB, Turkson J, Jove R (2005) Mechanisms of disease: insights into the emerging role of signal transducers and activators of transcription in cancer. Nat Clin Pract Oncol 2:315–324
Kim S, Takahashi H, Lin W-W, Descargues P, Grivennikov S, Kim Y, Luo J-L, Karin M (2009) Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457:102–106
Lin WW, Karin M (2007) A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest 117:1175–1183
Hunter CA, Jones SA (2015) IL-6 as a keystone cytokine in health and disease. Nat Immunol 16:448–457
Mauer J, Chaurasia B, Goldau J, Vogt MC, Ruud J, Nguyen KD, Theurich S, Hausen AC, Schmitz J, Bronneke HS, Estevez E, Allen TL, Mesaros A, Partridge L, Febbraio MA, Chawla A, Wunderlich FT, Bruning JC (2014) Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat Immunol 15:423–430
Naugler WE, Karin M (2008) The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med 14:109–119
Hong DS, Angelo LS, Kurzrock R (2007) Interleukin-6 and its receptor in cancer: implications for translational therapeutics. Cancer 110:1911–1928
Ji J, Shi J, Budhu A, Yu Z, Forgues M, Roessler S, Ambs S, Chen Y, Meltzer PS, Croce CM, Qin LX, Man K, Lo CM, Lee J, Ng IO, Fan J, Tang ZY, Sun HC, Wang XW (2009) MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med 361:1437–1447
Ratta M, Fagnoni F, Curti A, Vescovini R, Sansoni P, Oliviero B, Fogli M, Ferri E, Della Cuna GR, Tura S, Baccarani M, Lemoli RM (2002) Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood 100:230–237
Hegde S, Pahne J, Smola-Hess S (2004) Novel immunosuppressive properties of interleukin-6 in dendritic cells: inhibition of NF-kappaB binding activity and CCR7 expression. FASEB J 18:1439–1441
Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, Kamimura D, Ueda N, Iwakura Y, Ishihara K, Murakami M, Hirano T (2004) IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol 173:3844–3854
Kitamura H, Kamon H, Sawa S, Park SJ, Katunuma N, Ishihara K, Murakami M, Hirano T (2005) IL-6-STAT3 controls intracellular MHC class II alphabeta dimer level through cathepsin S activity in dendritic cells. Immunity 23:491–502
Chomarat P, Banchereau J, Davoust J, Palucka AK (2000) IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol 1:510–514
Bleier JI, Pillarisetty VG, Shah AB, DeMatteo RP (2004) Increased and long-term generation of dendritic cells with reduced function from IL-6-deficient bone marrow. J Immunol 172:7408–7416
Jenkins BJ, Grail D, Inglese M, Quilici C, Bozinovski S, Wong P, Ernst M (2004) Imbalanced gp130-dependent signaling in macrophages alters macrophage colony-stimulating factor responsiveness via regulation of c-fms expression. Mol Cell Biol 24:1453–1463
O’Garra A, Barrat FJ, Castro AG, Vicari A, Hawrylowicz C (2008) Strategies for use of IL-10 or its antagonists in human disease. Immunol Rev 223:114–131
Sredni B, Weil M, Khomenok G, Lebenthal I, Teitz S, Mardor Y, Ram Z, Orenstein A, Kershenovich A, Michowiz S, Cohen YI, Rappaport ZH, Freidkin I, Albeck M, Longo DL, Kalechman Y (2004) Ammonium trichloro(dioxoethylene-o, o′)tellurate (AS101) sensitizes tumors to chemotherapy by inhibiting the tumor interleukin 10 autocrine loop. Cancer Res 64:1843–1852
Yang AS, Lattime EC (2003) Tumor-induced interleukin 10 suppresses the ability of splenic dendritic cells to stimulate CD4 and CD8 T-cell responses. Cancer Res 63:2150–2157
Huang LY, Reis e Sousa C, Itoh Y, Inman J, Scott DE (2001) IL-12 induction by a TH1-inducing adjuvant in vivo: dendritic cell subsets and regulation by IL-10. J Immunol 167:1423–1430
Steinbrink K, Jonuleit H, Muller G, Schuler G, Knop J, Enk AH (1999) Interleukin-10-treated human dendritic cells induce a melanoma-antigen- specific anergy in CD8(+) T cells resulting in a failure to lyse tumor cells. Blood 93:1634–1642
Vicari AP, Chiodoni C, Vaure C, Ait-Yahia S, Dercamp C, Matsos F, Reynard O, Taverne C, Merle P, Colombo MP, O’Garra A, Trinchieri G, Caux C (2002) Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med 196:541–549
Kundu N, Fulton AM (1997) Interleukin-10 inhibits tumor metastasis, downregulates MHC class I, and enhances NK lysis. Cell Immunol 180:55–61
Kohno T, Mizukami H, Suzuki M, Saga Y, Takei Y, Shimpo M, Matsushita T, Okada T, Hanazono Y, Kume A, Sato I, Ozawa K (2003) Interleukin-10-mediated inhibition of angiogenesis and tumor growth in mice bearing VEGF-producing ovarian cancer. Cancer Res 63:5091–5094
Vuillefroy de Silly R, Ducimetiere L, Yacoub Maroun C, Dietrich PY, Derouazi M, Walker PR (2015) Phenotypic switch of CD8(+) T cells reactivated under hypoxia toward IL-10 secreting, poorly proliferative effector cells. Eur J Immunol 45:2263–2275
Ferrara N, Gerber HP, LeCouter J (2003) The biology of VEGF and its receptors. Nat Med 9:669–676
Toi M, Matsumoto T, Bando H (2001) Vascular endothelial growth factor: its prognostic, predictive, and therapeutic implications. Lancet Oncol 2:667–673
Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP (1996) Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med 2:1096–1103
Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, Gabrilovich DI (1998) Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-kappa B activation in hemopoietic progenitor cells. J Immunol 160:1224–1232
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer 8:579–591
Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, Carbone DP (1998) Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 92:4150–4166
Gabrilovich DI, Ishida T, Nadaf S, Ohm JE, Carbone DP (1999) Antibodies to vascular endothelial growth factor enhance the efficacy of cancer immunotherapy by improving endogenous dendritic cell function. Clin Cancer Res 5:2963–2970
Bowman T, Garcia R, Turkson J, Jove R (2000) STATs in oncogenesis. Oncogene 19:2474–2488
Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE Jr (1999) Stat3 as an oncogene. Cell 98:295–303
Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H (2002) Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21:2000–2008
Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D (2004) Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol 172:464–474
Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 10:48–54
Yu H, Kortylewski M, Pardoll D (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7:41–51
Yu H, Lee H, Herrmann A, Buettner R, Jove R (2014) Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14:736–746
Braun DA, Fribourg M, Sealfon SC (2013) Cytokine response is determined by duration of receptor and signal transducers and activators of transcription 3 (STAT3) activation. J Biol Chem 288:2986–2993
Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M, Hirano T, Chien KR, Yoshimura A (2003) IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol 4:551–556
Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 11:1314–1321
Hodge DR, Hurt EM, Farrar WL (2005) The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 41:2502–2512
Tang M, Diao J, Gu H, Khatri I, Zhao J, Cattral MS (2015) Toll-like receptor 2 activation promotes tumor dendritic cell dysfunction by regulating IL-6 and IL-10 receptor signaling. Cell Rep 13:2851–2864
Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, Manlongat N, Bender O, Kamradt T, Kuchroo VK, Gutierrez-Ramos JC, Coyle AJ, Strom TB (2003) Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol 4:1093–1101
Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, Bruce JN, Kane LP, Kuchroo VK, Hafler DA (2007) Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318:1141–1143
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M (2012) Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 13:832–842
Nagahara K, Arikawa T, Oomizu S, Kontani K, Nobumoto A, Tateno H, Watanabe K, Niki T, Katoh S, Miyake M, Nagahata S, Hirabayashi J, Kuchroo VK, Yamauchi A, Hirashima M (2008) Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J Immunol 181:7660–7669
Maurya N, Gujar R, Gupta M, Yadav V, Verma S, Sen P (2014) Immunoregulation of dendritic cells by the receptor T cell Ig and mucin protein-3 via Bruton’s tyrosine kinase and c-Src. J Immunol 193:3417–3425
Maroof A, English NR, Bedford PA, Gabrilovich DI, Knight SC (2005) Developing dendritic cells become ‘lacy’ cells packed with fat and glycogen. Immunology 115:473–483
Gao F, Liu C, Guo J, Sun W, Xian L, Bai D, Liu H, Cheng Y, Li B, Cui J, Zhang C, Cai J (2015) Radiation-driven lipid accumulation and dendritic cell dysfunction in cancer. Sci Rep 5:9613
Cubillos-Ruiz Juan R, Silberman Pedro C, Rutkowski Melanie R, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole Sarah E, Gupta D, Holcomb K, Ellenson Lora H, Caputo T, Lee A-H, Conejo-Garcia Jose R, Glimcher Laurie H (2015) ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161:1527–1538
Gardner JK, Mamotte CD, Patel P, Yeoh TL, Jackaman C, Nelson DJ (2015) Mesothelioma tumor cells modulate dendritic cell lipid content, phenotype and function. PLoS One 10:e0123563
Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, Mohammadyani D, Johnson JJ, Zhang LM, Klein-Seetharaman J, Celis E, Kagan VE, Gabrilovich DI (2014) Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol 192:2920–2931
Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, Mai J, Shen H, Hu DZ, Adoro S, Hu B, Song M, Tan C, Landis MD, Ferrari M, Shin SJ, Brown M, Chang JC, Liu XS, Glimcher LH (2014) XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 508:103–107
Hetz C, Chevet E, Harding HP (2013) Targeting the unfolded protein response in disease. Nat Rev Drug Discov 12:703–719
Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH (2003) Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci USA 100:9946–9951
Tang CH, Ranatunga S, Kriss CL, Cubitt CL, Tao J, Pinilla-Ibarz JA, Del Valle JR, Hu CC (2014) Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J Clin Invest 124:2585–2598
Iwakoshi NN, Pypaert M, Glimcher LH (2007) The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med 204:2267–2275
Osorio F, Tavernier SJ, Hoffmann E, Saeys Y, Martens L, Vetters J, Delrue I, De Rycke R, Parthoens E, Pouliot P, Iwawaki T, Janssens S, Lambrecht BN (2014) The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha + dendritic cells. Nat Immunol 15:248–257
Birkle S, Zeng G, Gao L, Yu RK, Aubry J (2003) Role of tumor-associated gangliosides in cancer progression. Biochimie 85:455–463
Palmano K, Rowan A, Guillermo R, Guan J, McJarrow P (2015) The role of gangliosides in neurodevelopment. Nutrients 7:3891–3913
Peguet-Navarro J, Sportouch M, Popa I, Berthier O, Schmitt D, Portoukalian J (2003) Gangliosides from human melanoma tumors impair dendritic cell differentiation from monocytes and induce their apoptosis. J Immunol 170:3488–3494
Shurin GV, Shurin MR, Bykovskaia S, Shogan J, Lotze MT, Barksdale EM Jr (2001) Neuroblastoma-derived gangliosides inhibit dendritic cell generation and function. Cancer Res 61:363–369
Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D (2012) RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J Clin Invest 122:1503–1518
Gorter A, Zijlmans HJ, van Gent H, Trimbos JB, Fleuren GJ, Jordanova ES (2010) Versican expression is associated with tumor-infiltrating CD8-positive T cells and infiltration depth in cervical cancer. Mod Pathol 23:1605–1615
Touab M, Villena J, Barranco C, Arumi-Uria M, Bassols A (2002) Versican is differentially expressed in human melanoma and may play a role in tumor development. Am J Pathol 160:549–557
Yang HZ, Cui B, Liu HZ, Mi S, Yan J, Yan HM, Hua F, Lin H, Cai WF, Xie WJ, Lv XX, Wang XX, Xin BM, Zhan QM, Hu ZW (2009) Blocking TLR2 activity attenuates pulmonary metastases of tumor. PLoS One 4:e6520
Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rebe C, Ghiringhelli F (2010) Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 120:457–471
Schwabe RF, Jobin C (2013) The microbiome and cancer. Nat Rev Cancer 13:800–812
Kroemer G, Galluzzi L, Kepp O, Zitvogel L (2013) Immunogenic cell death in cancer therapy. Annu Rev Immunol 31:51–72
Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G (2015) Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 28:690–714
Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Torne C, Weichert W, Engels K, Solbach C, Schrader I, Dietel M, von Minckwitz G (2010) Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol 28:105–113
Halama N, Michel S, Kloor M, Zoernig I, Benner A, Spille A, Pommerencke T, von Knebel DM, Folprecht G, Luber B, Feyen N, Martens UM, Beckhove P, Gnjatic S, Schirmacher P, Herpel E, Weitz J, Grabe N, Jaeger D (2011) Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res 71:5670–5677
Zitvogel L, Kroemer G (2015) Subversion of anticancer immunosurveillance by radiotherapy. Nat Immunol 16:1005–1007
Vacchelli E, Ma Y, Baracco EE, Sistigu A, Enot DP, Pietrocola F, Yang H, Adjemian S, Chaba K, Semeraro M, Signore M, Ninno AD, Lucarini V, Peschiaroli F, Businaro L, Gerardino A, Manic G, Ulas T, Günther P, Schultze JL, Kepp O, Stoll G, Lefebvre C, Mulot C, Castoldi F, Rusakiewicz S, Ladoire S, Apetoh L, Pedro JMB-S, Lucattelli M, Delarasse C, Boige V, Ducreux M, Delaloge S, Borg C, André F, Schiavoni G, Vitale I, Laurent-Puig P, Mattei F, Zitvogel L, Kroemer G (2015) Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 350:972–978
Berhanu A, Huang J, Alber SM, Watkins SC, Storkus WJ (2006) Combinational FLt3 ligand and granulocyte macrophage colony-stimulating factor treatment promotes enhanced tumor infiltration by dendritic cells and antitumor CD8(+) T-cell cross-priming but is ineffective as a therapy. Cancer Res 66:4895–4903
Miller G, Pillarisetty VG, Shah AB, Lahrs S, DeMatteo RP (2003) Murine Flt3 ligand expands distinct dendritic cells with both tolerogenic and immunogenic properties. J Immunol 170:3554–3564
Palucka K, Banchereau J (2013) Dendritic-cell-based therapeutic cancer vaccines. Immunity 39:38–48
Acknowledgments
We thank R. Gorczynski for helpful comments and advice. The authors declare no competing financial interests. This work was supported by the Canadian Institutes for Health Research (130438 to M.S.C.), Astellas Canada Inc., and the Toronto General Hospital Transplant Program. M.S.C. is a recipient of CIHR/Astellas Research Chair. M.T. is a recipient of a CIHR training award (121831).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tang, M., Diao, J. & Cattral, M.S. Molecular mechanisms involved in dendritic cell dysfunction in cancer. Cell. Mol. Life Sci. 74, 761–776 (2017). https://doi.org/10.1007/s00018-016-2317-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2317-8